

Abstract
Fragile X syndrome (FXS) is caused by the loss of the fragile X mental retardation protein (FMRP), an RNA binding protein that regulates translation of numerous target mRNAs, some of which are dendritically localized. Our previous biochemical studies using synaptoneurosomes demonstrate a role for FMRP and miR-125a in regulating the translation of PSD-95 mRNA. However, the local translation of PSD-95 mRNA within dendrites and spines, as well as the roles of FMRP or miR-125a, have not been directly studied. Herein, local synthesis of a Venus-PSD-95 fusion protein was directly visualized in dendrites and spines using single-molecule imaging of a diffusion-restricted Venus-PSD-95 reporter under control of the PSD-95 3′UTR. The basal translation rates of Venus-PSD-95 mRNA was increased in cultured hippocampal neurons from Fmr1 KO mice compared with WT neurons, which correlated with a transient elevation of endogenous PSD-95 within dendrites. Following mGluR stimulation with (S)-3,5-dihydroxyphenylglycine, the rate of Venus-PSD-95 mRNA translation increased rapidly in dendrites of WT hippocampal neurons, but not in those of Fmr1 KO neurons or when the binding site of miR125a, previously shown to bind PSD-95 3′UTR, was mutated. This study provides direct support for the hypothesis that local translation within dendrites and spines is dysregulated in FXS. Impairments in the regulated local synthesis of PSD-95, a critical regulator of synaptic structure and function, may affect the spatiotemporal control of PSD-95 levels and affect dendritic spine development and synaptic plasticity in FXS.
Introduction
Fragile X syndrome (FXS) is the most common inherited cause of intellectual disability and the most common monogenic cause of autism. FXS is caused by the loss of the fragile X mental retardation protein (FMRP), an mRNA binding protein that binds numerous mRNAs and often represses their translation, in addition to other modes of mRNA regulation. One of the targets of FMRP is PSD-95 mRNA, which is dendritically localized (Muddashetty et al., 2007; Zalfa et al., 2007; Subramanian et al., 2011). PSD-95 is the most abundant protein within the postsynaptic density (PSD), and it is thought to be a major organizer at the synapse (Gray et al., 2006). PSD-95 levels are dynamically regulated by new protein synthesis (Butko et al., 2012) and degradation (Colledge et al., 2003). Impaired synthesis and/or homeostasis of PSD-95 and other proteins could contribute to impaired synapse elimination and pruning (Tsai et al., 2012) or increased turnover and delayed stabilization of spines in Fmr1 KO mice (Cruz-Martín et al., 2010; Pan et al., 2010). Recently, it has been found that PSD-95 degradation, mediated by the ubiquitin proteasome system, is reduced in Fmr1 KO mice, suggesting altered PSD-95 dynamics in FXS. Analysis of PSD-95 mRNA levels in actively translating polysomes in synaptic fractions suggests an increase in basal translation in Fmr1 KO neurons (Muddashetty et al., 2007, 2011). Because PSD-95 mRNA is dendritically localized (Muddashetty et al., 2007; Zalfa et al., 2007; Subramanian et al., 2011), these studies suggest translational dysregulation of PSD-95 mRNA at synapses in FXS. Local translation of PSD-95 within dendrites has not been directly visualized, nor has it been possible using biochemical methods to analyze the spatiotemporal dynamics of PSD-95 synthesis in spines and assess possible impairments in FXS. In this study, we have used a single-molecule imaging approach that uses a Venus fluorescent protein (Venus)-based translation reporter to directly visualize the dendritic translation of Venus-PSD-95 mRNA as described for activity-regulated cytoskeleton-associated protein, Arc (Tatavarty et al., 2012; Barbarese et al., 2013). Using this single-molecule imaging assay, PSD-95 mRNA translation was visualized in dendrites and spines of live neurons. In mouse primary hippocampal neurons, basal translation of Venus-PSD-95 mRNA was increased in Fmr1 KO neurons compared with WT neurons, which correlated with transiently increased levels of endogenous PSD-95 within dendrites. In WT neurons, the rate of PSD-95 mRNA translation in dendrites was rapidly increased following mGluR stimulation with (S)-3,5-dihydroxyphenylglycine (DHPG), but this response was occluded in Fmr1 KO neurons or when the miR125a binding site in PSD-95 mRNA 3′UTR was mutated. These data directly reveal that PSD-95 mRNA is locally translated in dendrites, induced by mGluR activation and dysregulated in Fmr1 KO neurons, and further corroborate the roles of FMRP and miR125a in regulating PSD-95 mRNA translation (Muddashetty et al., 2011).
Materials and Methods
Constructs.
Venus PSD-95 plasmid, containing mouse PSD-95 5′UTR, open reading frame (ORF), 3′UTR, and Venus fluorescent protein ORF, situated upstream of PSD-95 ORF, was subcloned starting from the ECFP-boxB-3bact plasmid, which was kindly provided by Dr. Wilfried Rossoll, Emory University. ECFP was replaced with Venus fluorescent protein, which was inserted between AgeI and NotI (see Fig. 1A). The primers used were as follows: forward, 5′-CAGATCCGCTAGCGTCGCTACCGGTCGCCACCATGGTGAGCAAGGGCGAGG-3′; reverse, 5′-TTCAGGGCGGATCCCACTAGTTCATGCGGCGGCCGCGCCCTTGTCAGCTCGTCCATGC-3′. PSD-95 3′UTR was inserted between BglII and BspMII. PSD-95 ORF was inserted between NotI and SpeI. PSD-95 5′UTR was cloned by PCR from mouse DNA (the sequence was confirmed by sequencing), and it was inserted between NheI and AgeI. Because the PSD-95 N terminus contains two cysteines that are palmitoylated, a process that is necessary for PSD-95 membrane targeting (Topinka and Bredt, 1998), we inserted the first 10 amino acids, including cysteines 3 and 5, of PSD-95 ORF (ATGGACTGTCTCTGTATAGTGACAACCAAG), upstream of the Venus. The sequence was inserted by mutagenesis using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer's instructions using the following primers: forward, 5′-CCA ACA CCG GTC GCC ACC ATG GAC TGT CTC TGT ATA GTG ACA ACC AAG ATG GTG AGC AAG GGC GAG G-3′; reverse, 5′-CCT CGC CCT TGC TCA CCA TCT TGG TTG TCA CTA TAC AGA GAC AGT CCA TGG TGG CGA CCG GTG TTG G-3′.
λN22-mCherry3-M9 vector was subcloned from λN22-GFP3-M9 plasmid (Daigle and Ellenberg, 2007), by excising the three copies of GFP between AgeI and Bsp1407I and inserting sequentially three copies of mCherry between AgeI and NotI, NotI and SpeI, and SpeI and Bsp1407I, respectively (SpeI restriction site was removed from mCherry by silenced mutations affecting codon 2 and 3).
The following primers were used to introduce NotI and SpeI restriction sites: forward, 5′-GGATCCA CCG GTCGCCACCATGACCAGCAGCAAGGGCGAGGAGG-3′; reverse, 5′-TCTCTTGTACACGTATGACTAGTGCTTAGCGGCCGCGTACACTTATAGAGCTCGTCCATGCCG-3′).
Similarly, λN22-CFPX3-M9 vector was subcloned from λN22-GFP3-M9 plasmid (Daigle and Ellenberg, 2007) by inserting sequentially three copies of CFP between AgeI and NotI, NotI and SpeI, and SpeI and Bsp1407I, respectively.
Flag-mCherry-FMRP vector was previously reported (Gross et al., 2010).
His-Venus vector was made by subcloning Venus into pcDNA 3.1 between KpnI and NamHI restriction sites. The primers used were as follows: forward, 5′-GCT GCC ACC ATG CAT CAT CAC CAT GGT GAG CAA GGG-3′; reverse, 5′-AGC GGT GGA TCC TCA CTT GTA CAG CTC GTC CAT GCC G-3′.
miR125aMut was generated from Venus PSD-95 plasmid by mutating the seed sequence of miR125a binding site in PSD-95 3′UTR to disrupt miR125a binding, as shown previously (Muddashetty et al., 2011) using the following primers: forward, 5′-GGG GAG GGA TGG GTC TGG GGA GTG GGG AAT GTG GG-3′; reverse, 5′-CCC ACA TTC CCC ACT CCC CAG ACC CAT CCC TCC CC-3′.
Mutagenesis was performed using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer's instructions. All constructs were confirmed by sequencing (Beckman Coulter).
His-Venus purification and in vitro imaging.
N2A cells (50% confluent) were transfected with His-Venus pcDNA 3.1 using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions and incubated at 37°C overnight to allow for His-Venus expression. Cells were lysed and His-Venus was purified using Ni-NTA resin (QIAGEN) according to the manufacturer's instructions.
