

Abstract
Emerging evidence suggests that central synaptic inputs onto motor neurons (MNs) play an important role in developmental regulation of the final number of MNs and their muscle innervation for a particular motor pool. Here, we describe the effect of genetic deletion of glycinergic neurotransmission on single MN structure and on functional excitatory and inhibitory inputs to MNs. We measured synaptic currents in E18.5 hypoglossal MNs from brain slices using whole-cell patch-clamp recording, followed by dye-filling these same cells with Neurobiotin, to define their morphology by high-resolution confocal imaging and 3D reconstruction. We show that hypoglossal MNs of mice lacking gephyrin display increased dendritic arbor length and branching, increased spiny processes, decreased inhibitory neurotransmission, and increased excitatory neurotransmission. These findings suggest that central glycinergic synaptic activity plays a vital role in regulating MN morphology and glutamatergic central synaptic inputs during late embryonic development.
SIGNIFICANCE STATEMENT MNs within the brainstem and spinal cord are responsible for integrating a diverse array of synaptic inputs into discrete contractions of skeletal muscle to achieve coordinated behaviors, such as breathing, vocalization, and locomotion. The last trimester in utero is critical in neuromotor development, as this is when central and peripheral synaptic connections are made onto and from MNs. At this time-point, using transgenic mice with negligible glycinergic postsynaptic responses, we show that this deficiency leads to abnormally high excitatory neurotransmission and alters the dendritic architecture responsible for coherently integrating these inputs. This study compliments the emerging concept that neurodevelopmental disorders (including autism, epilepsy, and amyotrophic lateral sclerosis) are underpinned by synaptic dysfunction and therefore will be useful to neuroscientists and neurologists alike.
Introduction
During development, the neuromotor system undergoes a period of programmed neuronal cell death in the final trimester in utero, when over half of all motor neurons (MNs) generated during neurogenesis are lost (Lance-Jones, 1982; Oppenheim, 1991). During this period, peripheral reduction in skeletal muscle activity causes increased MN survival and skeletal muscle innervation (Landmesser, 1992; Usiak and Landmesser, 1999), whereas increased skeletal muscle activity causes decreased MN survival and muscle innervation (Oppenheim and Núñez, 1982). When the central activation of MNs by other neurons has been perturbed in mice, similar alterations in MN survival and muscle innervation have been shown to occur (Oppenheim et al., 2003; Banks et al., 2005; Fogarty et al., 2013b, 2015b) and are thought to depend on the level of MN activity passed onto muscle via intact neuromuscular synapses (Banks et al., 2003, 2005; Oppenheim et al., 2003). This has been demonstrated for central synaptic inputs releasing the amino acid neurotransmitters GABA and glycine, which provide an important source (along with glutamate) of excitation during MN development (Nishimaru et al., 1996; Singer and Berger, 2000; Ben-Ari, 2002). Close to birth and during early postnatal life, the postsynaptic action of GABA and glycine shifts from depolarizing to hyperpolarizing (Banks et al., 2005; Akerman and Cline, 2007). Mice lacking gephyrin, a glycine receptor-clustering molecule (Feng et al., 1998), showed changes in MN number and muscle innervation that were correlated with the measured levels of MN activity; namely, lower levels of hindlimb MN activity increased MN survival and hindlimb muscle innervation. By contrast, higher levels of respiratory MN activity (XII and phrenic) decreased MN survival and muscle innervation (Banks et al., 2005). Similar alterations of MN survival and axonal branching in mice lacking GABA (Fogarty et al., 2013b) and combined GABAergic and glycinergic neurotransmission loss (Fogarty et al., 2015b) further support the hypothesis that central synaptic inputs to MNs play a vital role in regulating MN survival and muscle innervation.
What remains unknown is how the level of central synaptic activation of MNs drives programmed neuronal death during development. One possible mechanism is that the embryonic loss of glycinergic depolarization of MNs results in a compensatory increase in glutamatergic excitatory inputs onto mutant MNs, overloading embryonic MNs with glutamate, a known mediator of excitotoxic neuronal death (Choi, 1992, 1995) and early dendritic growth (Kalb, 1994; Inglis et al., 2002; Metzger, 2010; Koleske, 2013). This mechanism predicts that genetic deletion of glycinergic central synaptic activity will induce developmental plasticity of the MN dendritic arbor and an imbalance in the levels of glycinergic and glutamatergic synaptic neurotransmission to MNs, in a similar manner to developmental synaptic homeostasis seen in other motor systems (Rich and Wenner, 2007; Wenner, 2014).
We have tested these predictions by quantifying alterations to the dendritic arbor, spine density, IPSCs, and EPSCs in individual XII MNs from gephyrin-deficient mice, using patch-clamp electrophysiology to record synaptic inputs, followed by filling recorded MNs with Neurobiotin for morphometric measurement.
Materials and Methods
Mice.
We used 9 gephyrin-deficient (−/−) and 6 litter-matched wild-type (+/+) pups at embryonic (E) day 18 and birth, termed E18.5. Gephyrin-deficient mice and wild-type littermates were generated and genotyped in accordance with previous studies (Feng et al., 1998). All procedures were approved by the University of Queensland Animal Ethics Committee and complied with international and national ethical guidelines.
