

Abstract
Photosensitization, an exaggerated sensitivity to harmless light, occurs genetically in rare diseases, such as porphyrias, and in photodynamic therapy where short-term toxicity is intended. A common feature is the experience of pain from bright light. In human subjects, skin exposure to 405 nm light induced moderate pain, which was intensified by pretreatment with aminolevulinic acid. In heterologous expression systems and cultured sensory neurons, exposure to blue light activated TRPA1 and, to a lesser extent, TRPV1 channels in the absence of additional photosensitization. Pretreatment with aminolevulinic acid or with protoporphyrin IX dramatically increased the light sensitivity of both TRPA1 and TRPV1 via generation of reactive oxygen species. Artificial lipid bilayers equipped with purified human TRPA1 showed substantial single-channel activity only in the presence of protoporphyrin IX and blue light. Photosensitivity and photosensitization could be demonstrated in freshly isolated mouse tissues and led to TRP channel-dependent release of proinflammatory neuropeptides upon illumination. With antagonists in clinical development, these findings may help to alleviate pain during photodynamic therapy and also allow for disease modification in porphyria patients.
SIGNIFICANCE STATEMENT Cutaneous porphyria patients suffer from burning pain upon exposure to sunlight and other patients undergoing photodynamic therapy experience similar pain, which can limit the therapeutic efforts. This study elucidates the underlying molecular transduction mechanism and identifies potential targets of therapy. Ultraviolet and blue light generates singlet oxygen, which oxidizes and activates the ion channels TRPA1 and TRPV1. The disease and the therapeutic options could be reproduced in models ranging from isolated ion channels to human subjects, applying protoporphyrin IX or its precursor aminolevulinic acid. There is an unmet medical need, and our results suggest a therapeutic use of the pertinent antagonists in clinical development.
Introduction
A key role in the detrimental action of blue to near ultraviolet (UVA) light to human cells is performed by endogenous photosensitizers through photosensitizing reactions of Type I, a direct electron transfer between photosensitizer and substrate, or Type II, an energy transfer to molecular oxygen producing singlet oxygen and reactive oxygen species (ROS). Porphyrins are common photosensitizers. This includes uroporphyrin, coproporphyrin, and protoporphyrin IX (PpIX), which are intermediates in the ubiquitous heme biosynthetic pathway (Wondrak et al., 2006). This pathway is initiated by δ-aminolevulinic acid (ALA)-synthase in mitochondria with the production of ALA from glycine and succinyl-CoA. Subsequent chemical reactions catalyzed by various enzymes culminate in the action of ferrochelatase, an enzyme that adds ferrous iron to PpIX, resulting in heme formation (Ajioka et al., 2006). Porphyrias are rare diseases caused by loss of function mutations in the genes of enzymes, which participate in the heme biosynthesis pathway. Deficiencies in early steps of the pathway, as for example in acute intermittent porphyria (Herrick and McColl, 2005), lead to accumulation of ALA and/or porphobilinogen and cause neurovisceral symptoms that are not accompanied by cutaneous photosensitivity. However, accumulation of later intermediates, including PpIX, results in severe burning pain (“ice-cold fire on the skin”), itching redness, and eventually blistering and swelling of the skin upon sun or even neon light exposure (Poblete-Gutiérrez et al., 2006; Puy et al., 2010; Balwani and Desnick, 2012). While it is widely accepted that cutaneous photosensitivity in these maladies is due to accumulation of porphyrins in the skin, which enhances the production of free radicals upon exposure to light, the actual molecular mechanism responsible for the pain reported, in particular, by erythropoietic protoporphyria patients within minutes of light exposure (Lecha et al., 2009) has not been investigated in depth.
