

Abstract
The neurotrophin brain-derived neurotrophic factor (BDNF) mediates activity-dependent long-term changes of synaptic strength in the CNS. The effects of BDNF are partly mediated by stimulation of local translation, with consequent alterations in the synaptic proteome. The ubiquitin-proteasome system (UPS) also plays an important role in protein homeostasis at the synapse by regulating synaptic activity. However, whether BDNF acts on the UPS to mediate the effects on long-term synaptic potentiation (LTP) has not been investigated. In the present study, we show similar and nonadditive effects of BDNF and proteasome inhibition on the early phase of synaptic potentiation (E-LTP) induced by theta-burst stimulation of rat hippocampal CA1 synapses. The effects of BDNF were blocked by the proteasome activator IU1, suggesting that the neurotrophin acts by decreasing proteasome activity. Accordingly, BDNF downregulated the proteasome activity in cultured hippocampal neurons and in hippocampal synaptoneurosomes. Furthermore, BDNF increased the activity of the deubiquitinating enzyme UchL1 in synaptoneurosomes and upregulated free ubiquitin. In contrast to the effects on posttetanic potentiation, proteasome activity was required for BDNF-mediated LTP. These results show a novel role for BDNF in UPS regulation at the synapse, which is likely to act together with the increased translation activity in the regulation of the synaptic proteome during E-LTP.
Introduction
The neurotrophin brain-derived neurotrophic factor (BDNF) is released to the synapse in response to neuronal activity (Hartmann et al., 2001; Matsuda et al., 2009) and activation of TrkB receptors for BDNF plays a role in the early and late phases of synaptic potentiation induced by high-frequency stimulation of hippocampal Schaffer collateral-CA1 synapses (Korte et al., 1995; Korte et al., 1996; Minichiello et al., 1999). Translation inhibitors abrogate BDNF-induced facilitation of CA1 synapses (Kang and Schuman, 1996) and protein synthesis is also required for induction and consolidation of long-term potentiation (LTP) elicited by local infusion of BDNF in the dentate gyrus of anesthetized rats (Messaoudi et al., 2007). However, application of mature BDNF to hippocampal slices was sufficient to maintain late LTP (L-LTP) in the presence of the protein synthesis inhibitor anisomycin (Pang et al., 2004), suggesting that translation-independent mechanisms may also be involved in synaptic potentiation by BDNF (Mei et al., 2011).
The ubiquitin-proteasome system (UPS) plays a key role in the modulation of synaptic transmission and synaptic plasticity in the CNS, showing that a fine tuning of protein half-life is instrumental in activity-dependent synaptic changes (Tai and Schuman, 2008; Mabb and Ehlers, 2010; Jarome and Helmstetter, 2013; Tsai, 2014). Inhibition of the proteasome was shown to downregulate the late L-LTP in CA1 synapses (Fonseca et al., 2006; Karpova et al., 2006; Dong et al., 2008), which is also dependent on transcription (Dong et al., 2008) and translation activity (Fonseca et al., 2006; Karpova et al., 2006; Dong et al., 2014). The interplay between synaptic activity and the UPS is bidirectional because changes in neuronal activity are also known to regulate proteasome activity (Bingol and Schuman, 2006; Djakovic et al., 2009; Djakovic et al., 2012; Caldeira et al., 2013) and protein ubiquitination (Ehlers, 2003; Hou et al., 2011), leading to changes in abundance of key proteins at the synapse (Colledge et al., 2003; Ehlers, 2003; Bingol and Schuman, 2004).
Although the effects of BDNF on LTP have been attributed to posttranslational modifications of existing proteins and to an upregulation of translation activity, the effect on protein degradation has not yet been addressed. In this study, we show similar and nonadditive effects of proteasome inhibition and BDNF on the early phase of LTP (E-LTP), suggesting that BDNF may act by downregulating the overall activity of the UPS. Accordingly, the effects of BDNF on the L-LTP were abrogated upon activation of the proteasome. Furthermore, biochemical experiments showed a BDNF-induced transient reduction of the proteasome activity in cultured hippocampal neurons and in hippocampal synaptoneurosomes, together with a downregulation of free ubiquitin levels and an increase in the deubiquitinating activity of ubiquitin C-terminal hydrolase-L1 (UchL1). In addition to the role in E-LTP, proteasome activity was found to be required for the facilitatory effect of BDNF on the late-phase of synaptic LTP. Altogether, these findings show a novel role for BDNF in regulating the UPS at the synapse, which is likely to affect the synaptic proteome during the E-LTP with impact on L-LTP maintenance.
Materials and Methods
Ex vivo electrophysiological recordings
Hippocampi from 3-week-old Wistar male rats were dissected in ice-cold Krebs solution composed of the following (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 MgSO4, 2 CaCl2, and 10 glucose, pH 7.4, previously gassed with 95% O2 and 5% CO2. Slices (400 μm thick) were cut perpendicularly to the long axis of the hippocampus with a McIlwain tissue chopper and allowed to recover functionally and energetically for 1 h in a resting chamber filled with the same solution at room temperature. Slices were transferred to a recording chamber for submerged slices and continuously superfused at 3 ml/min with gassed bathing solution at 32°C; drugs were added to this superfusion solution in a closed circuit. Recordings were obtained with an Axoclamp 2B amplifier and digitized (Molecular Devices). Individual responses were monitored and averages of eight consecutive responses were continuously stored on a personal computer with the LTP software (Anderson and Collingridge, 2001).