The glass coverslip of a glass-bottom culture dish (MatTek), treated for single-molecule imaging as described below, was covered with a diluted aqueous solution of His-Venus (∼2.5 μg/ml) and incubated for 1 min at room temperature. The His-Venus solution was removed, and the coverslip was rinsed five times with Milli-Q water (Millipore). Under these conditions, a small number of molecules still remain attached to the glass coverslip. The dish was dried and imaged immediately under the same settings used for single-molecule imaging in cells.
Cell culture and transfections.
Male Fmr1 KO and WT mice in C57BL/6J background were used (The Jackson Laboratory). Hippocampal neurons were isolated from WT or Fmr1 KO E16.5 mouse embryos as previously described (Gao et al., 2008) and plated on 35 mm glass-bottom culture dishes (MatTek) coated with poly-d-lysine (Sigma-Aldrich). For all single-molecule imaging experiments, dishes were cleaned as previously described (Tatavarty et al., 2009, 2012). Briefly, dishes were sonicated 30 min in EtOH, 30 min in 10% NaOH, and 30 min in Milli-Q water (Millipore). The dishes were coated with 0.1 mg/ml poly-d-lysine (Sigma-Aldrich) in borate buffer, incubated overnight at 37°C, rinsed three times with Milli-Q water, and dried. Neurons were maintained at 37°C and 5% CO2 for 12–16 d until transfection. Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After transfections, neurons were maintained overnight at 37°C and 5% CO2. Just before starting the imaging, the neuro-basal medium was replaced with Hibernate E low fluorescence (Brain Bits).
Single-molecule microscopy and imaging.
Single-molecule imaging of Venus-PSD-95 construct was done as previously described (Tatavarty et al., 2012; Barbarese et al., 2013). Single-molecule imaging was performed with a modified epifluorescence microscope Olympus IX71 (Olympus), equipped with 60×, NA = 1.45 microscope objective (Olympus), and an iXon EMCCD camera (Andor Technology). For single-molecule imaging of translation in live cells (SMI-TLC), Venus protein was excited with a 514 nm laser line (∼5 mW laser power) from a diode-pumped solid state laser (Spectra-Physics). The single-molecule imaging was done in epifluorescence mode. Following whole-cell photo-bleaching, single-molecule time-lapse images of Venus protein were collected continuously within a defined ROI at an exposure time of 150 ms per frame. To minimize the effects of photo-damage on the cells an illumination area of ∼10 μm diameter was used. Also, in drug treatment experiments in which a significant time (15 or 30 min) was required for the drug effect, the preexisting fluorescence was bleached and a 5–10 min continuous imaging-photo-bleaching protocol was used. Then the imaging (and laser exposure) was paused, the respective drug was added to the medium, incubated for 15 or 30 min, and the imaging restarted for another 5–10 min of the continuous imaging-photo-bleaching protocol. Because there is an increase of reporter signal in the illumination area during the incubation period, the first 1–2 min of time-lapse (until complete bleaching of preexisting fluorescence) was excluded from analysis. In contrast, in experiments where an acute (2–10 min) effect was investigated (DGPH treatment experiments), a continuous imaging-photo-bleaching protocol was used through the whole duration of the experiment.
Venus fluorescent protein is a fast folding variant of YFP (Nagai et al., 2002), which has biophysical properties that make it suitable for single-molecule imaging (Yu et al., 2006). Venus has a fast folding of ∼2 min in vitro and ∼7 min in bacteria (Xie et al., 2008), making it suitable for local mRNA translation events because the fast folding favors detection of Venus soon after translation and therefore close to the translation site. Venus also has fast bleaching kinetics (Xie et al., 2008), allowing for the maintenance of a low fluorescence level in the cell necessary for the detection of individual newly synthesized Venus molecules.
Image analysis.
Data analysis of time-lapse images was done as previously described (Yu et al., 2006; Tatavarty et al., 2012). Briefly, the centroid coordinates of individual molecules in each frame were determined and linked in time to construct temporal trajectories, using a standard single-particle tracking algorithm. Each Venus molecule detected was tracked from appearance (maturation) to disappearance (bleaching). For each molecule detected, the centroid coordinates and timing of its first appearance were recorded (see Fig. 1E). Preexisting Venus-PSD-95 fluorescent signal was used to manually define an ROI that contained neuronal dendrites, but not the cell body, for each dataset. From each dataset, translation events situated inside the ROI were analyzed with MATLAB (MathWorks). The analysis software is available at http://www.ccam.uchc.edu/yu. To visualize the collective locations where translation events were clustered, the centroid coordinates at the time of the first appearance for each newly detected Venus-PSD-95 molecule, apparent as light flashes, were recorded and plotted together to generate an event map (see Fig. 1F) (Tatavarty et al., 2012). To visualize the timing of appearance of new translation events, an event schedule (see Fig. 1G) was built by plotting the number of events detected in an ROI over time (Tatavarty et al., 2012). From these data, the translation rate in a dendritic area can be quantified.
To determine the spatial relationship between Venus-PSD-95 translation events and synapses, single-molecule imaging was performed, event maps were created, and subsequently the cells were fixed and immunocytochemistry was performed for synapsin and Venus. Synapsin immunocytochemistry images were used to create a mask that included synapsin puncta but excluded diffuse synapsin staining. Starting from this mask, ROIs extending from the periphery of the synapsin puncta in increments of 0.25 μm were drawn, up to 1 μm from synapsin puncta periphery. The events were classified based on whether they overlap with synapsin puncta or not and on the distance from each translation event to the periphery of the closest synapsin puncta. All the translation events detected at >1 μm from the synapsin puncta periphery were pooled together.
Dendritic fluorescence intensity for PSD-95, FMRP and synapsin was determined by averaging the fluorescence intensity along a line extending from the cell body to 50 μm from the cell body along the dendrite.
Immunocytochemistry.
Primary neurons in culture were fixed in 4% PFA for 20 min at room temperature, permeabilized with 0.1% NP40 (Sigma) for 90 s, and incubated in 10% horse serum for blocking and subsequently incubated with primary and secondary antibodies. Antibodies used were as follows: anti-FMRP (clone 1C3), anti-PSD-95 (clone 6G6-1C9) (Millipore), anti-synapsin (rabbit) (Sigma), and anti-GFP (chicken) (Aves Laboratories). Secondary antibodies were the following: goat anti-mouse IgG (H+L), DyLight 488 conjugate (Thermo Scientific), donkey anti-mouse AlexaFluor-647 conjugate, donkey anti-chicken AlexaFluor-488 conjugate (Jackson ImmunoResearch Laboratories), and donkey anti-rabbit, Alexa-546 conjugate (Thermo Scientific). Nuclei were stained with DAPI (1:1000) (Sigma).
Images were collected with a Nikon Eclipse epifluorescence microscope (Nikon).
Drug treatments.
The 50 μm S-DHPG (Tocris Bioscience) was used to activate mGluR in neurons. mRNA translation was inhibited by treatment with 40 μm anisomycin (Sigma) for 30 min, as previously described (Dieterich et al., 2010). TTX (Tocris Bioscience), used at a final concentration of 100 μm, was added to the medium to inhibit spontaneous action potentials.
Statistical analysis.
Each experiment was replicated at least three independent times. Normal distribution within all datasets was assessed using the Shapiro–Wilk's Test. Datasets that showed normal distribution was analyzed with Student's t test, general linear model (repeated measurements), or ANOVA (one-way, two-way). Datasets that did not display normal distribution were analyzed by the nonparametric Mann–Whitney or Kruskal–Wallis Tests. The statistical test used specifically to analyze each experiment is given in the corresponding figure legend. Significance threshold was set as p ≤ 0.05. All datasets are displayed as mean ± SEM. All datasets were analyzed using SPSS (IBM).
FISH.
FISH for PSD-95 ORF and GFP/Venus ORF was performed using Stellaris probes (Biosearch Technologies) according to the manufacturer's protocol and as previously reported (Chen et al., 2014) with slight modifications. Two pools of oligonucleotides (listed below) complementary to either a different region of PSD-95 ORF (48 oligonucleotides, each tagged with a single Quasar 670 dye on the 3′ end) or to GFP ORF (32 oligonucleotides, each tagged with a single TAMRA dye on the 3′ end) were designed using FISH design software and manufactured by Biosearch Technologies.