Slice preparation and electrophysiology.
For E18 mice, time-mated pregnant females containing E18 embryos were killed by cervical dislocation, and embryos were harvested by cesarean section; P0 mice were collected immediately after birth. All pups were anesthetized by hypothermia in an ice-cold dissection tray. Dissection and brain slicing was performed as previously described (Fogarty et al., 2013a; Kanjhan and Bellingham, 2013), with MNs randomly selected from left or right XII motor nucleus, and from dorsal, ventral, or lateral subdivisions of the XII nucleus to ensure little dendritic overlap and unbiased sampling (Kanjhan and Bellingham, 2013).
Patch electrodes were pulled from borosilicate glass capillaries (Edwards Medical), giving a tip resistance of 3–4 mΩ and filled with intracellular solution containing the following (in mm): 135 Cs+MeSO4, 6 KCl, 1 EGTA, 2 MgCl2, 5 Na-HEPES, 3 ATP-Mg2+, 0.3 GTP-Tris, pH 7.25, osmolarity 305 ± 5 mOsm (Kanjhan and Bellingham, 2013). The pipette was back-filled with 1–2 μl of intracellular solution containing 2% Neurobiotin (NB, Vector Laboratories). Recording procedures and dye electroporation parameters were unchanged from previous studies (Kanjhan and Vaney, 2008; Kanjhan and Bellingham, 2013). After membrane seal rupture by voltage pulses or suction, spontaneous IPSCs were recorded as outward currents at a holding potential of 0 mV, and spontaneous EPSCs were recorded as inward currents at a holding potential of −60 mV. Spontaneous synaptic events were detected over 75 s of continuous recording, chosen randomly from 2 min epochs, using an optimally scaled sliding template with defined rise and decay kinetics (Clements and Bekkers, 1997) in Axograph X (Axograph Scientific). Peak-to-peak noise ranged from 2 to 10 pA, and only events with peak amplitudes 2.5 times greater than peak-to-peak noise were analyzed. To pharmacologically characterize outward currents, 5 μm bicuculline (Sigma-Aldrich) was superfused into the bath chamber to antagonize postsynaptic GABAA receptors, as described previously (van Zundert et al., 2008). Intrinsic membrane properties, whole-cell capacitance, input resistance at −60 mV, and resting membrane potential were measured as described previously (van Zundert et al., 2008).
Immunocytochemistry.
After filling, slices were left for >5 min to allow NB diffusion, then fixed in 4% PFA in 0.1 m PBS, pH 7.4, for 30 min, washed in PBS, and incubated for 4 h in PBS containing 4% BSA and 0.5% Triton X-100 at 4°C. Slices were then incubated for 4 h at 4°C in Cy3-streptavidin (Sigma; 1:500 in 4% BSA in PBS) to visualize NB. Slices were washed in PBS and mounted on glass slides.
Imaging and morphologic quantification.
Morphological properties of NB-filled XII MNs were analyzed using Neurolucida (MBF Bioscience) and manual soma (volume and surface area), dendrite and spine tracing as for previous studies (Fogarty et al., 2015a). Low-powered (20× objective) z-series (with a 1.0 μm z-step) from a Zeiss LSM 510 META scanning confocal microscope (Carl Zeiss) using 555 nm laser excitation/detection filters were used to create images of the MN and its entire dendritic arbor. For quantifying spine density, a high-powered (63× oil, NA 1.4) objective was used with a 2.5× zoom to collect z-series with a 0.33 μm z-step. Dendritic processes were classified as spines only if they were <3 μm long and <0.8 μm in cross-sectional diameter, in accordance with past studies (Harris, 1999; Fogarty et al., 2015a). Proximal and distal dendrites were classified according to centrifugal branch orders where branches extending from the cell soma are considered first order, branch ramifications from this dendritic segment second order, and ramifications from these segments being classified as third order. Proximal dendrites consisted of first- and second-order branches, whereas distal dendrites consisted of third-order branches and beyond, as previously established (Klenowski et al., 2015). All axons, identified by their characteristic ventrolateral projection, lack of bifurcations, and absence of tapering were excluded from morphometric analysis. A total MN dendrite length of 54,384 μm was traced for morphometric analysis in 19 neurons. XII motor nucleus volume was measured using a Cavalieri volume estimate obtained on the slices recovered for XII MN dendritic morphology using previously established methods (Fogarty et al., 2013b, 2015b) and an average of 4 sequential transverse slices containing XII motor nucleus per animal (Kanjhan and Bellingham, 2013).
Statistical analysis.
Data were analyzed with Prism 6 (GraphPad) and expressed as mean ± SEM. Statistically significant differences were determined using Student's two-tailed unpaired t test, or a paired t test where appropriate. Correlations were performed with Pearson coefficients. Percentage differences were expressed as the change in relation to the mean of the wild-type control.