Exogenously, photosensitization can occur unintentionally by contact with plants that contain photosensitizers (e.g., Berloque dermatitis) (Wang et al., 2002). The vital stain methylene blue can be considered as a model compound as it excites nociceptive nerve terminals in isolated rat skin only when illuminated (Kress et al., 1997). In photodynamic therapy (PDT), photosensitization is induced by excess levels of PpIX, generating singlet oxygen upon illumination, which is used in treating precancerous lesions, nonmelanoma skin cancers, and other skin conditions (Ishizuka et al., 2011). Endoscopic PDT is used to treat neoplasms in the esophagus, bile duct, and bronchi; and in ophthalmology, laser-PDT is used to fight neovascularization in case of “wet” macular degeneration (Muguruma et al., 2012; Nowak-Sliwinska et al., 2013). In general, PDT relies on the administration of photosensitizing drugs that accumulate in tumor cells, followed by illumination with visible light of the appropriate wavelength, leading to generation of ROS and oxidative cell damage. This can be achieved by topical administration of the heme precursor ALA, which is internalized and metabolized to PpIX by target cells, or by systemic medication of PpIX derivatives in nondermatological practice. One of the frequent adverse effects of PDT is pain, which limits the therapeutic use of this approach (Warren et al., 2009; Gholam et al., 2010; Fink et al., 2015). Again, while excess PpIX and ROS generation during illumination is likely to be the underlying cause of pain in PDT, the precise mechanism involved in the activation and sensitization of nociceptors has not been unraveled.
In this study, we focus on TRPA1 and TRPV1, polymodal receptor channels in the pain pathway that are highly expressed in cutaneous nociceptive nerve endings (Caterina et al., 1997; Story et al., 2003). Based on the activation of both TRPA1 and TRPV1 by ROS (Andersson et al., 2008; Chuang and Lin, 2009) and the activation of TRPA1 by exposure to UV light (Hill and Schaefer, 2009), we hypothesized that these channels may mediate the exacerbated pain associated with cutaneous photosensitive porphyrias (particularly erythropoietic porphyria) and PTD. We provide evidence for such a role by investigating the mechanisms underlying photosensitivity and ALA- and PpIX-induced sensitization in heterologous expression systems, cultured primary afferent neurons, and nociceptive nerve endings in intact tissue preparations from wild-type (WT) and null mutant mice, and in a human translational model.
Materials and Methods
Chemicals and solutions.
The standard external solution (ES) used in calcium imaging experiments contained the following (in mm): NaCl 140, KCl 4, CaCl2 1.25, MgCl2 1, HEPES 10, glucose 5; the pH was adjusted to 7.4 with NaOH. The extracellular calcium-free solution used in patch-clamp experiments and some calcium imaging experiments contained the following (in mm): NaCl 140, KCl 4, MgCl2 2, HEPES 10, glucose 5; the pH was adjusted to 7.4 with NaOH. The pipette solution for patch-clamp recordings contained the following (in mm): potassium gluconate 135, MgCl2 3, EGTA 5, Na2ATP 2, Na3GTP 0.3, NaCl 4, HEPES 5; the pH was adjusted to 7.25 with KOH. Contents of synthetic interstitial fluid (SIF, in mm) used for ex vivo experiments were as follows: NaCl 108, KCl 3.5, MgSO4 3.5, NaHCO3 26, NaH2PO4 1.7, CaCl2 1.5, sodium gluconate 9.6, glucose 5.5, and sucrose 7.6 (Bretag, 1969). All reagents were purchased from Sigma-Aldrich. PpIX was obtained from Frontier Scientific or Sigma-Aldrich. Fresh stock solutions of ALA (100 mm in external solution), PpIX (10 mm in DMSO), and deferoxamine (DEF, 50 mm in external solution) were prepared on the day of the experiment. Carvacrol (100 mm, DMSO), allyl isothiocyanate (1 m in ethanol), capsaicin (1 mm in ethanol), succinyl acetone (10 mm in H2O), HC-030031 (10 mm in DMSO), and 4-(3-Chloro-2-pyridinyl)-N-[4-(1,1-dimethylethyl)phenyl]-1-piperazinecarboxamide (BCTC) (10 mm in ethanol) were kept as stock solution at −20°C and diluted to the working concentration immediately before the experiment. In all experiments, the final concentration of DMSO or ethanol was ≤0.1%.