Hippocampal neurons were plated on glass coverslips that were treated as described above and coated with poly-d-lysine. Briefly, 14 DIV hippocampal neurons transfected with Venus-PSD-95 reporter, EGFP-C1 plasmid or mock-transfected were fixed with 4% PFA for 10 min and then washed 3 times in 1× PBS for 5 min each, once in 1× SSC buffer for 10 min, and once in prewarmed 10% formamide in 2× SSC for 5 min. The cells were incubated with prehybridization solution at 37°C for 1.5 h in a moisture chamber. The prehybridization solution is composed of equal parts of hybridization solution (for 50 μl hybridization solution: 10 μl dextran sulfate, 10 μl 20 mg/ml BSA, 10 μl 20× SSC, 5 μl RVC, 0.5 μl 10× PBS, 14.5 μl H2O) and carrier (for 15 μl carrier: 2 μl salmon sperm DNA, 2 μl tRNA, 3 μl formamide, 8 μl H2O). The cells were incubated with hybridization solution plus probes on parafilm (28 μl) at 37°C overnight in a moisture chamber. The hybridization solution is composed of equal parts of hybridization solution (as above) and carrier + probes (for 15 μl carrier + probes: 1 μl GFP “Cy3” probes, 1:20 in TE buffer, pH 8; 1 μ- PSD-95 “Cy5” probes, 1:20 in TE buffer, pH 8; 2 μl salmon sperm DNA; 2 μl tRNA; 3 μl formamide; 6 μl H2O). Cells were washed 2 times with prewarmed 10% formamide in 2× SSC for 20 min at 37°C, washed briefly three times with 2× SSC, followed by two 5 min washes in 2× SSC while rotating and then mounted on a coverslip using ProLong Gold antifade reagent (Invitrogen) as mounting medium.
PSD-95 Stellaris probes, labeled with Quasar 670 dye were (5′ to 3′) as follows: ctctatgatcttctcagctg, taggccctttgataagcttg, tgcgatgctgaagccaagtc, ctattatctccagggatgtg, ccttcgatgatcttggttac, aggatcttgtctccgatctg, tcatgcatgacatcctctag, atatgtgttcttcagggctg, ccacctttaggtacacaacg, catagctgtcactcaggtag, tatgaggttgtgatgtctgg, tagctgctatgactgatctc, tcaacaccattgaccgacag, tgttcatgactggcattgcg, tactgagcgatgatcgtgac, cgaatcggctatactcttct, ataagctgttcccgaagatc, tgatatagaagccccgcttg, ttgtcgtagtcaaacagggc, tcaagaaaccgcagtccttg, gctggcgtcaattacatgaa, catcggtctcactgtcagag, tttgctgggaatgaagccaa, tctcatagctcagaaccgag, aaggatgatgatggggcgag, agaagatcatcgttggcacg, aaacttgtcggggaactcgg, tcgtatgagggacacaggat, tatctcatattcccgcttag, cgggaggagacaaagtggta, tgaatgtccttctccatttt, cagcctcaatgaacttgtgc, tagaggtggctgttgtactg, gcattggctgagacatcaag, ggatgaagatggcgataggg, cgcttattgatctctagcac, agatctcttcaaagctgtcg, cttcgatgacacgtttcact; GFP Stellaris probes (used to detect Venus mRNA) labeled with Tamra were (5′ to 3′) as follows: tcctcgcccttgctcaccat, gggcaccaccccggtgaaca, cgccgtccagctcgaccagg, ctgaacttgtggccgtttac, gccctcgccctcgccggaca, tcagcttgccgtaggtggca, gtggtgcagatgaacttcag, ccagggcacgggcagcttgc, tcagggtggtcacgagggtg, ctgaagcactgcacgccgta, cttcatgtggtcggggtagc, cggacttgaagaagtcgtgc, tggacgtagccttcgggcat, cttgaagaagatggtgcgct, gggtcttgtagttgccgtcg, ccctcgaacttcacctcggc, gatgcggttcaccagggtgt, ttgaagtcgatgcccttcag, ccccaggatgttgccgtcct, tgtagttgtactccagcttg, catgatatagacgttgtggc, tgccgttcttctgcttgtcg, cggatcttgaagttcacctt, gctgccgtcctcgatgttgt, tggtagtggtcggcgagctg, gtcgccgatgggggtgttct, ttgtcgggcagcagcacggg, ggactgggtgctcaggtagt, cgttggggtctttgctcagg, aggaccatgtgatcgcgctt, ggcggcggtcacgaactcca, tcgtccatgccgagagtgat.
Results
PSD-95 mRNA translation occurs in dendrites and in spines
Previous studies have shown that PSD-95 mRNA is dendritically localized (Muddashetty et al., 2007; Zalfa et al., 2007; Subramanian et al., 2011) and translated in synaptoneurosomes in response to treatment with DHPG (Muddashetty et al., 2007, 2011). However, PSD-95 mRNA translation and response to stimuli within dendrites and spines have not been directly visualized. To directly visualize PSD-95 mRNA translation in dendrites of mouse primary hippocampal neurons, single-molecule imaging was performed using a Venus fluorescent protein (Venus)-based translation reporter containing a Venus-PSD-95 fusion ORF, between the PSD-95 5′ and 3′UTRs (Fig. 1A) as previously described for imaging Arc mRNA translation (Tatavarty et al., 2012). The reporter also contains four copies of box B phage sequence allowing visualization of mRNA when the neurons are cotransfected with Venus-PSD-95 and λN22-mCherry-M9 constructs (Fig. 1A) as previously described (Daigle and Ellenberg, 2007; Subramanian et al., 2011). Using this method and FISH, we detected Venus-PSD-95 mRNA granules in dendrites (shown later in Fig. 4C,F), confirming that our Venus-PSD-95 mRNA reporter was localized to dendrites. The construct design favors detection of Venus during the translation of mRNA encoding a Venus-PSD-95 fusion protein. The insertion of Venus ORF at the amino terminus allows Venus to mature and fluoresce while the nascent protein is still anchored to the ribosome, while translation of PSD-95 ORF is occurring (Fig. 1B). In addition, the PSD-95 N terminus (the first 10 amino acids), which contains two cysteines that are palmitoylated, and serve to direct PSD-95 to the cell membrane (Topinka and Bredt, 1998), was inserted at the N terminus of Venus ORF. Our interpretation that the flashes of light detected with a sensitive EM-CCD (<514 nm laser illumination) represent translation events is supported by several factors: in our experimental setting, the vast majority of newly detected fluorescent events appear and disappear within 1 s (Fig. 1E,I; see Figs. 2A, 3J) and show a one step appearance and one step disappearance that is characteristic for single-molecule detection (Figs. 1E,I; see Fig. 2A). It is unlikely that these transient detection events represent the detection of PSD-95 molecules that were synthesized at other locations in the neuron, as it has been shown that the majority of PSD-95 molecules are stably localized at the synapse. Although there is a minor pool of mobile PSD-95 molecules that is shared by neighboring spines, the median residence time of PSD-95 in the spines is ∼30 min (Gray et al., 2006). These kinetics are much longer than the duration of our acute measurements (<10 min) and much longer than the time a Venus-PSD-95 molecule is visualized before it is photo-bleached (<1 s). Molecules belonging to the minor mobile pool of Venus-PSD-95 that were synthesized previously but enter the illumination field already mature would be detected (and recorded) at the periphery of the illumination field (inside the red ring area, Fig. 1C), as they are excited and fluoresce, before being photo-bleached. Because there is an increased chance that molecules detected at the periphery of the illumination field (Fig. 1C, red ring) were not synthesized locally, these events were excluded from further analysis and quantification. Only the events detected at least 1 μm away from the periphery, inside the illumination field, were used for quantification (Fig. 1C, white circle). Thus, only Venus-PSD-95 molecules that become fluorescent within the interior illumination field (Fig. 1C, white circle) (the vast majority of which represent local Venus-PSD-95 translation) were used for quantitative analysis. Together, the construct design and experimental conditions favor detection of translation events directly as they appear in real time; however, we cannot rule out the possibility that some of the events represent detection of newly synthesized PSD-95 molecules that were made within several microns distance. Nonetheless, this method affords very high spatiotemporal resolution that is far superior to previous methods using FRAP or photoconvertible protein reporters that can only make distinction between newly synthesized proteins made in the cell body versus the dendrite.