LTP induction and quantification
For recording of field EPSPs (fEPSPs) in the CA1 region of the hippocampus, a microelectrode (4 m NaCl, 2–6 MO resistance) was placed in stratum radiatum of CA1. LTP was induced by a θ-burst protocol consisting of 1 train of 15 bursts with 4 pulses each (100 Hz, 200 ms interburst interval). The intensity of the stimulus was adjusted to give ∼30% of the maximal fEPSP slope and maintained during the induction protocol. The extent of LTP was expressed as the percentage increase in the fEPSP slope relative to the baseline (10 min preceding LTP induction) at different time points, as mentioned in the figure captions. To test the effect of BDNF upon LTP, 20 ng/ml BDNF (Peprotech) was added to the superfused bath at least 30 min before induction of LTP and remained in the bath up to the end of the experiment. For experiments in which proteasome activity was inhibited using clasto-lactacystin β-lactone (25 μm in DMSO; Calbiochem), slices were preincubated with the drug for 30 min. Because the effect of clasto-lactacystin β-lactone is irreversible, the inhibition of proteasome activity remained until the end of the experiments. The final concentration of DMSO was kept at 0.02%. In the experiments testing the effect of the proteasome activator IU1 (1-[1-(4-fluorophenyl)-2,5-dimethylpyrrol-3 yl]-2-pyrrolidin-1-ylethanone) (100 μm), the slices were preincubated with the compound for 4 h. Because the effect of IU1 is reversible, this drug was superfused in the bath during the entire experiment.
Cell culture and subcellular fractionation
Hippocampal cultures.
Primary cultures of rat hippocampal neurons were prepared from the hippocampi of embryonic day 18 (E18)-E19 Wistar rat embryos after treatment with trypsin (1.5 mg/ml, 15 min, 37°C; Invitrogen) in Ca2+- and Mg2+-free Hank's balanced salt solution (HBSS) containing the following (in mm): 5.36 KCl, 0.44 KH2PO4, 137 NaCl, 4.16 NaHCO3, 0.34 Na2HPO4·2H2O, 5 glucose, 1 sodium pyruvate, and 10 HEPES plus 0.001% phenol red. The hippocampi were washed with HBSS containing 10% fetal bovine serum (Invitrogen) to stop trypsin activity, further washed with HBSS to avoid the development of glial cells, and finally transferred to Neurobasal medium (Invitrogen) supplemented with B27 supplement (1:50 dilution; Invitrogen), 25 μm glutamate, 0.5 mm glutamine, and 0.12 mg/ml gentamycin. For biochemistry studies, cells were cultured at a density of 90 × 103 cells/cm2 in 6-well plates. Cultures were maintained in Neurobasal medium supplemented with B27 supplement in a humidified incubator of 5% CO2/95% air at 37°C.
Cells were stimulated with 100 ng/ml BDNF for the indicated periods of time. This concentration of BDNF is typically used to study biological responses to this neurotrophin (Almeida et al., 2005; Jeanneteau et al., 2008). Where indicated, 200 nm K252a (Sigma) was added to the culture medium 30 min before stimulation.
Synaptoneurosome preparation.
Synaptoneurosomes were prepared as described previously with slight modifications (Yin et al., 2002). Briefly, 6–8 hippocampi were dissected from adult Wistar rats and the tissue was minced with scissors and homogenized with a glass homogenizer in a buffer containing 0.32 m sucrose, 10 mm HEPES-Tris, pH 7.4, and 0.1 mm EGTA. After centrifugation for 3 min at 1000 × g, the supernatant was collected and passed initially through nylon membranes (150 and 50 μm; VWR) and finally through an 8 μm pore size filter (Millipore). The flow-through was centrifuged for 15 min at 10,000 × g and the pellet was resuspended in incubation buffer containing the following (in mm): 8 KCl, 3 CaCl2, 5 Na2HPO4, 2 MgCl2, 33 Tris, 72 NaCl, and 100 sucrose, pH 7.5. The entire procedure was done at 4°C. Incubations were performed in a water bath at 30°C for the periods of time indicated. BDNF (100 ng/ml) was added to synaptoneurosomes and, for each time point considered, a control experiment was also performed. When indicated, synaptoneurosomes were preincubated with K252a (200 nm) for 10 min.
RNA extraction and quantitative PCR
Total RNA from 7 d in vitro cultured hippocampal neurons was extracted with TRIzol reagent (Invitrogen) following the manufacturer's specifications. Reverse transcription reaction, quantitative PCR, and analysis were performed as described previously (Santos and Duarte, 2008).
Proteasome activity assay
Cultured hippocampal neurons or synaptoneurosomes were lysed in 1 mm EDTA, 10 mm Tris-HCl, pH 7.5, 20% glycerol, 4 mm dithyothreitol (DTT), and 2 mm ATP. The samples were frozen in liquid nitrogen, thawed, and centrifuged at 12,100 × g for 10 min at 4°C. The amount of protein in the supernatant fractions was quantified using the Bradford method. Peptidase activities of the proteasome were assayed by monitoring the production of 7-amino-4-methylcoumarin (AMC) from fluorogenic peptides (Peptide Institute): Suc-LLVY-AMC (chymotrypsin-like activity), Z-LLE-AMC (caspase-like activity), and Boc-LRR-AMC (trypsin-like activity). Samples (20 μg for chymotrypsin-like and caspase-like activities; 30 μg for trypsin-like activity) were incubated with the fluorogenic substrates 50 μm Suc-LLVY-AMC, 400 μm Z-LLE-AMC, or 100 μm Boc-LLR-AMC in 50 mm Tris-HCl, pH 8.0, and 0.5 mm EDTA buffer, supplemented with 2 mm ATP, in a final volume of 100 μl. In parallel, the nonspecific cleavage of the substrates due to the activity of other enzymes was measured in the presence of 10 μm MG-132 (Calbiochem). The release of fluorescent AMC was measured at 37°C using a microplate reader (SPECTRAmax GeminiEM; Molecular Devices) at an excitation wavelength of 360 nm and an emission wavelength of 460 nm for 60 min at 5 min intervals. The slope was determined and the proteasome specific activity was calculated by subtracting the slope obtained in the presence of MG-132.
Site-directed inhibitor for deubiquitinase activity assay.