Animals.
Breeding and euthanasia and all procedures of animal handling were performed according to regulations of animal care and welfare (TierSchG). Experiments were performed in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC). TRPA1−/− mice were donated by Harvard University (Drs. Kelvin Kwan and David Corey, Howard Hughes Medical Institute, Department of Neurobiology, Harvard Medical School, Boston, Massachusetts) and TRPV1−/− mice by Dr. John Davis (formerly GlaxoSmithKline); both mouse lines were backcrossed onto C57BL/6 for >15 generations and used to breed the respective double knock-out mice. Mice of either sex were killed by inhalation of increasing levels of CO2.
Cell culture and transfection.
DRGs from all spinal levels were excised from adult male mice and transferred to DMEM containing 50 μg/ml gentamicin. DRGs were treated with 1 mg/ml collagenase and 0.1 mg/ml protease for 30 min, mechanically dissociated with a fire-polished silicone-coated Pasteur pipette and plated onto glass coverslips previously coated with poly-d-lysine (200 μg/ml). DRG neurons were cultured in serum-free TNB 100 cell culture medium supplemented with TNB 100 lipid-protein complex and streptomycin/penicillin (100 μg/ml, all from Biochrom). Mouse NGF was added at 100 ng/ml (Alomone Labs), and the cells were kept at 37°C and 5% CO2 for up to 24 h.
HEK293t cells were regularly passaged and controlled for mycoplasma contamination. HEK293t cells were transiently transfected with plasmids containing hTRPA1, hTRPV1, and hTRPA1 mutants hTRPA1-C621S/C641S/C665S (hTRPA1–3C) and hTRPA1-C633S/C651S (hTRPA1–2C) using jetPEI transfection reagent from Polyplus Transfection. The hTRPA1–2C mutant was generated by site-directed mutagenesis (QuikChange II XL, Agilent Technologies). Cells were plated onto poly-d-lysine-coated glass coverslips and used for experiments within 24 h.
Calcium and ROS imaging.
HEK293t expressing the human isoform of TRPA1 or TRPV1 (hTRPA1, hTRPV1) and DRG neurons plated on glass coverslips were loaded with fura-2 AM (3 μm, ∼30 min at 37°C, containing also 0.02% Pluronic, both from Invitrogen) and washed for ∼10 min before recording. Coverslips were then mounted on an Olympus IX71 inverted microscope and imaged using a 10× objective with 0.4 numerical aperture. Cells were permanently superfused with ES using a software-controlled 7-channel gravity-driven common-outlet system (Dittert et al., 2006). Fura-2 was excited at 340 and 380 nm with a Polychrome V monochromator (Till Photonics), with a FWHM wavelength range of ∼10 nm (e.g., 380 nm should be understood as 380 ± 5 nm). Fluorescence emission was long-passed at 495 nm, unless otherwise specified, pairs of images were acquired at a rate of 1 Hz with an exposure time of 4 ms with a 12 bit CCD camera (Imago Sensicam QE, Till Photonics). Data were processed off-line using the TILLvisION 4 (Till Photonics). The background intensity was subtracted before calculating the ratio between the fluorescence emitted when the dye was excited at 340 nm and at 380 nm (F340/380 nm). The time course of this ratio was analyzed for regions of interest adapted to individual cells. Response amplitudes were calculated as the integral of the ratio change for a period matching the duration of the stimulus but at least for 30 s. An average increase of the F340/380 ratio >0.1 in the analysis period was considered a response. This reflects a calcium increase >100 nm. To monitor ROS production, HEK293t cells or DRG neurons were incubated with hydroethidine (20 μm in ES, 10 min), 123 dihydrorhodamine (DHR, 20 μm in ES, 10 min) or carboxy-dichlorodihydrofluorescein (H2DCF, 25 μm in ES, 20 min, all from Invitrogen) and then left for 15 min to recover, changing the medium every 5 min. These dyes were excited at 480 nm, the emitted fluorescence was recorded using a 510 nm long pass filter. For light stimulation in addition to fluorescence imaging, cells were exposed to the respective wavelength for 940/1000 ms.