Results
Decreased frequency of spontaneous inhibitory neurotransmission in XII MNs of gephyrin-deficient mice
Unsurprisingly, the loss of gephyrin resulted in decreased IPSC frequency and amplitude (Figs. 1; 2). IPSC frequency decreased by 81% in XII MNs from gephyrin-deficient (−/−) mice compared with wild-type (+/+) controls (+/+: 1.71 ± 0.3 Hz, n = 8; −/−: 0.33 ± 0.1 Hz, n = 18; p < 0.0001; Fig. 1E) and IPSC amplitude decreased by 40% in XII MNs from gephyrin-deficient mice (−/−) compared with wild-type (+/+) mice (+/+: 42.5 ± 6.5 pA, n = 8; −/−: 25.5 ± 2.2 pA, n = 11; p = 0.0134; Fig. 2A). IPSC rise-time was increased by 39% in XII MNs from gephyrin-deficient (−/−) mice compared with wild-type (+/+) controls (+/+: 3.3 ± 0.5 ms, n = 8; −/−: 4.6 ± 0.4 ms, n = 11; p = 0.0336; Fig. 2B). By contrast, IPSC half-width (+/+: 6.5 ± 0.8 ms, n = 8; −/−: 12.9 ± 4.1 ms, n = 11; p = 0.2139; Fig. 2C) remained unchanged. IPSC total peak-to-baseline decay time was increased by 125% in XII MNs from gephyrin-deficient (−/−) mice compared with wild-type (+/+) controls (+/+: 19.6 ± 1.7 ms, n = 8; −/−: 44.1 ± 9.4 ms, n = 11; p = 0.0433; Fig. 2D).

Decreased IPSC frequency and amplitude in gephyrin-deficient (−/−) XII MNs compared with wild-type controls (+/+) is abolished by bicuculline. A, B, Recordings of XII MNs voltage-clamped at 0 mV represent decreased IPSC frequency and amplitude in gephyrin-deficient (−/−) mice (B) compared with litter-matched wild-type (+/+) mice (A). C, Recording of a gephyrin-deficient XII MNs voltage-clamped at 0 mV under control conditions before all synaptic inhibitory activity is abolished using bicuculline (D). E, Scatterplot quantifying decreased IPSC frequency (Hz) of XII MNs from gephyrin-deficient (−/−) compared with wild-type (+/+) mice. +/+: n = 8; −/−: n = 11. ***p < 0.001 (Student's unpaired t test). IPSCs in a subset of gephyrin-deficient mice were almost abolished after application of bicuculline (−/− Bic, in E). −/−: n = 7; −/− Bic: n = 7. *p < 0.05 (Student's unpaired t test).

IPSC shape parameters differ for XII MNs from gephyrin-deficient (−/−) XII MNs compared with wild-type (+/+). A, Scatterplot represents decreased IPSC amplitude (pA) of XII MNs from gephyrin-deficient (−/−) compared with wild-type (+/+) mice. +/+: n = 8; −/−: n = 11. *p < 0.05 (Student's unpaired t test). B, Scatterplot represents increased IPSC 10%–90% rise-time (ms) of XII MNs from gephyrin-deficient (−/−) compared with wild-type (+/+) mice. +/+: n = 8; −/−: n = 11. *p < 0.05 (Student's unpaired t test). C, Scatterplot represents unchanged IPSC half-width (ms) of XII MNs from gephyrin-deficient (−/−) compared with wild-type (+/+) mice; the outlying data points are likely due to sampling very low-frequency events in gephyrin-deficient mice. +/+: n = 8; −/−: n = 11. p = 0.21 (Student's unpaired t test). D, Scatterplot represents increased IPSC total decay time (ms) of XII MNs from gephyrin-deficient (−/−) compared with wild-type (+/+) mice. +/+: n = 8; −/−: n = 11. *p < 0.05 (Student's unpaired t test).
Abnormalities of bicuculline-sensitive residual outward currents in XII MNs of gephyrin-deficient mice
At E18.5, all XII MNs recorded in whole-cell patch configuration exhibited robust inward currents at a holding voltage of −60 mV, indicating the presence of excitatory synaptic activity onto these neurons. All wild-type XII MNs had outward currents at a holding voltage of 0 mV, but the proportion of gephyrin-deficient XII MNs that had outward currents at holding voltages of 0 mV was decreased by 44% (+/+: 1.00, n = 8; −/−: 0.56, n = 18; p = 0.0403). Namely, 44% (8 of the 18) gephyrin-deficient XII MNs had no detectable outward currents, despite them having excitatory inputs, as evidenced by the presence of frequent inward currents (∼2 Hz).
A subset of gephyrin-deficient XII MNs with outward currents were assessed to determine whether these currents were GABAergic. The GABAA receptor antagonist bicuculline (5 μm; 3–5 min) was bath applied to test its effects on gephyrin-deficient XII MN outward currents under control conditions (0.24 ± 0.08 Hz, n = 7; Fig. 1C,E). Outward currents were almost completely abolished after bicuculline (0.004 ± 0.02 Hz, n = 7; Fig. 1D), and the mean frequency was 98% lower than the control condition (p = 0.0266, paired t test; Fig. 1E). These results in conjunction with shape parameter alterations indicate that the residual IPSCs seen in XII MNs from the gephyrin-deficient mice are generated almost entirely by GABAergic mechanisms, consistent with previous findings that glycine and GABA are frequently coreleased onto XII MNs (O'Brien and Berger, 1999).