PSD-95 mRNA translation occurs in dendrites and dendritic spines. A, Venus-PSD-95 translation reporter: Venus-PSD-95 fusion protein with PSD-95 ORF, 5′UTR, and 3′UTR. Blue circle represents miR-125a binding site. Ten amino acids representing the N terminus of PSD-95, which contains two cysteine residues shown to be palmitoylated and necessary for membrane targeting of PSD-95 (Topinka and Bredt, 1998), were inserted at the N terminus of Venus (red) to favor membrane targeting and thus limit diffusion. In the proximal 3′UTR, there are four copies of boxB phage sequence inserted for mRNA labeling by λN22 phage protein fused to three CFP and nuclear localization signal (M9) (see Fig. 4). B, Illustration depicting visualization of local translation of Venus-PSD-95 reporter within a dendrite. While PSD-95 ORF is still being translated, Venus ORF has time to start the maturation process to become fluorescent and thus increase the chances of Venus-PSD-95 reporter to be detected near its translation site. In addition, the N terminus of PSD-95, which contains two cysteine residues where PSD-95 is palmitoylated, could help to anchor Venus-PSD-95 to the cell membrane in the proximity of the translation site. C, Diagram depicting the experimental setup, with the illumination area (large outer circle) situated in an apical dendrite of a neuron expressing Venus-PSD-95. White interior circle represents the ROI used for analysis. To filter out detection of possible mobile Venus-PSD-95 molecules that might have been synthesized nearby, all Venus-PSD-95 molecules detected within the periphery of the illumination area (red ring) were excluded from analysis. D, Frame 1 of a representative time-lapse imaging of a dendrite of a 14 DIV hippocampal neuron transfected with Venus-PSD-95 translation reporter was used to determine the dendrite contour, before gradual photo-bleaching of preexisting Venus protein. A dendritic spine (white rectangle) was analyzed in panels E, G. Scale bar, 5 μm. E, Montage of the time-lapse imaging corresponding to the white rectangle in D. The four successive frames of time-lapse imaging were collected at ∼20 min after Frame 1 and are displayed in pseudo-color within a 36,300–35,000 pixel intensity value range, which allowed visualization of individual Venus-PSD-95 molecules as light flashes on a black background after the preexisting signal was photo-bleached (E, arrowheads). The full arrowheads indicate the appearance of three molecules in the same frame. Observe the single-molecule behavior of the particles: single-step appearance and single-step disappearance. Empty arrowhead at 300 ms indicates one of the molecules that appeared at 150 ms, which is assigned as being the same molecule even with a small displacement. The montage shows raw data images, without any processing, aside from contrast adjustment. Scale bar, 2 μm. F, Event map corresponding to the dendrite ROI shown in D. The positions of the first appearance of all Venus-PSD-95 molecules detected over a time interval (15 s here) were recorded and plotted generating an event map (Tatavarty et al., 2012). Each green dot indicates the location where each newly translated Venus-PSD-95 molecule was detected. Red dots (white arrows) indicate the individual Venus-PSD-95 molecules indicated by white arrows in E. The dendrite contour is outlined in white, based on the image shown in D. G, Enlarged inset of the event map corresponding to the white rectangle in D. H, Event schedule showing the detection of events in the dendrite ROI shown in D, F, over a 15 s period. Red bar represents the three molecules shown in E–G. I, Montage showing an ROI (1.5 μm/1.5 μm) from time-lapse imaging of a dendrite expressing Venus-PSD-95 over 100 frames (∼15 s at 0.15 ms interval) advances from left to right and top to bottom. The four discrete flashes of light detected represent single-Venus-PSD-95 molecules. Green squares represent the frame where the single molecule (light flash) was first detected, which was used to establish the position and the time of appearance of each Venus-PSD-95 molecule. The numbers on top and left side indicate the frame number within the montage. The montage shows raw data images, without any processing, aside from contrast adjustment. Scale bar, 1.5 μm.
Dendrites of neurons expressing Venus-PSD-95 were identified by the fluorescence of the preexisting Venus-PSD-95 (Fig. 1D). Before collecting time-lapse images at 150 ms per frame under the 514 nm laser illumination, preexisting Venus-PSD-95 was rapidly photo-bleached due to the fast bleaching kinetics of Venus fluorescent protein. Consequently, the fluorescence in the cell was markedly reduced allowing the detection and spatial mapping of newly synthesized Venus-PSD-95 molecules with single-molecule resolution (Fig. 1E,I). For each Venus-PSD-95 molecule that matures inside the observation field, the time of appearance (first frame in which the molecule was detected) (Fig. 1E, white arrowheads, I, green squares) and the x, y coordinates of its centroid (determined by fitting with a point spread function) were recorded (Fig. 1E). The x, y coordinates of each Venus-PSD-95 molecule were used to create an event map (Fig. 1F,G), which in turn allows for the visualization of the position where newly translated Venus-PSD-95 are detected, as previously described (Tatavarty et al., 2012). Plotting the time of events appearance over time was used to create a translation event schedule (Fig. 1H), as previously described (Tatavarty et al., 2012).
Single-molecule imaging of Venus-PSD-95 mRNA translation in 12–16 DIV mouse hippocampal neurons detected Venus-PSD-95 translation events in both dendrites and spines (Fig. 1F,G), suggesting that PSD-95 mRNA is translated in dendrites and spines.
To determine whether PSD-95 mRNA translation occurs at synapses, we performed single-molecule analysis of PSD-95 mRNA translation in apical dendrites of 12–16 DIV hippocampal neurons (Fig. 2A) and built an event map that indicated the location of translation sites (Fig. 2B, green). Subsequently, neurons were fixed and stained for the presynaptic marker synapsin (Fig. 2B, red) and Venus (Fig. 2B, blue). The Venus-PSD-95 event map and subsequent synapsin immunofluorescence images were aligned using the first Venus time-lapse image (Fig. 2B, blue) acquired at the beginning of the single-molecule imaging (before photo-bleaching), and the immunochemistry image of Venus acquired together with the immunofluorescence images (data not shown). The merged image (Fig. 2B, merged, ROI 1, ROI 2, ROI 3) revealed that many of the sites with high Venus-PSD-95 mRNA translation activity were in the proximity of synapsin clusters, which is a marker for synapses, suggesting that PSD-95 mRNA translation occurs in the proximity of synapses. To determine whether there is a relationship between the location of Venus-PSD-95 translation events and the synapses, we quantified the translation events situated in ROIs overlapping synapsin puncta or in ROI of 0.25 μm width concentrically drawn around synapsin puncta. All the translation events detected at >1 μm from the synapsin puncta periphery were pooled together. The quantification indicated that the vast majority (∼80%) of Venus-PSD-95 translation events were detected at <0.5 μm from a synapsin punctum (Fig. 2C), suggesting that PSD-95 mRNA translation occur near synapses.