Cultured hippocampal neurons or synaptoneurosomes were lysed in 50 mm Tris-HCl, pH 7.4, 250 mm sucrose, 5 mm MgCl2, 1 mm DTT, and 1 mm ATP. The samples were frozen in liquid nitrogen, thawed, and centrifuged at 12,100 × g for 10 min at 4°C. The amount of protein present in the supernatant fractions was quantified using the Bradford method. To assess the deubiquitinase activity, 20 μg of protein was incubated with 0.5 μg of HA-Ub-VS (Boston Biochem; Borodovsky et al., 2002), a deubiquitinase active site-directed irreversible inhibitor, for 5 min (cultured hippocampal cells) or 10 min (synaptoneurosomes) at 30°C. The samples were then subjected to Western blot analysis using an anti-UchL1 antibody. The inhibitor, a modified version of the ubiquitin C-terminal, binds irreversibly to active DUBs and the type of linkage formed is resistant to reducing sample buffers used in SDS-PAGE (Borodovsky et al., 2002). Therefore, in Western blot experiments, two bands are observed, with the upper band corresponding to the active form of the deubiquitinase.
Western blotting
Protein samples were separated by SDS-PAGE in 10% polyacrylamide gels, transferred to PVDF membranes (Millipore), and immunoblotted. To check for free ubiquitin levels, 16% tricine gels were used. Blots were incubated with primary antibodies (overnight at 4°C), washed, and exposed to alkaline phosphatase-conjugated secondary antibodies (1:20,000 dilution; 1 h at room temperature). Alkaline phosphatase activity was visualized by ECF on the Storm 860 Gel and blot Imaging System (GE Healthcare) and quantified using the ImageQuant program (GE Healthcare). The following primary antibodies were used: anti-ubiquitin (1:2000, Dako Cytomation; 1:1000, clone P4D1, Cell Signaling Technology), anti-Psma2 (1:2000; Abcam), anti-UchL1 (1:1000; Abcam), and anti-Usp14 (1:2000, a kind gift from Scott Wilson; Crimmins et al., 2006). An anti-β-actin (1:5000, Sigma) antibody was used as loading control when indicated.
Statistical analysis
Statistical analysis was performed using one-way ANOVA, followed by the appropriate post hoc test or the Student's t test, as indicated in the figure captions.
Results
BDNF and proteasome share common pathways in the modulation of LTP
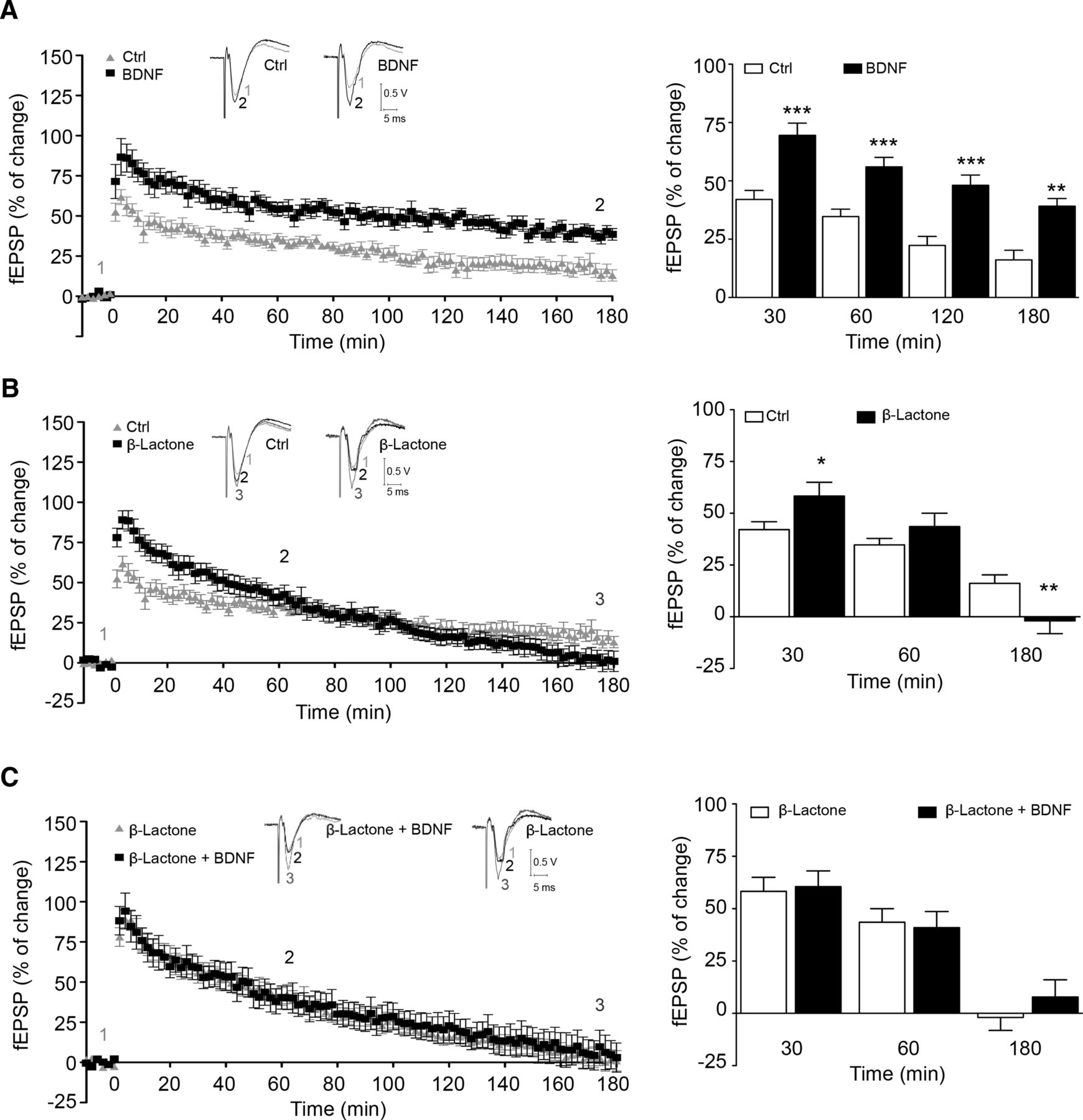
To assess the role of the proteasome in BDNF-induced long-term synaptic facilitation, we used a θ-burst protocol to induce LTP in the CA1 region of hippocampal slices. This protocol is thought to mimic the rhythm of hippocampal neurons in awake, behaving animals (Bland, 1986; Larson and Munkacsy, 2014). After an early and transient phase of synaptic potentiation known as short term-potentiation (STP), which lasts for a few minutes, hippocampal LTP can be divided into at least two phases that involve distinct mechanisms (Leal et al., 2014): E-LTP, usually quantified around 60 min after induction, and L-LTP, which lasts for several hours, being frequently quantified 3 h after induction. As shown in Figure 1A, the fEPSP slope decreased progressively after STP, reaching a first moderate plateau ∼60 min after LTP induction in control conditions. fEPSP slope values then progressively declined, again reaching a near stable value at 160–180 min after induction. A similar pattern of fEPSP decline after LTP induction has been reported by others using mice hippocampal slices (Dong et al., 2008; Dong et al., 2014). The decline in fEPSP slope was much more pronounced in slices incubated with the proteosome inhibitor β-lactone (25 μm; clasto-lactacystin β-lactone) and L-LTP was virtually blocked under such conditions (Fig. 1B; Dong et al., 2008). Previous studies have shown that BDNF upregulates LTP induced by θ-burst stimulation of CA1 synapses (Diógenes et al., 2011). Accordingly, preincubation of hippocampal slices with BDNF (20 ng/ml) increased the fEPSP slope after θ-burst stimulation, being the effect still visible 60 min after induction (E-LTP), and such an increment in synaptic strength was maintained throughout 180 min of recording (L-LTP) (Fig. 1A, 30 min: control, 42.1 ± 3.8%, n = 10; BDNF, 69.5 ± 5.2%, n = 8, p = 0.0005; 60 min: control, 34.7 ± 3.1%, n = 10; BDNF, 56.0 ± 4.0%, n = 8,p = 0.0006; 120 min: control, 22.3 ± 3.9%, n = 10; BDNF, 48.1 ± 4.4%, n = 8, p = 0.0005; 180 min: control, 16.1 ± 4.1%, n = 10; BDNF, 39.2 ± 3.3%, n = 8, p = 0.001).