Increased frequency of spontaneous excitatory neurotransmission in XII MNs of gephyrin-deficient mice
To assess whether the loss of glycinergic neurotransmission induced plasticity in XII MNs by increasing other types of synaptic inputs onto gephyrin-deficient mice, we quantified EPSC frequency and shape parameters in the mutant mice compared with wild-type controls (Fig. 3).

Increased EPSC frequency in gephyrin-deficient (−/−) XII MNs compared with wild-type controls (+/+) is sufficient to return net total synaptic activity to control levels. A, B, Recordings of XII MNs voltage-clamped at −60 mV show increased EPSC frequency in gephyrin-deficient (−/−) mice (B) compared with litter-matched wild-type (+/+) mice (A). C, Scatterplot represents increased EPSC frequency (Hz) of XII MNs from gephyrin-deficient (−/−) compared with wild-type (+/+) mice. +/+: n = 8; −/−: n = 11. **p < 0.01 (Student's unpaired t test). D, Scatterplot showing no significant correlation of IPSC (y-axis) and EPSC (x-axis) frequencies from the same XII MNs from wild-type (open circles) and gephyrin-deficient (filled circles) mice. E, Scatterplot represents unchanged total summed IPSC and EPSC frequency (Hz) of XII MNs from wild-type (+/+) and gephyrin-deficient (−/−) mice. +/+: n = 8; −/−: n = 8. p = 0.59 (Student's unpaired t test).
The mean frequency of spontaneous EPSCs was more than twofold greater (127% increase) in XII MNs from gephyrin-deficient mice (−/−) compared with wild-type (+/+) controls (+/+: 0.87 ± 0.18 Hz, n = 8; −/−: 1.98 ± 0.29 Hz, n = 11; p = 0.0093; Fig. 3C). By contrast, EPSC amplitude (+/+: −13.7 ± 3.4 pA, n = 8; −/−: − 21.4 ± 3.0 pA, n = 11; p = 0.096), 10%–90% rise-time (+/+: 4.8 ± 0.9 ms, n = 8; −/−: 3.5 ± 0.5 ms, n = 11; p = 0.1877), and total peak-to-baseline decay time (+/+: 22.3 ± 2.3 ms, n = 8; −/−: 18.5 ± 1.6 ms, n = 11; p = 0.1788) was unchanged. EPSC half-width was decreased by 51% in gephyrin-deficient XII MNs (+/+: 4.9 ± 0.6 ms, n = 8; −/−: 2.4 ± 0.6 ms, n = 11; p = 0.0117). Together, our patch-clamp data demonstrate that XII MNs from gephyrin-deficient mice receive decreased inhibitory inputs, with a concomitant increase in excitatory neurotransmission.
To determine whether there was a correlation between the frequency of IPSCs and EPSCs in the same neuron, we plotted XII MN IPSC frequencies (y-axis) against the EPSC frequency (x-axis) of that same cell. Although the spatial distribution of the wild-type XII MNs shared almost no overlap with gephyrin-deficient mice, there was no significant correlation of the relationship between IPSC and EPSC frequency in the same XII MNs in either wild-type (+/+: 0.36, p = 0.39, Spearman coefficient) or gephyrin-deficient (+/+: 0.23, p = 0.48, Spearman coefficient) mice (Fig. 3D).
To determine whether increased EPSC frequency in gephyrin-deficient XII MNs was returning overall synaptic inputs (both IPSC and EPSC) to a level seen in wild-type controls, we analyzed the sum of IPSC and EPSC frequencies within the same XII MNs across both genotypes. The sum of the IPSC and EPSC frequencies (i.e., net overall synaptic activity) was unchanged between the groups (+/+: 2.5 ± 0.2 Hz, n = 8; −/−: 2.4 ± 0.3 Hz, n = 11; p = 0.5915; Fig. 3E). Thus, although the amount of synaptic input is conserved between gephyrin-deficient XII MNs and their wild-type controls, there is a stark shift in the overall balance of these inputs from inhibitory to excitatory, with inhibitory inputs on gephyrin-deficient mice decreased to under one-fifth of wild-type controls, and excitatory inputs increasing by more than twofold in gephyrin-deficient mice compared with wild-type controls. This change in the frequency of inputs is likely to increase the probability of action potential firing in gephyrin-deficient XII MNs along axons to their target skeletal muscle.
We also investigated the intrinsic membrane properties (whole-cell capacitance, input resistance, and resting membrane potential) of XII MNs from gephyrin-deficient and wild-type mice. We observed no significant difference between genotype for these properties (Table 1). These electrophysiological measurements are predominantly determined by somatic size, as somatic electrode placement has limited ability to space clamp the distal dendritic membrane. Accordingly, we observed no significant change in XII MN soma volume and surface area between wild-type and gephyrin-deficient mice. A summary of these intrinsic membrane properties and soma morphology values are provided in Table 1.