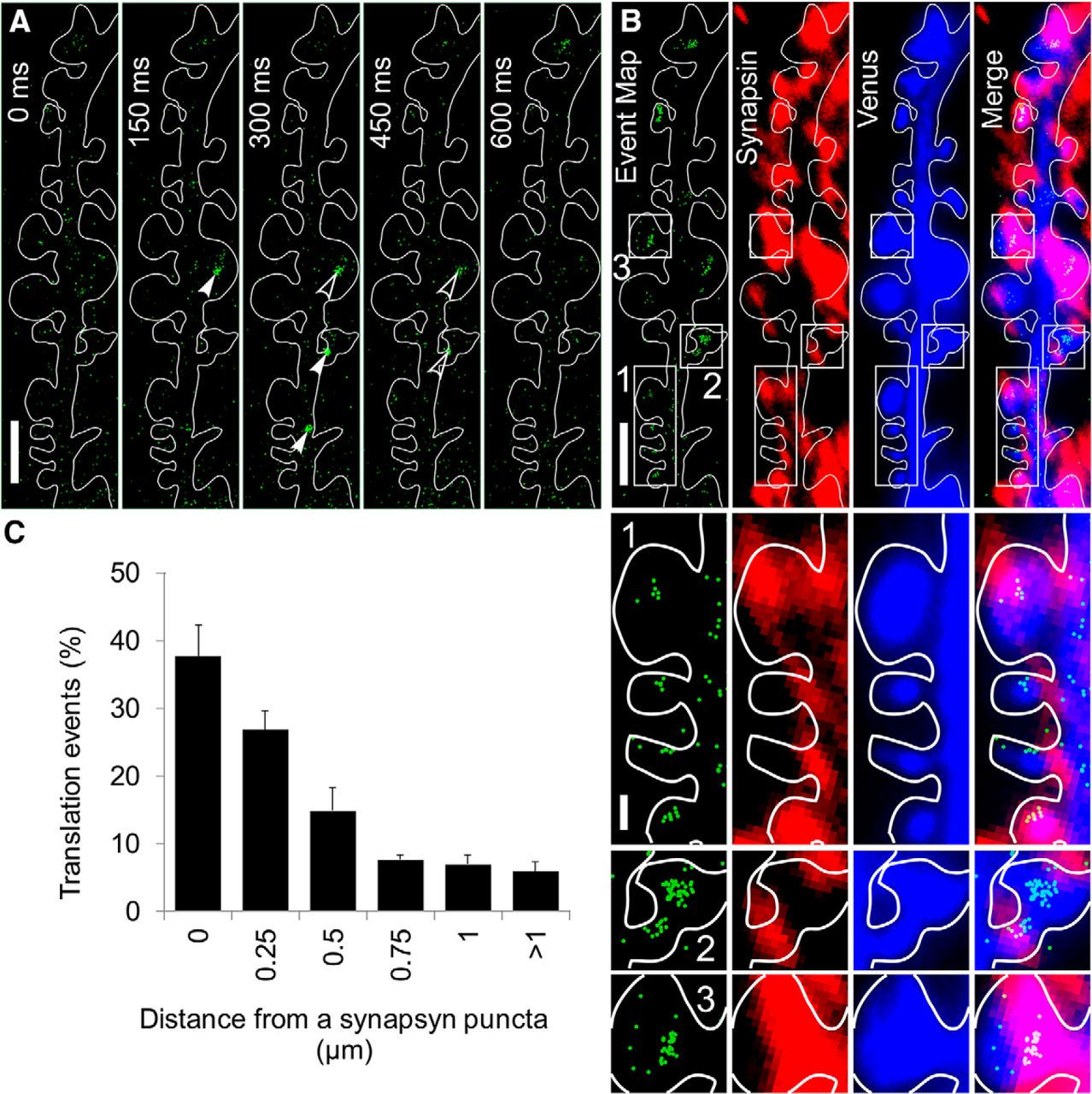
Venus-PSD-95 translation events are detected near synapses. A, Montage showing raw time-lapse images of a dendrite segment of a 14 DIV hippocampal neuron expressing Venus-PSD-95 reporter. Individual Venus-PSD-95 molecules are indicated by white arrowheads, in the first appearance frame, and by open arrowheads subsequently. The montage shows raw data images, without any processing, aside from contrast adjustment. Scale bar, 2.5 μm. B, Single-molecule analysis of Venus-PSD-95 translation was first performed in live cells to generate an event map (green) followed by immunostaining for synapsin (red) and Venus (blue). Venus signal in the first frame of the time-lapse and the Venus signal from the immunostaining image were used to determine the cell contour and to align and merge the event map (green) to the synapsin (red). Scale bar, 2.5 μm. Translation event clusters are visualized in dendritic shaft and in spines and near synapsin puncta. White rectangles labeled 1, 2, and 3 represent ROIs shown at higher magnification at B (bottom). Scale bar, 0.5 μm. C, Quantification of the spatial relationship between Venus-PSD-95 translation events and synapsin puncta. Venus-PSD-95 translation events were classified based on whether they overlap with synapsin puncta or not, and the distance from an event to the periphery of the closest synapsin punctum. The vast majority of the events is detected at <0.5 μm from a synapsin punctum. N = 5 dendrites, 3 different cultures, 2333 translation events quantified. Error bars indicate SEM.
To further validate that the flashes of light detected represent detection of Venus-PSD-95 mRNA single translation events, several controls were performed. Venus-PSD-95 translation was assessed before and after treatment with the translation inhibitor anisomycin. We observed the expected result of a decrease in translation after anisomycin treatment. Event maps (shown as heat maps), displaying Venus-PSD-95 mRNA translation over a period of 3 min, displayed a higher number of events before anisomycin than following 30 min treatment with anisomycin (Fig. 3A). The quantification of events over 3 min before anisomycin and after anisomycin treatment showed a decrease in the number of events detected, as expected, suggesting that the flashes of light detected represent bona-fide individual Venus-PSD-95 molecules (Fig. 3B). After anisomycin treatment, we still detected a significant number of events, which might represent anisomycin-insensitive translation since it has been previously shown that anisomycin does not completely abolish translation (Dieterich et al., 2010). Additionally, the noise level (events detected outside the cell, which might be produced by debris attached on the glass coverslip) was unchanged after anisomycin compared with the level before anisomycin (Fig. 3C). However, long exposure of dendrites to laser illumination could lead to photo damage of the dendrite, which would cause a decrease in the number of events observed in the cell, similar to the result observed after anisomycin treatment. To exclude this possibility, we performed the opposite experiment, in which we assess the number of events in cells treated with anisomycin (+ anisomycin) and after anisomycin (− anisomycin) was removed. After anisomycin was removed, we observed an increased number of events (Fig. 3D,E), arguing that the effect we see is due to anisomycin and not due to photo damage. In addition, to assess whether the laser exposure used has a significant photo-damaging effect, we recorded the translation events at time 0 (0 min) and 30 min later (30 min) (Fig. 3F). The number of events detected at 0 and 30 min was similar (Fig. 3F,G), suggesting that, in the imaging paradigm used, there is no significant photo damage. When spontaneous neuronal activity was blocked with TTX, a significant reduction in the number of events was detected (Fig. 3H,I), further suggesting that the flashes of light represent fluorescence of individual Venus-PSD-95 molecules, synthesized following spontaneous synaptic activity. The fluorescence intensity of the flashes of light detected in the transfected neurons was compared with that of purified individual Venus molecules imaged in vitro (on the glass coverslip of a MatTek dish) under the same image acquiring conditions as those used for live cells. The distribution of fluorescence was similar in both cases, further supporting that the flashes of light detected in neurons represent individual Venus-PSD-95 molecules (Fig. 3J). To further confirm that the flashes of light detected in neurons represent Venus-PSD-95 translation events, we measured the photo-bleaching time of the molecules detected <514 nm laser illumination that was used to excite Venus fluorescent protein. Venus-PSD-95 molecules displayed fast bleaching kinetics, most of them bleaching in <1 s, as have been previously shown for Venus (Yu et al., 2006; Tatavarty et al., 2012) (Fig. 3K). These lines of evidence show that the flashes of light detected in neurons represent individual Venus-PSD-95 molecules.

Venus-PSD-95 translation event rate decreases following treatment with anisomycin or TTX. A, Event maps (shown as heatmap) of events detected over 3 min, before addition of anisomycin (top) or 30 min after addition of anisomycin (bottom). Scale bar, 5 μm. B, Quantification of events (normalized) before addition of anisomycin (before anis) or 30 min after addition of anisomycin (after anis). N = 4. *p < 0.05 (t test). C, Quantification of extracellular noise (normalized) before addition of anisomycin (before anis) or 30 min after addition of anisomycin (after anis). N = 3. #p > 0.1 (t test). D, Event maps of events detected over 3 min, at 30 min after addition of anisomycin (+ anisomycin; top) or at 45 min after anisomycin was washed out (− anisomycin; bottom). Scale bar, 2 μm. E, Quantification of events (normalized) at 30 min after addition of anisomycin (+ anisomycin) and at 45 min after anisomycin was removed (− anisomycin). N = 3. *p < 0.05 (t test). F, Event maps of events detected over 3 min were created at time 0 (0 min) and after 30 min. DIC images were also collected at 0 and 30 min. Overlay of the first time-lapse frame (showing the dendrite outline; blue), the DIC image, and the corresponding event map, reveals similar event rate at 0 and 30 min (quantified in G), and no changes in the dendrite morphology as viewed in DIC. G, Quantification of events (normalized) detected in dendrites at time 0 (0 min) and after 30 min. N = 4. #p > 0.1 (t test). H, Event maps of events detected over 3 min were created before TTX and at 15 min after adding TTX to the medium (after TTX). DIC images were also collected before and after TTX. Overlay of the first time-lapse frame (showing the dendrite outline; blue), the DIC image, and the corresponding event map reveals decreased event rate after TTX (quantified in I), and no changes in the dendrite morphology as viewed in DIC. I, Quantification of events (normalized) before addition of TTX (before TTX) or 15 min after addition of TTX (after TTX). N = 3. *p < 0.05 (t test). J, Fluorescence intensity of individual particles detected in neurons was similar to that of purified individual Venus molecules imaged in vitro using identical parameters. K, Particles detected in neurons showed fast photo-bleaching kinetics, similar to that previously reported for Venus (Yu et al., 2006).
Venus-PSD-95 reporter protein and mRNA localize to dendrites
Venus-PSD-95 reporter, introduced in neurons by transfection, is expressed under the CMV promoter, creating the risk of overexpression at high levels and of mislocalization of both Venus-PSD-95 mRNA and protein. To determine whether the Venus-PSD-95 reporter accurately replicates the localization of the endogenous PSD-95, the localization and expression levels of Venus-PSD-95 mRNA and protein were compared with the localization and expression levels of endogenous PSD-95 mRNA and protein.