Whole-cell patch-clamp recordings.
Voltage-clamp experiments were performed on HEK293t cells transiently transfected with a hTRPV1::YFP fusion plasmid or with hTRPA1 equipped with a IRES-based YFP coexpression for selecting successfully transfected cells. Currents were acquired using an Axopatch 200B patch-clamp amplifier (Molecular Devices), low-passed at 1 kHz, and sampled at 2 kHz; pCLAMP 10 was used for voltage control, data acquisition, and off-line analysis. Patch pipettes were pulled from borosilicate glass tubes (TW150F-3, World Precision Instruments) and heat-polished to a final resistance of 1.7–4.0 mΩ. Cells were held at −60 mV and probed every 4 s by voltage ramps from −100 to 100 mV of 400 ms duration. Illumination of cells during patch-clamp recordings was performed using a mercury arc lamp and the optical filters of the imaging system (410 nm short pass filter for UV stimulation or a 460–490 nm bandpass filter).
Single-channel activity in planar lipid bilayers.
The expression and purification of hTRPA1 were performed as described previously (Moparthi et al., 2014). Briefly, purified hTRPA1 was reconstituted into preformed planar lipid bilayers composed of 1,2-diphytanoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids) and cholesterol (Sigma-Aldrich) in a 9:1 ratio and produced by using the Vesicle Prep Pro Station (Nanion Technologies). Under these conditions, a uniform protein orientation is favored with N- and C termini facing the recording chamber (i.e., the “cytosolic compartment”) (Moparthi et al., 2014). PpIX (1 μm) was added to this compartment when photosensitization was tested. The 405 nm laser illumination had a light intensity of 0.45 mW/mm2 (LT-2010, www.miniinthebox.com). This laser wavelength was closest to the observed maximum sensitivity, measured in calcium imaging experiments using the monochromator. Ion channel activity was recorded using the Port-a-Patch (Nanion Technologies) at both positive and negative test potentials in a symmetrical K+ solution (150 mm KCl, 10 mm HEPES) adjusted to pH 7.2 with KOH. Signals were acquired with an EPC 10 amplifier and PatchMaster software (HEKA) at a sampling rate of 50 kHz. The patch-clamp experiments were performed at room temperature. Electrophysiological data were analyzed using Clampfit 9 (Molecular Devices) and Igor Pro (WaveMetrics). Data were processed by a Gaussian low-pass filter at 1000 for analysis and 100 or 500 Hz for traces. The single-channel mean conductance (Gs) was obtained from Gaussian fit of all-points amplitude histograms. The single-channel mean open probability (Po) was calculated from time constant values, which were obtained from exponential standard fits of dwell time histograms.
Calibration of cellular light stimulation.
The intensity of the light beam in the inverted microscope was calibrated using a 468 nm light emitting diode (LED) with a peak emission of 650 mcd over a solid angle of 0.076 steradian (and therefore a luminous flux of 0.05 lumen). The luminous flux of the LED was converted using the luminosity function to a power of 0.87 mW. The LED and the monochromator light intensities were measured with a S2386 photodiode (Hamamatsu Photonics), and the measurements were corrected for wavelength-dependent sensitivity. The Polychrome V monochromator (see above) illuminated an area of 6 mm2 in a largely homogeneous fashion and had an almost constant power output of 0.4 mW/mm2 in the range 360–400 nm. This translates to 0.0076 mW/mm2 when only fluorescence at 340 and 380 nm was measured once per second for calcium measurement and ∼0.36 mW/mm2 for light stimulation (940/1000 ms) in addition to fluorescence imaging.