Membrane characteristics and somatic measurements are unchanged between gephyrin-deficient (−/−) and wild-type (+/+) XII MNsa
Our synaptic electrophysiological data are consistent with our previous work on these gephyrin-deficient mice, where we have shown increased spontaneous neural activity from the axon bundles exiting the brainstem (i.e., increased frequency of spontaneous activity from XII nerve recordings) (Banks et al., 2005).
Increased dendritic arbor length and complexity in XII MNs from gephyrin-deficient mice
These functional alterations in excitatory and inhibitory synaptic neurotransmitter inputs onto XII MNs are likely to affect MN morphology and consequently nerve output to muscle. We therefore assessed XII MN morphology in 3D, including dendritic architecture and spines from wild-type and gephyrin-deficient mice at E18.5 (Fig. 4). First, to ensure that there was no gross anatomical disturbance between genotype, we examined the total XII motor nucleus volume (mm3) and found it to be unchanged between wild-type and gephyrin-deficient mice at E18.5 (+/+: 1.45 ± 0.11 mm3, n = 6; −/−: 1.32 ± 0.12 mm3, n = 6; p = 0.4495).

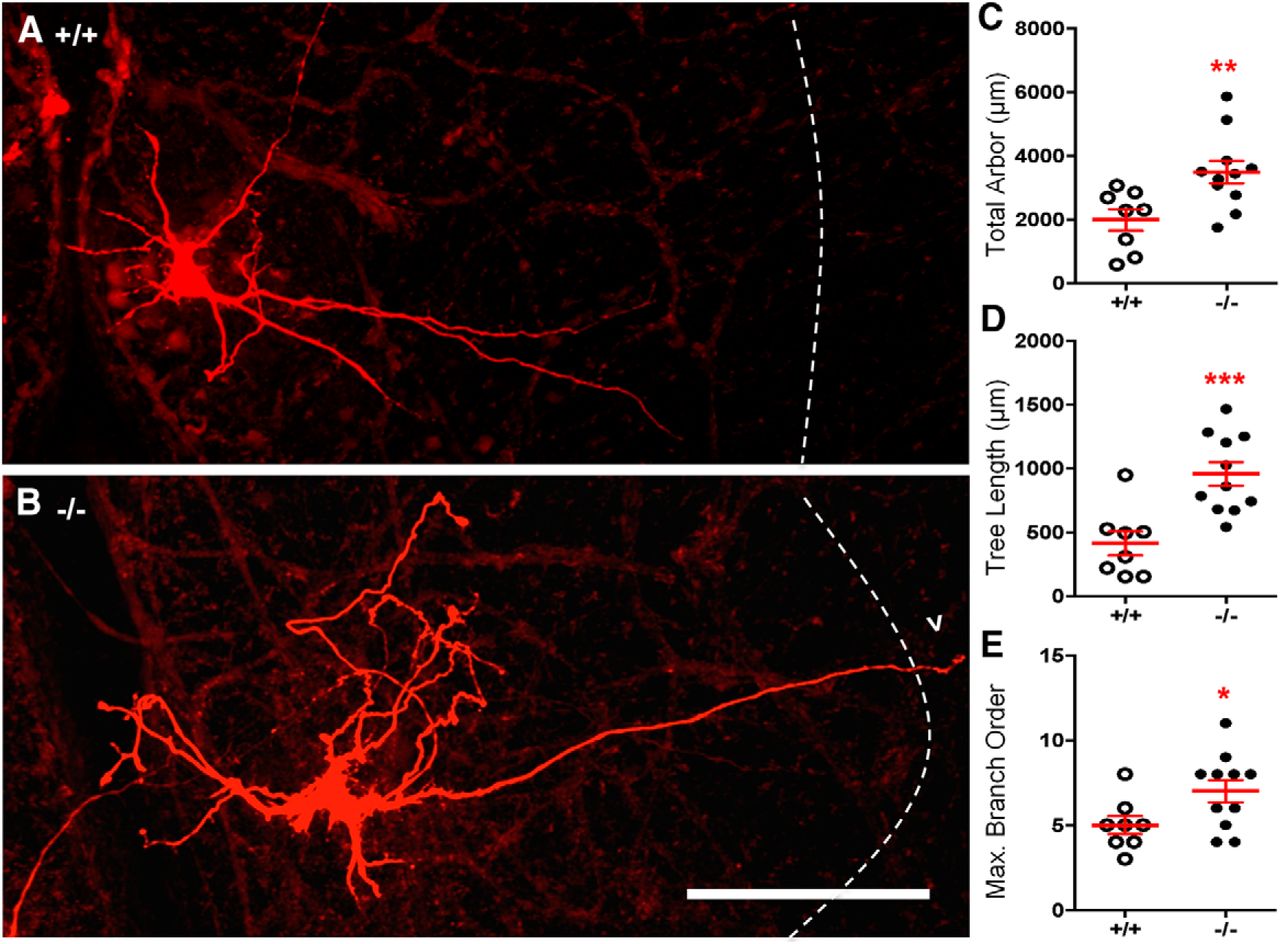
Increased length and complexity of dendritic arbor in XII MNs from gephyrin-deficient mice. A, B, Dendritic arbors of NB-filled XII MNs from wild-type (+/+) and gephyrin-deficient (−/−) mice. Dashed line indicates the lateral border of the XII motor nucleus. Also shown is an example of a dendrite projecting outside of the XII motor nucleus (arrowhead). Scatterplots represent morphological measurements of XII MNs from gephyrin-deficient (−/−) and wild-type (+/+) mice, showing increased total dendritic arbor length (C), increased individual dendritic tree length (D), and increased dendritic branch order ramifications (E). +/+: n = 8; −/−: n = 11. *p < 0.05 (Student's unpaired t test). **p < 0.01 (Student's unpaired t test). ***p < 0.001 (Student's unpaired t test). Scale bar, 100 μm.