Proteasome inhibition and BDNF facilitate θ-burst induction of LTP. Left panels in A–C show averaged time course changes in fEPSP slope induced by a θ-burst stimulation (% of change) in the absence (control) or presence of BDNF (A, C) and/or β-lactone (B, C). The ordinates represent normalized fEPSP slopes, where the averaged slopes recorded for 10 min before θ-burst stimulation was set to 0% and the abscissa represents the time of recording. Representative traces are shown above A–C: each trace is the average of eight consecutive responses obtained at the indicated periods of time after LTP induction and is composed of the stimulus artifact followed by the presynaptic volley and the fEPSP. Bar graphs on the right depict the magnitude of LTP (change in the fEPSP slope over time) induced by θ-burst stimulation in relation to pre-θ-burst values (0%) in hippocampal slices. The results are the average ± SEM of 6–10 independent experiments, one slice per experiment, performed in different preparations. Statistical analysis was performed by the Student's t test. *p < 0.05; **p < 0.01; ***p < 0.001.
The role of the proteasome in LTP induced by θ-burst stimulation was tested using the irreversible proteasome inhibitor β-lactone (25 μm). Figure 1B shows that proteasome inhibition differentially regulated the induction and maintenance phases of LTP. Thus, β-lactone significantly increased posttetanic potentiation and the increment was still visible at very early LTP, 30 minutes after LTP induction, when mechanisms related to L-LTP maintenance are far from occurring (Fig. 1B; 30 min: control, 42.0 ± 3.8%, n = 6; β-lactone, 58.3 ± 6.7%, n = 8, p = 0.02083). However, at 3 h after LTP induction, the proteasome inhibitor decreased synaptic strength (Fig. 1B, 180 min: control, 16.1 ± 4.1%, n = 6; β-lactone, −1.9 ± 6.2%, n = 8, p = 0.023), highlighting the importance of protein degradation for synaptic consolidation. The effect of β-lactone in proteasome activity and accumulation of ubiquitinated proteins was confirmed in extracts prepared from the slices that were incubated with the inhibitor for 30 min and further incubated in a saline solution for 180 min (data not shown). The effect of β-lactone in LTP induced by θ-burst stimulation observed here is in agreement with the results of a previous report (Dong et al., 2008) showing a time-dependent effect of proteasome inhibition upon LTP.
We next applied BDNF to β-lactone-inhibited hippocampal slices and recorded the synaptic strength after LTP induction. BDNF incubation in the presence of β-lactone did not further increase the early synaptic response to θ-burst stimulation (Fig. 1C; 30 min: β-lactone, 58.3 ± 6.7%, n = 8; β-lactone + BDNF 60.6 ± 7.5%, n = 6, p = 0.82). Furthermore, BDNF was unable to overcome the decay on synaptic strength induced by proteasome inhibition, losing the ability to facilitate L-LTP (180 min: β-lactone, −1.9 ± 6.2%, n = 8; β-lactone + BDNF 7.8 ± 8.3%, n = 6, p = 0.35).
The overlapping effects of BDNF and β-lactone on the induction and early phases of LTP suggest that the effects of the neurotrophin are mediated by transient inhibition of the proteasome. To further investigate the role of proteasome in the response to BDNF, hippocampal slices were stimulated with the neurotrophin in the presence of a proteasome activator, IU1 (Lee et al., 2010). IU1 (100 μm) abrogated the effects of BDNF on E-LTP induced by θ-burst stimulation (Fig. 2; 30 min, IU1 29.5 ± 4.2%, n = 4; BDNF + IU1 21.4 ± 10%, n = 4, p = 0.41). IU1 did not appreciably modify the magnitude of LTP as observed by the comparable magnitudes of LTP in control slices and in slices from the same animal that were preincubated with IU1 (Fig. 2A,B; control 34.6 ± 8.0%, n = 6; IU1 29.5 ± 4.2%, n = 4, p = 0.74).