Neuropeptide release.
The experiments measuring release of CGRP were performed as previously described (Averbeck and Reeh, 2001; Kichko et al., 2015). Briefly, hindpaw skin flaps of adult mice were carefully excised and washed for 30 min in carbogen-saturated synthetic interstitial fluid with or without PpIX (5 μm). Experiments in a 25°C water bath consisted of three 10 min incubation steps in glass tubes filled with 700 μl SIF. In the second step, the preparations were continuously illuminated through a triple light guide by a standard cold light source (150 W, KL1500, Schott) immersed in the bath. Thermal effects of the light source were determined by a thermocouple to ≤0.2°C increase. The mouse trachea was excised together with the two main bronchi, cut open along the dorsal midline, and mounted upside down on a triangular aluminum block (22°C) for continuous gravity-driven superfusion in the natural direction of the mucus flow. Light stimulation was performed for a period of 5 min using a 405 nm laser, defocused to provide a homogeneous rectangular illuminated area of 15 × 10 mm from a distance of 130 mm. The respective light intensity measured with a calibrated power meter at this distance was 0.45 mW/mm2 (VEGA with 3A-ROHS sensor, Ophir Photonics). During light stimulation, the surface temperature of the trachea rose from 22 ± 0.5°C to 23 ± 0.6°C as determined by a clinical infrared thermometer (FTN, Medisana). The CGRP content of the incubation fluid was measured using a commercial enzyme immunoassay kit (Bertin Pharma) as previously described (Averbeck and Reeh, 2001). The enzyme immunoassay plates were analyzed photometrically using a microplate reader (Dynatech).
Human psychophysical experiments.
Human experiments have been conducted according to the Declaration of Helsinki. The experiments and the subject's consent forms were approved by the institutional Ethics Committee of the University of Erlangen. Seven healthy volunteers (aged 39–66 years, 2 female), educated about the purpose of the study, signed the informed consent form at least one day before the experiments and completed a medical history questionnaire. On the day of the experiments, subjects were seated comfortably in a dentistry chair and injected with 100 μl of sterile SIF or ALA (1, 3.1, and 10 mm) diluted in SIF in a double-blinded design. Solutions were randomized and blinded by an independent person not involved in the experiment and only unblinded after the experiment. Injection sites were covered to avoid light exposure. Three hours after injection into the volar side of the forearm, light stimulation was first tested at untreated skin as training, and then the four test sites were stimulated at an interval of 5 min. For light stimulation, the defocused 405 nm laser mentioned above was positioned directly on the skin with a 5-mm-thick rubber foam spacer, generating a rectangular light spot of 6 × 4 mm with an intensity of 3.2 mW/mm2 at the skin surface. The pain intensity was reported by the subjects every 5 s by means of a numeric rating scale (NRS, 0–100) with the fixed points “no sensation” (0), “minimal pain” (10), and “maximum imaginable pain” (100). Stimulation was terminated after 60 s or when reaching a rating of 50. Subjects were instructed to indicate the rating of 50 at any time; the exact time was then recorded. For averaging, a value of 50 was assumed for all time points after termination before 60 s. Subjects were instructed to avoid light exposure within 48 h of the injection.
Statistics.
Data are presented as mean ± SEM. Two experimental groups were compared by t tests for dependent or independent samples. Multiple groups were evaluated by ANOVA followed by a HSD post hoc test. Analysis was performed using Statistica 7 (StatSoft). All statistical tests were two-sided. A value of p < 0.05 was considered statistically significant and is reported with three valid digits.
Study approval.
Animal experiments were approved by the Animal Authority of the District Government of Mittelfranken (Ansbach, Germany). Human studies were approved by the University's Ethics committee (375-14 B).