Next, we assessed the fine morphological details of XII MNs. Total MN dendritic length was increased by 75% in gephyrin-deficient mice (−/−) compared with wild-type (+/+) controls (+/+: 1995 ± 337 μm, n = 8; −/−: 3493 ± 356 μm, n = 11; p = 0.009; Fig. 4C). As total dendrite length is strongly influenced by the number of dendritic trees present on each XII MN (+/+: 5.38 ± 0.50, n = 8; −/−: 4.18 ± 0.3, n = 11; p = 0.038), we also quantified dendritic length on a per tree basis to derive a more accurate estimate of changes in dendritic arbors, without any bias due to the number of individual dendritic trees each MN may contain. Mean MN dendritic tree length was increased by 131% in gephyrin-deficient mice (−/−) compared with wild-type (+/+) controls (+/+: 413.9 ± 94.2 μm, n = 8; −/−: 955.1 ± 91.9 μm, n = 11; p = 0.0009; Fig. 4D).
As dendritic lengthening may be caused by increased length of dendrites already present in the arbor, or from increased branching complexity, we quantified the maximum number of centrifugal branch orders for each dendritic tree, to give a measure of dendritic tree complexity. Using this method, the first branch order is the dendrites coming off the soma, second-order branches begin when a first-order branch bifurcates into two, third-order branches commence when a second-order branch divides and so on. Compared with wild-type controls (+/+), the maximum MN branch order was increased by 40% in gephyrin-deficient (−/−) mice (+/+: 5.00 ± 0.53, n = 8; −/−: 7.00 ± 0.66, n = 11; p = 0.04; Fig. 4E).
Additional morphologic changes were present in XII MN dendrites from gephyrin-deficient mice. An increased proportion of XII MNs had dendrites crossing the midline or dendrites extending beyond the border of the hypoglossal motor nucleus (Fig. 4B, dashed line, arrowhead). The proportion of hypoglossal MNs with dendrites showing midline crossing increased by 118% in gephyrin-deficient mice (−/−, 9 of 11 XII MNs) compared with wild-type (+/+, 3 of 8 XII MNs) controls (+/+: 0.375, n = 8; −/−: 0.818, n = 11; p = 0.0487). The proportion of hypoglossal MNs with dendrites showing extranuclear projections increased by 118% in gephyrin-deficient mice (−/−, 8 of 11 XII MNs) compared with wild-type (+/+, 2 of 8 XII MNs) controls (+/+: 0.25, n = 8; −/−: 0.73, n = 11; p = 0.0413).
Together, these results indicate that the developmental absence of effective glycinergic neurotransmission to XII MNs leads to a significant increase of overall dendritic arborization, increased dendritic tree lengths, and increased dendritic arbor complexity. As glycinergic transmission in the prenatal nervous system is depolarizing (Singer and Berger, 2000; Banks et al., 2005; Fogarty et al., 2013a), this genetically induced loss of depolarizing synaptic drive to MNs may result in increased dendritic arborization, as a compensatory mechanism that increases the probability of receiving excitatory glutamatergic synaptic inputs. As dendritic spines are a morphological component of the glutamatergic postsynaptic domain (Harris, 1999), we next quantified dendritic spines on the soma, proximal dendrites (first- and second-order branches), and distal dendrites (third branch order and beyond) of XII MNs.
Increased somatic and dendritic spine densities of gephyrin-deficient XII MNs
High-magnification confocal z-stacks were used to quantify the total number of somatic spines and spine density of proximal (Fig. 5) and distal dendrites (Fig. 6). The mean somatic spine number was increased by 48% in gephyrin-deficient mice (−/−) compared with wild-type (+/+) controls (+/+: 116.4 ± 16.7, n = 8; −/−: 172.5 ± 13.6, n = 11; p = 0.0187; Fig. 5C). The mean proximal dendritic spine density per 100 μm was increased by 77% in gephyrin-deficient mice (−/−) compared with wild-type (+/+) controls (+/+: 12.5 ± 2.8, n = 8; −/−: 22.1 ± 2.7, n = 11; p = 0.0273; Fig. 5D). The mean distal dendritic spine density per 100 μm was increased by 108% in gephyrin-deficient mice (−/−) compared with wild-type (+/+) controls (+/+: 12.6 ± 1.9, n = 8; −/−: 26.2 ± 3.5, n = 11; p = 0.0068; Fig. 6C).