BDNF-induced enhancement of LTP is antagonized by proteasome activation with IU1. A, B, Left, Averaged time course changes in fEPSP slope induced by a θ-burst stimulation (% of change) in the absence (gray triangles) or in the presence of 20 ng/ml BDNF (black squares) and/or 100 μm IU1 (A, n = 6; B, n = 4). When the proteasome activator was tested, rat hippocampal slices were preincubation with IU1 for 4 h in aCSF solution (B, n = 4); data in A include the positive controls for the effect of BDNF in slices from the same animals as in B. The ordinates represent normalized fEPSP slopes, where the averaged slopes recorded for 10 min before θ-burst stimulation was set to 0%, and the abscissa represents the time of recording. Representative traces are shown above A and B: each trace is the average of eight consecutive responses obtained at the indicated periods of time before (1 and 3) and 50–60 min after (2 and 4) LTP induction, and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP. Traces 1 and 2 were obtained in the absence of BDNF and traces 3 and 4 in its presence from a second pathway in the same slice. Traces recorded from the same pathway before and after LTP induction are superimposed. Bar graphs on the right depict the magnitude of LTP (change in the fEPSP slope over time) induced by θ-burst stimulation in relation to pre-θ-burst values (0%) in hippocampal slices. The results are the average ± SEM of 4–6 independent experiments (the same as in the time course panels on the left), one slice per experiment, performed in different preparations. Statistical analysis was performed by the Student's t test. *p < 0.05.
Altogether, these results suggest that the effect of BDNF on the induction and early phases of LTP (30 min) are mediated by transient inhibition of the proteasome activity. However, our observations also anticipate that long-term effects of BDNF and the proteasome activity may converge on the same pathway upon θ-burst stimulation to induce LTP in the CA1 region of hippocampal slices so that the activity of the proteosome is required for the influence of BDNF in L-LTP.
BDNF reduces proteasome activity in hippocampal neurons and in synaptoneurosomes
The similarity of the effects of BDNF and proteasome inhibition in the early phase of synaptic potentiation induced by θ-burst stimulation led us to further investigate the effect of BDNF on the catalytic activity of the proteasome. BDNF incubation (100 ng/ml) transiently decreased the proteasome activity in cultured hippocampal neurons and in hippocampal synaptoneurosomes, a subcellular fraction containing the presynaptic and postsynaptic regions (Fig. 3). Both chymotrypsin- and caspase-like activities of the proteasome were transiently reduced after incubation of hippocampal neurons with BDNF, with the lowest activity observed at 15–30 min of incubation (chymotrypsin-like activity: 83.4 ± 4.6%, 15 min; 85.7 ± 3.9%, 30 min, p = 0.017, one-way ANOVA; caspase-like activity: 83.7 ± 5.8%, 15 min; 85.7 ± 4.2%, 30 min, p < 0.0001, one-way ANOVA). BDNF also induced a transient decrease of the trypsin-like activity when tested for 30–60 min of incubation, but the effect was not statistically significant. In fact, an upregulation of this specific proteasome activity was observed in extracts prepared from hippocampal neurons incubated with BDNF for 180 min (124.7 ± 10.5% of the control after 180 min of incubation, p = 0.0002, one-way ANOVA).

BDNF decreases proteasome activity and Psma2 protein levels in hippocampal neurons and synaptoneurosomes. Extracts were prepared from cultured hippocampal neurons (A) and synaptoneurosomes (B) incubated with 100 ng/ml BDNF for the indicated periods of time; 20 ng/ml BDNF was used in hippocampal slices (C). Proteasome activity in cell extracts was measured in the presence of specific fluorogenic substrates to determine chymotrypsin-, caspase-, and trypsin-like activity of the proteasome. In synaptoneurosomes and hippocampal slices, only the chymotrypsin-like activity was investigated (B, C). In B, the effect of K252a (preincubation of 15 min) on the BDNF-induced downregulation of proteasome activity was tested in synaptoneurosomes incubated with the neurotrophin for 15 min. The effect of BDNF stimulation (100 ng/ml) on the Psma2 expression in cultured hippocampal neurons was determined by quantitative PCR after incubation with the neurotrophin for the indicated periods of time (D). The results were normalized with three internal control genes, namely Ppia (peptidylprolyl isomerase A), Pgk1 (phosphoglycerate kinase 1), and Hprt1 (hypoxanthine guanine phosphoribosyl transferase I), which have a stable and invariable expression in hippocampal neurons stimulated with BDNF (Santos and Duarte, 2008). E, F, Effect of BDNF stimulation (100 ng/ml) on Psma2 protein levels in cultured hippocampal neurons and in hippocampal synaptoneurosomes. The preparations were stimulated with BDNF for the indicated period of time and Pmsa2 protein levels were determined by Western blot with a specific antibody. The results are the average ± SEM of 4–9 independent experiments performed in different preparations. Statistical analysis was performed by one-way ANOVA, followed by the Dunnett's test, except in B, where a paired one-way ANOVA followed by the Bonferroni's test was applied. *p < 0.05; **p < 0.01; #p < 0.05 comparing BDNF stimulation alone with preincubation with K252a (B).
We next investigated the effect of BDNF on the proteasome activity of isolated synaptoneurosomes to elucidate the effect of BDNF at the synapse. Synaptoneurosomes were incubated with BDNF for different time points and the chymotrypsin-like activity was measured. In agreement with the results obtained in cultured hippocampal neurons, BDNF stimulation also downregulated the chymotrypsin-like activity of the proteasome (Fig. 3B) at the synapse (to 85.6 ± 2.9% and 88.7 ± 3.3% of the control, at 15 and 30 min, respectively, p = 0.004, one-way ANOVA) and this effect was sensitive to the Trk receptor inhibitor K252a (94.3 ± 2.9%, p = 0.004, one-way ANOVA). The concentration of BDNF used in the electrophysiological experiments (20 ng/ml) described above was confirmed to decrease the chymotrypsin-like activity of the proteasome in hippocampal slices (p = 0.0161, one-way ANOVA), demonstrating that even lower concentrations of BDNF reduce proteasome activity (Fig. 3C).