Results
ALA enhances the pain produced by 405 nm stimulation in human subjects
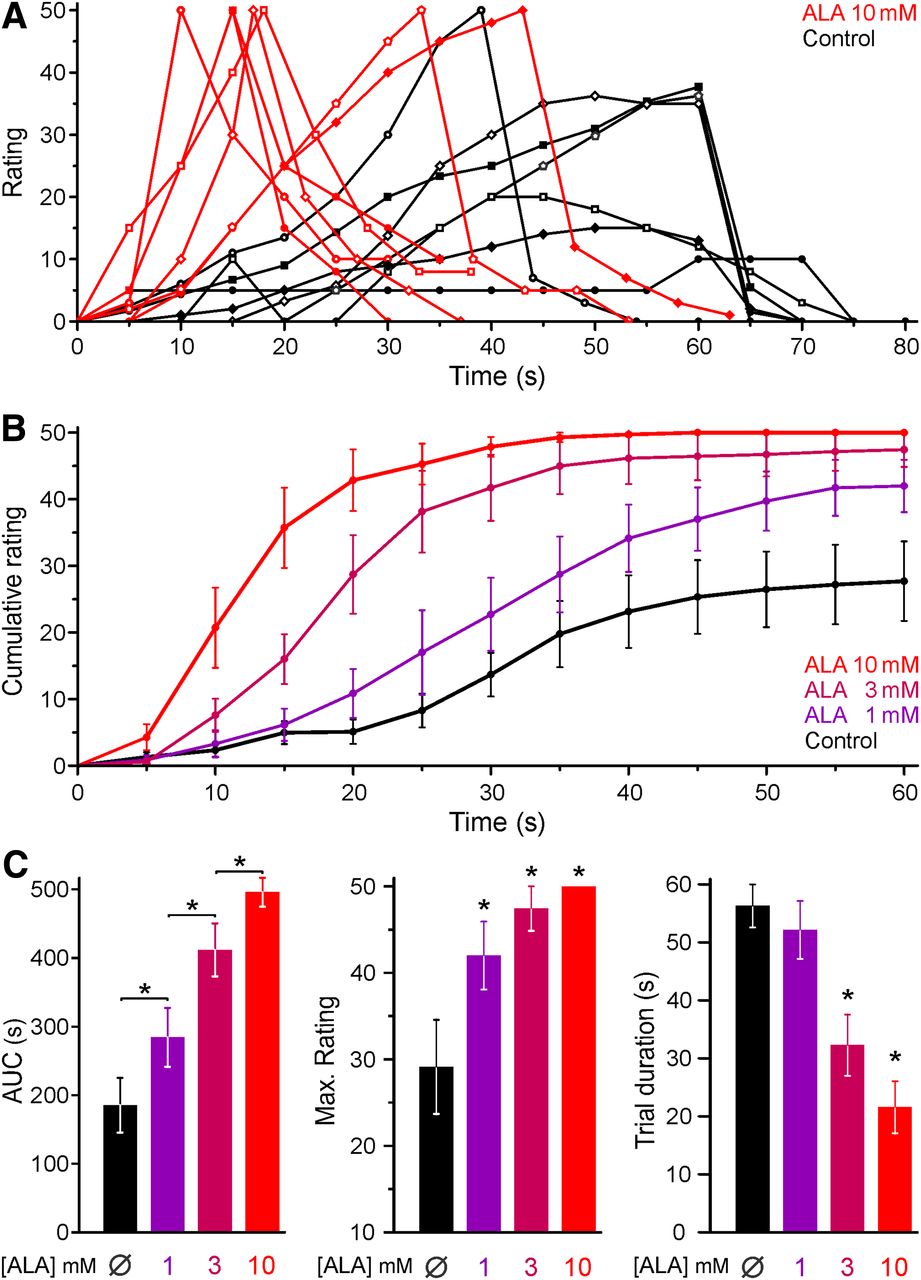
PDT with ALA is known to cause photosensitization and burning pain during therapeutic irradiation with red light used for the sake of deeper skin penetration. We asked whether the illumination of the skin with a violet laser, providing the optimal wavelength (405 nm) to excite endogenous PpIX, would also evoke pain in the absence of photosensitization. Accounting for the minor penetration depth of violet as opposed to red light and the keratinized epidermis, the laser was adjusted to deliver 3.2 mW/mm2 at the skin surface. In pilot experiments, this stimulus applied to the un-tanned volar forearm caused pin-prick sensations within ∼30 s that developed into moderate burning pain at ∼60 s and could easily be tolerated for another minute; the temperature of the skin rose by 2.2 ± 0.3°C (n = 7) in these 2 min. Based on this experience, 7 healthy volunteers were recruited and double-blind experiments performed with intradermal injections (100 μl) of various ALA concentrations (1, 3, and 10 mm) and vehicle. After an uneventful period of 3 h following injections, the 405 nm illumination revealed a painful concentration-dependent photosensitization. This is apparent in the individual trials (Fig. 1A) and in the average reported pain rating (Fig. 1B). The area under the curve and the maximum pain rating increased in a concentration-dependent manner (ANOVA, planned comparisons for the area under the curve of adjacent concentrations and HSD-post hoc tests compared with control). The cutoff was reached earlier at higher ALA concentrations, which decreased the average trial duration versus control for ALA 3 mm and above (HSD-post hoc tests, p < 0.05, Fig. 1C). It should be noted that injections of ALA could not be distinguished from vehicle and did not cause any pain in the 3 h interval between injection and illumination, during which the injected skin was protected from ambient light. Having demonstrated that blue light irradiation itself causes pain, which is strongly enhanced following treatment with ALA, our aim was to identify the molecular targets responsible for this photoactivation and photosensitization of nociceptors.