Increased somatic spines and proximal dendritic spine density in XII MNs from gephyrin-deficient mice. A, B, Soma and proximal dendrites of NB-filled XII MNs from wild-type (+/+) and gephyrin-deficient (−/−) mice from a single 0.3 μm optical z-slice. The discontinuity (loss of fluorescence) in some of the proximal dendrites is due to the dendritic tree winding in and out of the focal plane. White arrows indicate spiny processes on somas and proximal dendrites. Scatterplots represent morphological measurements of XII MNs from gephyrin-deficient (−/−) and wild-type (+/+) mice, showing increased somatic spines (C) and increased proximal dendritic spine density per 100 μm of first- and second-order dendrite (D). +/+: n = 8; −/−: n = 11. *p < 0.05 (Student's unpaired t test). Scale bar, 10 μm.

Increased distal dendritic spine density in XII MNs from gephyrin-deficient mice. A, B, Distal dendrites of NB-filled XII MNs from wild-type (+/+) and gephyrin-deficient (−/−) mice. C, Scatterplot represents increased distal dendritic spine density per 100 μm of third-order and above dendrites of XII MNs from gephyrin-deficient (−/−) and wild-type (+/+) mice. +/+: n = 8; −/−: n = 11. **p < 0.01 (Student's unpaired t test). Scale bar, 5 μm.
Discussion
The level of MN activity during prenatal development is well established as a critical determinant of postnatal MN number and neuromuscular innervation (Oppenheim and Núñez, 1982; Landmesser, 1992; Usiak and Landmesser, 1999; Banks et al., 2005). Consequently, the formation of effective neuromotor circuits at birth is likely to be strongly influenced by the transformation of synaptic activity into MN activity during embryonic development (Oppenheim, 1991). Here, we have shown that genetic deletion of gephyrin causes upregulation of glutamatergic neurotransmission and marked morphological and functional changes in single XII MNs, consistent with our previous report of an increase in XII motor activity, coupled with a 32% decrease in XII MN number (Banks et al., 2005).
In interpreting our data, it is vital to note that loss of gephyrin will disrupt glycine and, to a lesser extent, GABAA, receptor clustering at synapses (Feng et al., 1998). These Cl−-permeable channels are depolarizing, rather than hyperpolarizing, throughout prenatal and into postnatal development, due to high intracellular Cl− concentration, which declines with maturation (Singer and Berger, 2000; Ben-Ari, 2002; Ren and Greer, 2003; Akerman and Cline, 2007). Thus, the loss of gephyrin will result in a decrease in depolarizing synaptic drive to XII MNs during a critical phase of development in the neuromotor system. Perturbation of GABAA or AMPA receptor-mediated neurotransmission in the embryonic spinal cord of other systems causes compensatory homeostatic alterations in both synaptic strength and neuronal excitability (Wilhelm and Wenner, 2008; Gonzalez-Islas et al., 2009, 2010; Wilhelm et al., 2009; Wenner, 2014), indicating that this process may be a nuanced balance of differing mechanisms, presynaptic vesicular filling, and changes in the number of postsynaptic receptors (Rich and Wenner, 2007; Wenner, 2014). An additional caveat is the common occurrence of developmental GABA/glycine corelease onto MNs, before one neurotransmitter predominates at particular synaptic sites (O'Brien and Berger, 1999; Russier et al., 2002; Nabekura et al., 2004; Wojcik et al., 2006).
Our data indicate that loss of glycinergic neurotransmission is coupled with an increase in glutamatergic excitation of XII MNs, as shown by an increase in the frequency and amplitude of EPSCs, and an increased prevalence of dendritic spines, which are considered a canonical part of excitatory synapses (Spruston, 2008). These changes are in the absence of any difference in intrinsic membrane properties or somatic size between genotypes and were not due to altered gross morphology of the XII nucleus, as shown by our estimates of Cavalieri volume. Excitatory glutamatergic signaling is also a key regulator of dendritic length and spine growth during development (Kalb, 1994; Metzger, 2010), suggesting that our observed increased dendritic length may be a consequence of enhanced glutamatergic neurotransmission early in neuromotor development. These changes may lead to a positive feedback loop, where the increased receptive area generated by dendritic growth leads to further glutamatergic excitation, spine development, and dendrite branching.
Our experiments do not directly investigate the mechanism of glutamatergic synaptogenesis, but we believe it is likely to be similar to Ca2+ and/or glutamate-dependent dendritic plasticity mechanisms characterized in other neuronal network perturbations. For example, previous studies have shown that glutamatergic synaptic plasticity is a consequence of Ca2+ influx mediated by NMDA and/or AMPA receptors, modulating spine growth in a concentration-dependent manner (Segal et al., 2000; Hering and Sheng, 2001).