Two independent proteomic studies showed that BDNF regulates the abundance of several components of the ubiquitin-proteasome system in cultured cerebrocortical (Liao et al., 2007) and hippocampal (Manadas et al., 2009) neurons. We therefore investigated the effect of BDNF on total protein and mRNA levels of Psma2, a subunit of the 20S proteasome. BDNF stimulation decreased Pmsa2 protein levels both in cultured hippocampal neurons and in hippocampal synaptoneurosomes (Fig. 3E,F). Furthermore, incubation of hippocampal neurons with BDNF for 3 h decreased Psma2 mRNA levels to ∼60% of the control (Fig. 3D, p = 0.0019, one-way ANOVA). These results indicate that BDNF stimulation targets the proteasome at different levels, altering both the mRNA and protein level of proteasome components and downregulating the various proteasome activities.
UchL1 activity is regulated by BDNF
The proteomic study performed in our laboratory (Manadas et al., 2009) also showed a regulation of the abundance of deubiquitinating enzymes by BDNF in cultured hippocampal neurons. Indeed, UchL1 and Usp14 (Ubiquitin C-terminal hydrolase 14) were specifically altered upon BDNF stimulation. Both enzymes cleave ubiquitin moieties from proteins targeted for degradation in the proteasome and therefore contribute to the maintenance of the pool of free ubiquitin that can be integrated in a new cycle of ubiquitination. Furthermore, changes in the abundance of UchL1 and USP14 are expected to affect the half-life of their target proteins. Loss of Usp14 results in a developmental disorder ataxia in mice (Wilson et al., 2002) and UchL1 has been implicated in LTP and memory formation (Gong et al., 2006; Sakurai et al., 2008; Cartier et al., 2009), showing the importance of both proteins in ubiquitin homeostasis and synaptic plasticity.
We first investigated the effect of BDNF on general deubiquitinating activity, but no changes were observed in hippocampal neurons stimulated with BDNF (data not shown). To specifically address the effect of BDNF on deubiquitinating activity, we took advantage of a specific inhibitor (Ha-Ub-Vs) that binds irreversibly to the active site of the enzymes, allowing discriminating between active and inactive forms of UchL1. Extracts prepared from cultured hippocampal neurons and hippocampal synaptoneurosomes stimulated with BDNF were incubated with the Ha-Ub-Vs inhibitor and proteins were separated by SDS-PAGE. Because the inhibitor binds irreversibly to active deubiquitinating enzymes, a 10 kDa shift is observed in Western blot experiments. As shown by a representative Western blot image in Figure 4A, the activity of UchL1 in cultured hippocampal neurons was reduced after 3 to 6 h of BDNF stimulation (3 h: 43.0 ± 7.9%; 6 h: 63.8 ± 14.0%, compared with the control, p = 0.0014, one-way ANOVA). Interestingly, BDNF showed a specific effect at the synapse, as indicated by the upregulation of UchL1 activity after stimulation of hippocampal synaptoneurosomes with BDNF for 45 min (Fig. 4B, 133.2 ± 11.16% of the control, p = 0.0212, one-way ANOVA). We next investigated the total protein levels and mRNA expression of UchL1 and observed a differential effect in cultured hippocampal neurons and in hippocampal synaptoneurosomes. Thus, BDNF downregulated UchL1 total protein levels in cultured neurons (Fig. 4C, 3 h: 67.7 ± 7.7%, 6 h: 69.6 ± 13.4% compared with the control, p = 0.0017, one-way ANOVA), whereas an increase in UchL1 protein levels was observed in hippocampal synaptoneurosomes (Fig. 4D, 15 min: 117.3 ± 4.4% of the control, p = 0.0022, one-way ANOVA). Interestingly, mRNA expression of UchL1 was also reduced upon stimulation of hippocampal neurons with BDNF (Fig. 4E).

Effect of BDNF on the deubiquitinating activity of UchL1. Cultured hippocampal neurons (A, C, E, F, H) and hippocampal synaptoneurosomes (B, D, G) were stimulated with BDNF (100 ng/ml) for the indicated periods of time before preparation of cell extracts. To specifically address the effect of BDNF on UchL1 deubiquitinating activity, 20 μg of total protein was incubated with 0.5 μg of HA-Ub-Vs for 5 min (cultured hippocampal neurons) (A) or 10 min (synaptoneurosomes) (B) at 30°C. In control experiments (far left lane in the representative Western blot images), the extracts were not incubated with HA-Ub-Vs. The extracts were subject to SDS-PAGE and blotted against UchL1 antibody. The inhibitor binds irreversibly to the catalytic site of deubiquitinating enzymes, thereby shifting up the UchL1-immunoreactive band in SDS-PAGE by ∼10 kDa. The bands corresponding to the active and nonactive UchL1 were quantified, normalized with actin, and the ratio was calculated (n = 6–9 independent experiments). The effect of BDNF (100 ng/ml) on UchL1 and Usp14 total protein levels in cultured hippocampal neurons (C, F) and in hippocampal synaptoneurosomes (D, G) was determined by Western blot using a specific antibody (n = 5–8 independent experiments). E, H, Effect of BDNF stimulation (100 ng/ml) on UchL1 and Usp14 mRNA levels in cultured hippocampal neurons, respectively, as determined by quantitative PCR (n = 5–8 independent experiments). Statistical analysis was performed by one-way ANOVA, followed by the Dunnett's test. *p < 0.05; **p < 0.01; n.s., nonsignificant.