To determine the localization and expression levels of Venus-PSD-95 protein, 14 DIV hippocampal neurons were transfected with Venus-PSD-95 reporter or mock-transfected. After ∼15 h incubation at 37°C, 5% CO2, the cells were fixed, permeabilized, and immunochemistry was performed for PSD-95 and Venus (using antibodies against GFP). In cells expressing the Venus-PSD-95 reporter (Fig. 4A, right), an increased level of total PSD-95 protein was detected and the fluorescence intensity of PSD-95 puncta was increased (∼3-fold) compared with mock-transfected neurons (Fig. 4A,B). Additionally, Venus-PSD-95 protein was detected in puncta situated along the dendritic shaft and also in spines (Fig. 4A, right, white arrowheads), suggesting that the reporter is not mislocalized.

Expression and localization of Venus-PSD-95 in dendrites relative to endogenous PSD-95. A, Representative images of 14 DIV hippocampal neurons mock-transfected or transfected with Venus-PSD-95 reporter on which immunocytochemistry for PSD-95 and Venus (GFP) was performed. PSD-95 puncta intensity is quantified in B. Arrowheads indicate PSD-95 puncta where PSD-95 and Venus (GFP) signal overlap. Raw data epifluorescence images are shown, without any processing aside from contrast adjustment. Scale bars, 20 μm. B, Quantification of PSD-95 puncta intensity in mock- and Venus-PSD-95 reporter-transfected 14 DIV neurons. Three independent experiments done in three different cultures were quantified. N (Venus-PSD-95/mock) = 14/12 (neurons), 766/526 (PSD-95 puncta). *p < 0.05 (t test). C, Representative images of 14 DIV hippocampal neurons mock-transfected or transfected with Venus-PSD-95 reporter on which FISH for PSD-95 mRNA and Venus (GFP) mRNA was performed using Stellaris probes. PSD-95 mRNA FISH signal dendritic intensity at >50 μm from the cell body is quantified for both conditions in D. Raw data epifluorescence images are shown, without any processing, aside from contrast adjustment. Scale bars, 20 μm. D, Quantification of PSD-95 FISH signal intensity in mock- and Venus-PSD-95 reporter-transfected 14 DIV neurons. Three independent experiments done in three different cultures were quantified. N (Venus-PSD-95/mock) = 6/23 (neurons), 29/38 (dendrites). *p < 0.05 (t test). E, Diagrams of Venus-PSD-95 (top), λN22-CFP (middle), and mCherry-FMRP (bottom) constructs. Venus-PSD-95 mRNA contains four boxB sequences, which are bound by λN22-CFP (containing M9 nuclear localization signal). F, Dendrite of a 14 DIV neuron cotransfected with the constructs shown in E. Green represents Venus-PSD-95 mRNA labeled by λN22-CFP. Red represents mCherry-FMRP. Arrowheads indicate the Venus-PSD-95 mRNA granules that colocalize with mCherry-FMRP.
To determine the localization and expression levels of Venus-PSD-95 reporter mRNA, 14 DIV hippocampal neurons were transfected with Venus-PSD-95 reporter or mock-transfected. After ∼15 h incubation at 37°C, 5% CO2, the cell were fixed, and FISH was performed for PSD-95 ORF and Venus ORF mRNAs. The transfected cells were identified using the florescent signal of Venus-PSD-95 protein. In the transfected cells, fluorescent RNA granules labeled with both probes for PSD-95 ORF, and GFP probes (for Venus ORF) were detected in distal dendrites (Fig. 4C, right), indicating that Venus-PSD-95 reporter mRNA is localized to the dendrites. Because our single-molecule experiments were performed in distal dendrites, at >50 μm from the cell body, the fluorescence of PSD-95 FISH signal was quantified in distal dendritic segment at >50 μm from the cell body. In the distal dendrites (50–100 μm from the cell body) of neurons transfected with the Venus-PSD-95 reporter (Fig. 4C, right), PSD-95 mRNA FISH signal was increased ∼2.8-fold (quantified in Fig. 4D) compared with neurons that were mock-transfected (Fig. 4C, left), a level of overexpression that we consider would not significantly perturb the mRNA trafficking and the translation regulation.
PSD-95 mRNA has been shown to be localized in dendrites (Muddashetty et al., 2007; Zalfa et al., 2007), and FMRP is present in dendritic mRNA granules (Antar et al., 2005). However, FMRP has not been shown to colocalize with PSD-95 mRNA. To determine whether FMRP is present in PSD-95 mRNA granules, we cotransfected 12–15 DIV hippocampal neurons with plasmids expressing mCherry-FMRP, Venus-PSD-95 translation reporter, and λN22-CFP-M9, which binds the box B sequence in the Venus-PSD-95 reporter allowing for mRNA visualization (Fig. 4E) as previously described (Daigle and Ellenberg, 2007). Imaging of mCherry-FMRP and CFP-labeled Venus-PSD-95 mRNA granules in dendrites revealed colocalization (Fig. 4F), suggesting that FMRP could bind PSD-95 mRNA in dendrites and thus control PSD-95 mRNA translation in dendrites.
Loss of FMRP alters the developmental expression pattern of endogenous PSD-95 protein in hippocampal neurons
To determine the PSD-95 protein expression pattern in hippocampal neurons and assess the effect that loss of FMRP has on PSD-95 expression, the levels of endogenous PSD-95 and FMRP protein were determined by immunofluorescence at 7, 14, and 21 DIV in WT and Fmr1 KO hippocampal neurons. PSD-95 and FMRP fluorescence signal was measured in the proximal dendrites (first 50 μm from the cell body). In WT neurons, PSD-95 expression level increased moderately at 14 DIV followed by a robust increase at 21 DIV, compared with the lower expression level at 7 DIV. In Fmr1 KO neurons, PSD-95 expression levels are similar to WT neurons at 7 DIV, yet at 14 DIV display a precocious robust increase in PSD-95 expression, and at 21 DIV have expression levels that were similar to WT neurons (Fig. 5A,B).

Developmental pattern of endogenous PSD-95 protein expression is perturbed in Fmr1 KO hippocampal neurons. Hippocampal neurons at 7, 14, or 21 DIV were fixed and immunocytochemistry was performed as indicated. To determine the developmental level of expression of the indicated proteins, the dendritic average fluorescence intensity was measured along the proximal 50 μm of dendrites using a line scan. All the experiments were repeated at least three times using at least three different cultures. N represents the total number of neurons quantified per condition. A, Representative images of 7, 14, and 21 DIV WT and Fmr1 KO cultured hippocampal neurons from PSD-95 immunofluorescence. Scale bar, 10 μm. PSD-95 fluorescence intensity is shown as a heat map. B, Quantification of a representative PSD-95 immunofluorescence experiment. The experiments were repeated three times using three different cultures and compared by two-way ANOVA (p < 0.05). NWT (7/14/21 DIV) = 60/77/47; NKO (7/14/21 DIV) = 63/87/55. C, Dendritic FMRP levels in 7, 14, and 21 DIV WT hippocampal neurons. Representative images are shown. Scale bar, 10 μm. FMRP immunofluorescence intensity is shown as a heat map. D, Quantification of a representative FMRP immunofluorescence experiment is shown. The experiments were repeated three times, using different cell cultures. N (7/14/21 DIV) = 37/28/37 neurons in total. E, Dendritic synapsin (red) and PSD-95 (green) levels in 7, 14, and 21 DIV WT hippocampal neurons. Representative images are shown. Scale bar, 25 μm. F, Average of three synapsin immunofluorescence experiments from three different cell cultures. N (7/14/21 DIV) = 26/41/33 neurons in total. G, Higher magnification of the white rectangles shown in E, depicting synapsin (red) and PSD-95 (green) expression levels at 7, 14, and 21 DIV, respectively. Scale bar, 10 μm.
To assess how PSD-95 expression might be influenced by FMRP, we measured FMRP expression in WT hippocampal neurons. In WT hippocampal neurons at 7, 14, and 21 DIV, immunofluorescence was performed for FMRP and the fluorescence signal in the proximal dendrites (proximal 50 μm) and cell bodies was measured. At 7 DIV, FMRP expression was low, confined mostly to the cell body. At 14 DIV, there was a robust increase, which further increased moderately at 21 DIV (Fig. 5C,D). It has been shown that, during early neural development, PSD-95 expression is repressed post-transcriptionally by PTB1 and PTB2, which subsequently decline in expression (Zheng et al., 2012). This level of control over PSD-95 expression could explain the low PSD-95 protein levels observed at 7 DIV.