ALA sensitizes human skin to blue light illumination. In healthy human volunteers, ALA was injected into the epidermal layer of the skin. After a period of 3 h, the skin was illuminated with a 405 nm laser. Subjects rated the painful sensation on a numerical scale (1–100) every 5 s; the illumination was terminated after 60 s or when reaching a rating of 50. A, Time course of individual pain ratings induced by blue light after ALA 10 mm or control solution. The trials of the same subject are indicated by the same symbol. Recovery was recorded for 20 s after the end of the light stimulation. B, The averaged pain rating of 7 volunteers shows a concentration-dependent increase in the pain rating after ALA compared with control. For trials terminated earlier, a rating of 50 was extrapolated for all subsequent time points. C, ALA pretreatment photosensitized, which manifests as an increased area under the curve (AUC) of the rating, an increased maximum rating reported in a trial, and a decreased trial duration. *p < 0.05.
Activation of TRPA1 by near UV light is mediated by formation of ROS
Human TRPA1 and TRPV1 were transiently expressed in HEK293t cells and exposed to UVA light (390 nm, 85 s exposure in 90 s). Cells expressing hTRPA1 displayed large calcium transients in response to this light stimulus, compared with minimal responses in hTRPV1-expressing cells and no responses in untransfected HEK293t cells (Fig. 2A). To obtain the action spectrum of light-dependent activation of hTRPA1, cells were stimulated by monochromatic light in the range of 360–470 nm for 2 min in separate experiments. Activation of hTRPA1 occurred in a wide wavelength range, but with a clear optimum in the range of 410–430 nm (Fig. 2B). Using the other optical cubes of the microscope, no activation was observed by 2 min of stimulation at 488 or 562 nm.