XII MNs receive rhythmic glutamatergic synaptic drive generated by the respiratory central pattern generator in the ventrolateral medulla (Funk et al., 1993; Ireland et al., 2012), and the frequency and amplitude of respiratory rhythm are strongly reduced by glycinergic pacemaker neurons within this area (Ren and Greer, 2003; Sherman et al., 2015). As the genetic deletion of gephyrin will also cause a deficit in glycinergic transmission within the respiratory central pattern generator, respiratory drive to XII MNs will be increased in frequency and strength, as we have previously shown in the rhythmically active in vitro brainstem-spinal cord (Banks et al., 2005). As other studies have shown that overstimulation of neurons can drive increases in spine number and structure (Engert and Bonhoeffer, 1999), this enhancement of respiratory rhythm is likely to contribute to overstimulation of XII MNs.
Gephyrin is essential for clustering glycine receptors in the postsynaptic membrane (Feng et al., 1998), and its genetic deletion results in the absence of glycinergic neurotransmission (Banks et al., 2005). Gephyrin is also important for clustering specific subsets of GABAA receptors (Kneussel et al., 1999). However, we have previously shown that XII MNs from gephyrin-deficient mice remain pharmacologically sensitive to GABA (Banks et al., 2005). Our observations here further demonstrate that the residual synaptic inhibitory outward currents in XII MNs from gephyrin-deficient mice are almost entirely due to bicuculline-sensitive GABAA receptors (Fig. 1F). These results together indicate that, although GABAergic neurotransmission persists, it has not been upregulated to compensate for the complete loss of glycinergic transmission, as spontaneous IPSCs are less frequent and of lower amplitude (Figs. 1E, 2A).
Although there was no correlation between the frequency of IPSC and EPSC synaptic inputs received by individual XII MNs in either genotype studied (Fig. 3D), the overall net synaptic inputs (glycinergic, glutamatergic, and GABAergic) onto XII MNs were unchanged (Fig. 3E). We interpret this as indirect evidence that compensation for glycinergic neurotransmission loss in gephyrin-deficient mice is predominantly through enhanced glutamatergic synaptogenesis, as opposed to GABAergic mechanisms. Our data also show that 44% of gephyrin-deficient XII MNs failed to have any detectable outward currents, an observation that further supports our interpretation. While we cannot rule out that the absence of glycine and/or GABAA postsynaptic receptor clustering is altering the maturation of presynaptic inhibitory neurotransmitter release sites from a mixed release of glycine and GABA (O'Brien and Berger, 1999; Russier et al., 2002; Nabekura et al., 2004; Wojcik et al., 2006) or even corelease of glutamate and GABA (Gillespie et al., 2005; El Mestikawy et al., 2011; Spitzer, 2015), our conclusion is that the increased XII MN output previously reported by us (see Banks et al., 2005) is driven by an increase in glutamatergic synaptic inputs to XII MNs. Indeed, in experiments where spontaneous synaptic activity was pharmacologically reduced by lidocaine in utero, compensatory gains were seen in excitatory AMPA currents and not GABA currents (Gonzalez-Islas and Wenner, 2006). However, these changes were not seen when blocking ionic glutamatergic, GABAergic, or glycinergic ionotropic channels specifically (Wilhelm and Wenner, 2008), indicating robust neurotransmitter compensation at early embryonic ages.
The type and number of synaptic inputs onto MNs during embryonic development are major determinants of the overall activity of the MN, contributing to the formation of effective neuromotor circuits for motor behaviors vital to survival at birth, such as breathing, suckling, and vocalization. We have previously shown that glycinergic neurotransmission helps regulate final MN numbers and peripheral muscle innervation (Banks et al., 2005). The present study demonstrates that glycinergic neurotransmission to MNs has novel roles, which extend beyond activity-dependent mechanisms for MN survival and nerve-muscle innervation. XII MNs from gephyrin-deficient mice displayed increased dendritic arbors, increased dendritic complexity, increased somatic and dendritic spines, and increased excitatory synaptic neurotransmission, coupled with decreased inhibitory synaptic neurotransmission. Glycinergic neurotransmission thus plays a critical role in determining the pattern of XII MN dendritic growth, and the optimal balance of excitatory and inhibitory synaptic inputs during development. In the absence of glycinergic inputs, increased glutamatergic transmission may promote increased dendritic growth and spine production, and contribute to activity-dependent apoptotic MN death.
Footnotes
This work was supported by National Health and Medical Research Council Project Grant 1065884 (Motor Neuron Disease Research Institute Australia) to P.G.N. and M.C.B. We thank Dr. Heinrich Betz kindly for providing Gephyrin founder mice and for continued support of this work; M. Shayegh and M. White for genotyping; and L. Hammond for microscopy assistance (ARC LIEF Grant LE100100074).
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Peter G. Noakes, School of Biomedical Sciences, University of Queensland, St. Lucia, 4072 QLD, Australia. p.noakes{at}uq.edu.au





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}