Considering the compensatory mechanism between UchL1 and Usp14 in the brain (Walters et al., 2008), we analyzed the effect of BDNF on Usp14 protein and mRNA expression. Interestingly, we found that UchL1 and Usp14 are differentially regulated by BDNF. The BDNF-induced transient downregulation of UchL1 observed in cultured hippocampal neurons contrasts with the transient increase in Usp14 protein levels under the same conditions, with maximal protein levels observed after 6 h of incubation with the neurotrophin (156.7 ± 10.7% of the control, p = 0.0009, one-way ANOVA). Furthermore, although BDNF upregulated Usp14 and UchL1 protein levels in synaptoneurosomes, the effects on Usp14 were observed at a much later time point. In addition, BDNF did not affect Usp14 mRNA (Fig. 4H), whereas a downregulation of UchL1 expression was observed after 3 h of incubation with the neurotrophin (Fig. 4E).
BDNF decreases free ubiquitin levels
The transient inhibition of the proteasome by BDNF, together with the increase on UchL1 activity at the synapse, suggests that the neurotrophin may have a significant impact in the levels of free ubiquitin. Accordingly, BDNF transiently decreased free ubiquitin levels in isolated synaptoneurosomes by ∼25% (Fig. 5A, p = 0.007, one-way ANOVA). After 45 min of stimulation, such effect was no longer observed and this change was correlated with the increase in UchL1 activity (Fig. 4B) upon BDNF stimulation. The effect of BDNF on free ubiquitin was sensitive to K252a, a Trk receptor inhibitor (Fig. 5B).

BDNF changes the levels of monomeric ubiquitin in hippocampal synaptoneurosomes. The effect of BDNF on free ubiquitin levels in synaptoneurosomes incubated with BDNF for the indicated periods of time is represented in A. Synaptoneurosomes were incubated with BDNF for 15 min (B) in the presence or in the absence of the Trk inhibitor K252a (200 nm). When the effect of the inhibitor was tested, synaptoneurosomes were preincubated with K252a for 10 min before stimulation with BDNF. The results are the average ± SEM of four independent experiments performed in different synaptoneurosomal preparations. In A, statistical analysis was performed by one-way ANOVA, followed by the Dunnett's test (n = 4 independent experiments). In B, statistical analysis was performed by paired one-way ANOVA, followed by the Bonferroni's test. *p < 0.05; ##p < 0.001 (n = 5 independent experiments).
Discussion
In this work, we show that the early and late phases of BDNF-induced facilitation of LTP in the CA1 region of the hippocampus are differentially affected by the proteasome. Incubation of hippocampal slices with BDNF or with the proteasome inhibitor β-lactone upregulated to a similar extent the E-LTP induced by θ-burst stimulation and no additional effect was obtained when BDNF and β-lactone were tested together. The observed proteasome inhibition in synaptoneurosomes incubated with BDNF suggests that this mechanism may account for the early effects of BDNF in LTP. Accordingly, activation of the proteasome with IU1 fully abrogated the BDNF-induced upregulation of the E-LTP induced by θ-burst stimulation. In contrast to the role of the proteasome in E-LTP induced by BDNF, activity of this proteolytic system is required for the expression of the L-LTP and for the effect of BDNF upon LTP consolidation. This role of the proteasome adds a new layer of complexity to the mechanisms involved in the effects of BDNF on LTP in the hippocampus and indicates that the upregulation of translation activity acts together with the regulation of the rate of protein degradation to tightly control the synaptic proteome in this form of plasticity.
Proteasome inhibition upregulated the E-LTP induced by θ-burst stimulation, whereas activation of the proteasome with IU1 was without effect on synaptic potentiation. These results indicate that, under the experimental conditions used, the proteasome activity exerts a maximal effect in refraining the early response to high-frequency stimulation and conditions decreasing the proteasome activity below this threshold level (e.g., after the addition of BDNF) facilitate synaptic potentiation. The effect of β-lactone on the E-LTP induced by high-frequency stimulation was previously attributed to the local accumulation of newly synthesized positive modulators of synaptic plasticity (Dong et al., 2008; Dong et al., 2014). Although the effects of BDNF on proteasome activity are not as significant as those induced by chemical inhibitors, BDNF also regulated the abundance and activity of specific deubiquitinating enzymes, which may further increase the stability of target proteins. The transient inhibition of a pool of deubiquitinating enzymes was correlated with a transient decrease in total free ubiquitin protein levels in hippocampal synaptoneurosomes stimulated with BDNF. This decrease in the levels of free ubiquitin available to be conjugated to target proteins is likely to further contribute to reduce protein degradation by the UPS and to upregulate key target proteins. However, the effect of BDNF on free ubiquitin levels is transient, at least in part due to the enhancement on UchL1 activity. Interestingly, in the presence of protein synthesis inhibitors, BDNF was shown to increase PKMζ protein levels when applied together with induced electrical neuronal activity (Mei et al., 2011), suggesting that BDNF can regulate protein degradation. A BDNF-induced upregulation of the ubiquitination of synaptic proteins was also reported in cultured cerebrocortical neurons, but in this system, the effects of the neurotrophin were attributed to the enhancement of ubiquitin conjugation because no alteration in the proteasome activity was detected (Jia et al., 2008). This difference, when compared with our results in hippocampal neurons, may suggest that the effects of BDNF on the activity of the proteasome may be distinct depending on the brain region. Alternatively, the effects of BDNF in cortical neurons may follow distinct kinetics.
Some of the proteins that can be accumulated after inhibition of the proteasome include postsynaptic density (PSD) proteins, the aurora kinase, and/or components of the translation machinery. Accordingly, stimulation of cerebrocortical neurons with bicuculline was shown to induce the degradation of PSD proteins by a mechanism sensitive to proteasome inhibitors (Ehlers, 2003). Key proteins in the expression of synaptic plasticity, including PSD-95 (Colledge et al., 2003), Arc (Greer et al., 2010), glutamate receptors (Nelson et al., 2006; Jurd et al., 2008), and other synaptic proteins (Hung et al., 2010), have been shown to be proteasome targets. The aurora kinase, a proteasome substrate (Huang et al., 2002), phosphorylates CPEB (cytoplasmic polyadenylation element binding protein), a protein that plays an important role in the translation of transcripts containing the cytoplasmic polyadenylation element, such as the CaMKII-α. Furthermore, because the ubiquitin-proteasome system also degrades several key players involved in translation, inhibition of the proteasome may indirectly upregulate the translation activity at the synapse (Murata and Shimotohno, 2006). Nonproteolytic functions of ubiquitin, namely through monoubiquitination of cytoplasmic polyadenylation element-binding protein 3 (CPEB3) and regulation of CPEB3-dependent genes, have also been associated with hippocampal plasticity and memory storage (Pavlopoulos et al., 2011).