PSD-95 expression remained reduced at 14 DIV in WT neurons, whereas in Fmr1 KO neurons PSD-95 expression increased robustly at 14 DIV (Fig. 5A,B), suggesting that PSD-95 expression is repressed by FMRP. We further observed that, in contrast to PSD-95, synapsin levels increase significantly between 7 DIV and 14 DIV, suggesting that a large number of synapses form during this interval, whereas the levels of PSD-95 increase only modestly (Fig. 5E–G), likely due to repression by FMRP.
Basal translation of PSD-95 mRNA is increased in Fmr1 KO neurons
To directly visualize the spatiotemporal dynamics of PSD-95 synthesis, PSD-95 mRNA translation events were directly visualized by single-molecule imaging of Venus-PSD-95 translation reporter. In WT and Fmr1 KO mouse hippocampal neurons at 12–16 DIV, the basal PSD-95 mRNA translation rate was observed to be increased in Fmr1 KO neurons (Fig. 6B,D) compared with WT neurons (Fig. 6A,C). The number of translational events per minute in spines and at the base of spines was quantified using ROIs of equal size so that the translation rate could be compared between cells. The basal PSD-95 mRNA translation rate was increased in Fmr1 KO neurons in both spines and at the base of spines compared with WT neurons (Fig. 6E).

Basal translation of Venus-PSD-95 is increased in Fmr1 KO hippocampal neurons. Representative event maps showing the Venus-PSD-95 translation events detected over 5 min in a dendrite of a 12–14 DIV WT (A) and Fmr1 KO (B) hippocampal neuron. Each green dot represents the location where each newly translated Venus-PSD-95 molecule was first detected. The dendrite contour is outlined in white. Red circles represent the ROI where the translation rate was quantified in spines or in dendritic shaft. Scale bar, 1 μm. Representative event schedules showing Venus-PSD-95 translation in WT (C) and Fmr1 KO neurons (D). Spine (top) and base of spine (bottom). E, Quantification of Venus-PSD-95 translation rates (events/min) measured in spine and dendritic shaft for WT and Fmr1 KO hippocampal neurons at 13–15 DIV. *p < 0.05 (t test). WT events/min: spine, 0.6; spine base, 1.2; N = 13. KO events/min: spine, 1.4; spine base, 4.7; N = 14.
Stimulus-regulated PSD-95 mRNA translation is occluded in Fmr1 KO neurons
In a mouse model of FXS, in addition to increased basal translation rates of many FMRP target mRNAs, deficiency of FMRP leads to a loss of Group 1 metabotropic glutamate receptor (mGluR)-stimulated translation. PSD-95 mRNA translation has been shown to be dysregulated in synaptoneurosomes from Fmr1 KO mice (Muddashetty et al., 2011). However, the extent of translational dysregulation of PSD-95 mRNA translation within dendrites and spines in Fmr1 KO neurons has not been examined. To directly visualize the effect of mGluR activation on dendritic PSD-95 mRNA translation, single-molecule imaging was used to quantify the number of translational events before and after mGluR activation with DHPG. In WT mouse hippocampal neurons, the rate of PSD-95 mRNA translation in dendrites rapidly increased following mGluR stimulation with DHPG (Fig. 7B–F). Of note, we observed that the sites of DHPG-induced synthesis of PSD-95 (Fig. 7B, red spots) were often not colocalized with preexisting translation sites (Fig. 7B, green spots). By contrast, in Fmr1 KO hippocampal neurons, mGluR-stimulated translation was occluded (Fig. 7G–I). Increased basal translation rate and occluded mGluR-stimulated translation suggest that PSD-95 mRNA translation is dysregulated in Fmr1 KO neurons. Recent studies using biochemical methods in mouse synaptoneurosomes indicate that miR125a is involved in PSD-95 mRNA translation regulation as part of the RISC complex, which also contains Ago2 (Muddashetty et al., 2011). To assess the effect of miR125a in the regulation of dendritic PSD-95 mRNA translation, the seed sequence of the miR125a binding site in PSD-95 3′UTR was mutated to prevent binding of miR125a to the 3′UTR of the Venus-PSD-95 translation reporter, as previously described (Muddashetty et al., 2011) (Fig. 7A). In WT mouse hippocampal neurons, the translation rate of Venus-PSD-95 mRNA reporter with the miR125a binding site mutated (miR125a-Mut) in dendrites was unchanged following mGluR stimulation with DHPG (Fig. 7J–L) as also seen for the Venus-PSD-95 reporter in Fmr1 KO neurons. Together, this suggests that both FMRP and miR125a are necessary to inhibit PSD-95 mRNA translation at basal state and lack of either FMRP of miR125a derepresses this translation.

Activity-regulated translation of PSD-95 mRNA is occluded in Fmr1 KO mouse hippocampal neurons. A, WT and miR125aMut constructs. The miR125a binding site in the PSD-95 3′UTR was mutated as previously described (Muddashetty et al., 2011). B, Dendrite of a 14 DIV neuron expressing Venus-PSD-95 translation reporter. Single-molecule imaging was performed before and after DHPG treatment. Translational events detected under control conditions (green) and after DHPG treatment (red) were overlaid on the first image of the time-lapse. The dendrite countour (blue) is delineated by the Venus-PSD-95 protein accumulated in the cells before photo-bleaching. After mGluR stimulation with DHPG, new translation events are detected in proximity of sites where translation events were detected before DHPG treatment (open arrowheads) but also at new sites (white arrowheads). C, Event schedule corresponding to the dendrite shown in B. Green represents events detected under control conditions. Red represents events detected after DHPG treatment. D–L, Single-molecule imaging was performed in WT and Fmr1 KO neurons expressing Venus-PSD-95 reporter, and in WT neurons expressing miR125aMut reporter, before and after adding DHPG. Representative event schedules for each condition, which were not normalized are shown in D, G, J. The event rate before DHPG was used as an internal control, and the rates after DHPG were normalized to the rate before DHPG for each cell (E, F, H, I, K, L). D, Representative event schedule of a WT neuron showing the translation rate before and after DHPG treatment. E, Treatment with DHPG induces a rapid increase in normalized translation rate of PSD-95 mRNA in dendrites of WT neurons (13–15 DIV). N = 7. F, Data as in E, binned at 3 min intervals. N = 7. *p < 0.05 (t test). G, Representative event schedule of a Fmr1 KO neuron showing the translation rate before and after DHPG treatment. H, Treatment with DHPG did not change normalized translation rate of PSD-95 mRNA in dendrites of Fmr1 KO neurons (13–15 DIV). N = 4. I, Data as in H, binned at 3 min intervals. N = 4. *p < 0.05 (Student's t test). J, Representative events schedule of a WT neuron transfected with the miR125aMut construct showing the translation rate before and after DHPG treatment. K, Treatment with DHPG does not enhance translation rate of miR125aMut Venus-PSD-95 mRNA reporter in dendrites of WT neurons (13–15 DIV). N = 7. L, Data as in K, binned at 3 min intervals. N = 7. NS In all panels, error bars indicate SEM. *p < 0.05 (t test). N is shown for each experiment. Repeated-measures analysis shows that DHPG response in WT neurons (E) is significantly different from that in Fmr1 KO neurons (H) or when miR125a mutant construct is used (K). *p < 0.05.
Discussion
This study advances use of single-molecule imaging of Venus fluorescent protein-based translation reporters (Tatavarty et al., 2012) to establish that PSD-95 mRNA is locally translated in dendrites and spines and can be rapidly induced by mGluR activation. These data are consistent with a previous study using TimeSTAMP reporters, which revealed that newly synthesized PSD-95 is preferentially localized to stimulated synapses (Butko et al., 2012), although this method could not directly visualize translation in dendrites or at synapses. Herein we further show that local translation of PSD-95 mRNA is dysregulated in dendrites and spines of a mouse model of FXS, as evident by increased basal translation and occlusion of activity-regulated translation. These data are consistent with our past findings of PSD-95 mRNA translational dysregulation in biochemical studies using synaptoneurosomes (Muddashetty et al., 2007, 2011). Dysregulation of local protein synthesis for PSD-95 and other postsynaptic components (e.g., Arc) (Tatavarty et al., 2012) in dendritic spines has important implications for understanding how synapse development and synaptic plasticity are altered in FXS.
Protein synthesis has long been known to be required for long-term memory (Flexner et al., 1963). Local protein synthesis is thought to ensure high spatial and temporal accuracy for gene expression, allowing experience-dependent synapse-specific changes in dendritic spine structure and function, which are necessary for certain long-lasting forms of synaptic plasticity underlying learning and memory (Steward and Schuman, 2001). Protein synthesis has been shown to occur locally in dendrites mechanically separated from the cell body, both in vitro and in vivo, in response to plasticity-inducing stimuli (Kang and Schuman, 1996; Huber et al., 2000; Aakalu et al., 2001), arguing that synapse-specific changes following synaptic activation may depend on local protein synthesis as originally hypothesized (Steward and Levy, 1982). Loss of FMRP is known to disrupt several forms of protein synthesis-dependent synaptic plasticity (Ronesi and Huber, 2008; Bhakar et al., 2012). Because FMRP binds many target mRNAs encoding diverse types of proteins (Darnell and Klann, 2013), it will be important to assess how the dysregulation of specific FMRP target mRNAs each contributes to alterations in synapse function and structure, which might underlie some of the neurophysiologic phenotypes seen in FXS (Bhakar et al., 2012).