BDNF induced a rapid (<15 min) and transient decrease in the chymotrypsin-like activity of the proteasome in hippocampal synaptoneurosomes, compatible with a role in the E-LTP. These effects may be attributed to at least two different mechanisms: posttranslational modification of the proteasome proteins and/or a decrease in the abundance of proteasome subunits. The latter hypothesis is supported by the results showing a decrease in Psma2 protein levels, a subunit of the 20S proteasome, in hippocampal synaptoneurosomes and in cultured hippocampal neurons stimulated with BDNF. However, there are also multiple pieces of evidence showing a regulation of the proteasome by phosphorylation (Cui et al., 2014). For example, phosphorylation of the Rpt6 proteasome subunit on Ser120 enhances its proteolytic activity and regulates synaptic strength in hippocampal neurons (Zhang et al., 2007; Djakovic et al., 2012). The rapid effects of BDNF on the activity of the proteasome in hippocampal synaptoneurosomes reported here contrast with the results showing an induction of proteasome activity in response to synaptic activity either via activation of NMDA receptors (Bingol and Schuman, 2006; Djakovic et al., 2009; Hamilton et al., 2012) or with bicuculline (Djakovic et al., 2009). Differences in the preparations used and/or in the protocol of stimulation may explain the differential effects on the activity of the proteasome.
In contrast to the effects of β-lactone in the induction phase of BDNF-induced facilitation of LTP, the proteasome inhibitor abrogated the late phase of potentiation. These results show that normal activity of the proteasome is required for the consolidation of LTP, in contrast to the effect of the proteasome in constraining the early response. This effect of β-lactone mimics the effect of proteasome inhibition on the L-LTP induced by high-frequency stimulation and similar mechanisms may be involved (Fonseca et al., 2006; Karpova et al., 2006; Dong et al., 2008). In this case, proteasome inhibitors were suggested to act by preventing the degradation of transcription repressors and/or by stabilizing translation repressors, which may block protein synthesis in the soma or at the synapse (Fonseca et al., 2006; Dong et al., 2008). The former hypothesis is in accordance with the proposed role for the proteasome in transcription regulation (Muratani and Tansey, 2003; Dong et al., 2008; Geng et al., 2012).
The effects of BDNF on the proteasome activity are likely to influence to a similar extent the rate of degradation of the different UPS targets. In contrast, the upregulation of UchL1 activity observed in hippocampal synaptoneurosomes stimulated with BDNF is expected to affect the half-life of a subpopulation of UPS targets that are regulated by this deubiquitinating enzyme. BDNF also increased transiently UchL1 protein levels in hippocampal synaptoneurosomes, suggesting that this enzyme may be locally synthesized at the synapse. However, the effects of BDNF on UchL1 activity and total protein levels did not follow the same kinetics, suggesting that they may be independent. It remains to be determined whether UchL1 may be regulated by phosphorylation. An upregulation of UchL1 activity with a consequent increase in free ubiquitin was also observed in cultured hippocampal neurons stimulated with NMDA (Cartier et al., 2009), suggesting a tight regulation of this deubiquitinating enzyme. UchL1 was suggested to play a role in synaptic remodeling in the hippocampus through modulation of free ubiquitin protein levels by an activity-dependent mechanism (Cartier et al., 2009).
In addition to the upregulation of UchL1 in hippocampal synaptoneurosomes, BDNF also increased Usp14 protein levels, a deubiquitinating enzyme involved in the regulation of free ubiquitin levels in the brain (Crimmins et al., 2006). These results suggest that Usp14 may be locally synthesized at the synapse, similarly to UchL1. The fast responses detected in synaptoneurosomes were correlated with the delayed effects of BDNF in cultured hippocampal neurons. This contrasts with the results obtained for UchL1, which was downregulated in hippocampal neurons stimulated with BDNF, pointing to a regional specificity in the mechanisms of regulation of the deubiquitinase in hippocampal neurons.
The present work shows a role for BDNF in the regulation of protein degradation by the UPS and proteasome activity was found to be a key regulator in the effects of BDNF on LTP in the hippocampal CA1 region. Although UPS inhibition was found to contribute to the early phase of BDNF-induced facilitation of LTP, the combined action of synthesis and degradation of plasticity-related proteins mediates the facilitatory effects of the neurotrophin in the late phase of LTP. It remains to be determined whether proteasome inhibition also plays a role in other forms of LTP in the hippocampus and the putative role in other brain regions.
Footnotes
This work was supported by FEDER (QREN) through Programa Mais Centro (Projects CENTRO-07-ST24-FEDER-002002, CENTRO-07-ST24-FEDER-002006, and CENTRO-07-ST24-FEDER-002008), through Programa Operacional Factores de Competitividade- COMPETE and National funds via Fundação para a Ciência e a Tecnologia (Projects Pest-C/SAU/LA0001/2013–2014, PTDC/SAU-NMC/120144/2010, and PTDC/SAU-NMC/0198/2012). We thank Ivan Lalanda (Center for Neuroscience and Cell Biology, University of Coimbra) for critical reading of the manuscript and Elisabete Carvalho for assistance in the preparation of cell cultures.
The authors declare no competing financial interests.
This work is dedicated to the memory of our friend and colleague Dr. Krisztina Szabadfi.
- Correspondence should be addressed to Carlos B. Duarte, Center for Neuroscience and Cell Biology, Faculty of Medicine (Polo I), University of Coimbra, Rua Larga, 3004-504 Coimbra, Portugal. cbduarte{at}ci.uc.pt





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}