PSD-95 is a major synapse organizer that is required for activity-regulated formation of the postsynaptic density, and the stabilization of newly formed spines and synapses (Ehrlich et al., 2007; De Roo et al., 2008). Overexpression of PSD-95 can lead to increased synapse number and an increase in dendritic spine size when coupled to activity (El-Husseini et al., 2000). We speculate that elevated basal translation of PSD-95 mRNA could lead to inappropriately higher levels of PSD-95, which could accelerate synapse development. This model is consistent with morphologic and neurophysiologic evidence for increased synapse number in Fmr1 KO neurons. Cultured neurons from Fmr1 KO mice have increased density of synapses, as evident by immunofluorescence of synaptic markers and electrophysiological findings of increased mEPSC frequency (Pfeiffer and Huber, 2007). Organotypic slice cultures from hippocampus of Fmr1 KO mice also show increased mEPSC frequency and increased evoked EPSCs (Zang et al., 2013). Although we speculate that elevated basal levels of PSD-95 in dendrites may contribute to precocious synapse development, the loss of activity-regulated synthesis of PSD-95 may impair activity-regulated dendritic spine stabilization and maturation, which could depend on the ability of a synapse to synthesize PSD-95 and other proteins with spatiotemporal control. Recent studies suggest that Fmr1 KO neurons have transient and unstable spines (Cruz-Martín et al., 2010; Pan et al., 2010) but loss of glutamate-dependent elongation (Cruz-Martín et al., 2012). Moreover, several studies have observed that spines are immature in FXS; there are more long and thin spines and fewer mushroom-shaped spines (Galvez and Greenough, 2005; de Vrij et al., 2008; Swanger et al., 2011; He and Portera-Cailliau, 2013). Because the presence of polyribosomes within spines is correlated with activity-dependent regulation of mature shaped spines (Ostroff et al., 2002), it will be interesting to assess how loss of the activity-regulated local synthesis of PSD-95 and other postsynaptic components may contribute to immature spine morphology in FXS. PSD-95 is also involved in regulated AMPA receptor endocytosis (El-Husseini Ael et al., 2002; Shepherd and Huganir, 2007; Shepherd and Bear, 2011). Excess internalization of AMPA receptors is observed in Fmr1 KO neurons (Nakamoto et al., 2007) and drives the exaggerated mGlu-LTD (Huber et al., 2002). The significance of mGlu-induced synthesis of PSD-95 for LTD remains unclear. It will be of interest to investigate whether loss of activity-regulated synthesis of PSD-95 in Fmr1 KO is compromised in LTP in general, such as during glycine-LTP, which is altered in Fmr1 KO (Shang et al., 2009).
The present study provides new insight into the molecular mechanism and dynamics of local protein synthesis in response to neuronal signaling. In order for local protein synthesis to provide spatial and temporal control of gene expression, specific molecular events have to occur to ensure that translation occurs only when and where it is required. Numerous studies on translation regulation mechanisms have identified the role of RNA binding proteins and microRNAs (Siegel et al., 2011; Swanger and Bassell, 2012; Xing et al., 2012) as major regulators of translation of mRNAs coding for synaptic glutamate receptors, ion channels, proteins with a role in actin dynamics, or scaffolding proteins. These mechanisms involve translation inhibition by microRNAs (Schratt et al., 2006; Ho et al., 2014), yet the mechanisms whereby specific factors disinhibit the microRNA remain unclear. Two studies have revealed interactions between mRNA binding proteins and microRNAs that regulate dendritic mRNA translation. FMRP was shown to cooperate with miR-125a in a reversibly regulated manner to regulate PSD-95 mRNA translation (Muddashetty et al., 2011), whereas competitive interactions between HuD and miR-129 promote translation of Kv1.1 mRNA in dendrites in response to mTOR inhibition (Sosanya et al., 2013). The present study using a Venus reporter provides direct evidence that mutation of a microRNA binding site can impair mRNA translation regulation in dendrites, which was also observed for Kv1.1 mRNA (Sosanya et al., 2013). Because FMRP, miR-125a, and PSD-95 mRNA are all present in dendrites, the data suggest that the mechanisms to remove translational inhibition occur rapidly within dendrites in response to mGluR activation (Fig. 8). Here we detected increased PSD-95 mRNA translation within 2 min of DHPG treatment. This is consistent with past studies showing that mGluR activation rapidly leads to FMRP dephoshorylation, within 30 s, as a translation switch to remove translational inhibition (Narayanan et al., 2007). There may be series of molecular events that follow FMRP dephosphorylation and remove translational inhibition, including FMRP ubiquitination and ubiquitin proteasome system-mediated degradation (Nalavadi et al., 2012) and removal of RISC/miR-125a from PSD-95 mRNA (Muddashetty et al., 2011). Recently, it was shown that mGluR activation could stimulate dendritic protein synthesis by reactivation of stalled ribosomes (Graber et al., 2013). Because FMRP can inhibit translation elongation (Darnell et al., 2011), it will be interesting to assess the role of mGluR-induced dephosphorylation of FMRP on this mechanism.

Model for regulation of PSD-95 mRNA translation at the synapse. At the synapse, PSD-95 mRNA is in two distinct states: (1) At resting state, PSD-95 mRNA is bound by FMRP, which represses its translation in conjunction with RISC complex (which contains AGO2 and miR-125a–5p). (2) Upon activation of mGluRs, FMRP is dephosphorylated and subsequently ubiquitinated and degraded. Dephosphorylation of FMRP leads to disassembly of RISC complex and derepression of PSD-95 mRNA translation. Translation of Fmr1 mRNA present at the synapse replenishes the pool of FMRP at the synapse, leading to return to resting state (1).
To understand the normal synaptic changes that occur during long-lasting forms of synaptic plasticity and how these are altered in FXS, there is a need to visualize local translation of dendritically localized mRNA species, under stimulation paradigms, for both WT and Fmr1 KO neurons. The analysis of local protein synthesis in neurons has been the focus of numerous studies, which used various methods to assess mRNA translation. However, most methods used present disadvantages. GFP-based translation reporters have the disadvantage that, given the long maturation time, GFP-fused molecules are going to be detected a long time after translation; therefore, they can move significantly from the translation site, yielding low spatial and temporal resolution. Detection of the location of protein synthesis can be achieved using a tetracysteine peptide at the amino terminus of a GFP-β-actin fusion protein, which can be subsequently bound by the biarsenical dye ReAsh (Rodriguez et al., 2006). However, this method does not allow for the quantification of the translation rate in live cells. In contrast, single-molecule imaging of translation in live cells (Tatavarty et al., 2012) using Venus fluorescent protein-based reporters is suitable for analysis of local mRNA translation in neurons because of its excellent spatial and temporal resolution and because it allows visualization of translation of a single molecular species of interest at a time and quantification of translation rate with single-molecule resolution. Here, we have used Venus reporters to demonstrate the role of FMRP and miR-125a in the translation of PSD-95 mRNA in dendrites, and we provide direct evidence for dysregulated translation of PSD-95 mRNA in dendrites and spines of FXS. The inability of synapses to have spatiotemporal control of protein synthesis likely contributes to more general impairments in activity-dependent control of synapse development and plasticity in FXS.
Footnotes
This work was supported by National Institutes of Health Grants R21NS091038 and 1R21DA033478 to G.J.B., in part by the Emory University Integrated Cellular Imaging Microscopy Core of the Emory Neuroscience, and National Institute of Neurological Disorders and Stroke Core Facilities Grant P30NS055077. We thank Dr. Ivan Rasnik for assistance with collecting single-molecule data; Dr. Ji Yu for providing the software used to analyze single-molecule data; Dr. Jan Ellenberg for kindly providing the λN22-3XGFP-M9 plasmid; Dr. Wilfried Rossoll for kindly providing the ECFP-boxB-bact3 plasmid; and Dr. Bart Anderson and Kristen Thomas for critically reading the manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Gary J. Bassell, Emory University School of Medicine, Whitehead Biomedical Research Building, Room 415, 615 Michael Street NE, Atlanta, GA 30322. gary.bassell{at}emory.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}