Characterization of UV-induced activation of hTRPA1 and hTRPV1. A, UV light (390 nm, 85 s illumination within 90 s) evokes calcium transients in hTRPA1-expressing HEK293t cells (n = 108) and, to a lesser extent, in cells expressing hTRPV1 (n = 170), but does not activate untransfected cells (n = 91). Carvacrol (Carv., 100 μm) and capsaicin (Caps., 300 nm) were used to confirm functional expression of hTRPA1 and hTRPV1. B, The action spectrum of hTRPA1 activation was tested using monochromatic light stimulation for 120 s at the given wavelength ±5 nm. C, Compared with control conditions (n = 92), light-induced activation was absent without extracellular calcium, but large calcium transients were observed when calcium was resupplied (n = 97). In the absence of UV stimulation, the rebound calcium transient was substantially smaller (n = 83). D, hTRPA1-expressing cells were stimulated with 390 nm in the absence (n = 289) and in the presence (n = 103) of the TRPA1 antagonist HC-030031 (10 μm HC). E, F, The UV-induced activation of TRPA1 depends on critical N-terminal cysteines. Compared with WT hTRPA1 (n = 289 and n = 71), the hTRPA1–3C mutant (hTRPA1–C621S/C641S/C665S; n = 222) and the hTRPA1–2C mutant (hTRPA1–C633S/C651S, n = 54) showed a reduced response UV. G, H, The UV-induced activation of TRPA1 is mediated by ROS production. HEK293-hTRPA1 were illuminated with UV light in control conditions (n = 178 and n = 77), or upon pretreatment with NAC (10 mm, 4 h; G, n = 129) or DTT (5 mm, 4 h; H, n = 110). Both NAC and DTT reduced the UV response amplitude, whereas the carvacrol response is unaffected. Data are mean ± SEM.
The source of the constitutive light-evoked calcium transients in hTRPA1-expressing cells was investigated. In the absence of extracellular calcium ions, exposing the cells to 390 nm failed to evoke an increase in intracellular calcium concentration ([Ca2+]i), demonstrating a stimulated calcium entry by UVA light (Fig. 2C). The involvement of TRPA1 in the generation of these calcium transients was tested using the selective TRPA1 antagonist HC-030031 (10 μm), which completely abolished the response to 390 nm (Fig. 2D). Interestingly, upon return to a calcium-containing external solution (Fig. 2C), or upon removal of the antagonist (Fig. 2D), large rebound calcium transients were generated even in the absence of illumination, suggesting either the accumulation of an intracellular chemical agonist produced by 390 nm light, or an outlasting modification of TRPA1, which are both able to trigger calcium entry once it becomes available or the antagonist is removed. Activation of TRPA1 by electrophilic compounds involves certain critical cysteine residues located in the cytoplasmic N-terminal domain of the protein (Macpherson et al., 2007). The role of these residues in the activation of hTRPA1 by UVA light was investigated in two mutants: hTRPA1-3C, in which cysteines were substituted by serines in positions 621, 641, and 665; and hTRPA1-2C, in which the cysteines C633 and C651 involved in disulfide bonds with the cysteines of the hTRPA1-3C mutant were replaced by serine (Eberhardt et al., 2012, 2014). Cells transfected with either mutant displayed substantially diminished responses to UVA light: the response of the hTRPA1-3C mutant to 390 nm (90 s) was reduced by ∼70% (Fig. 2E), the response of the hTRPA1-2C mutant to the same stimulus was completely abolished (Fig. 2F); both mutants were well activated by carvacrol.
The activation of hTRPA1 by 390 nm light was antagonized by the antioxidants NAC and dithiothreitol (DTT) acting as ROS scavengers. Compared with the light-induced rise of [Ca2+]i of hTRPA1-expressing cells in control experiments, the response was substantially reduced after pretreatment with either NAC (10 mm, 4 h, 55% decrease, t-test independent samples, p < 0.001, n = 178 for control and 129 for NAC-treated cells; Fig. 2G) or DTT (5 mm, 4 h, 67% decrease, p < 0.001, n = 77 for control and 110 for DTT-treated cells; Fig. 2H). This suggests that oxidative processes, likely the formation of disulfides between cysteine thiols, are involved in the activation of TRPA1 at 390 nm.
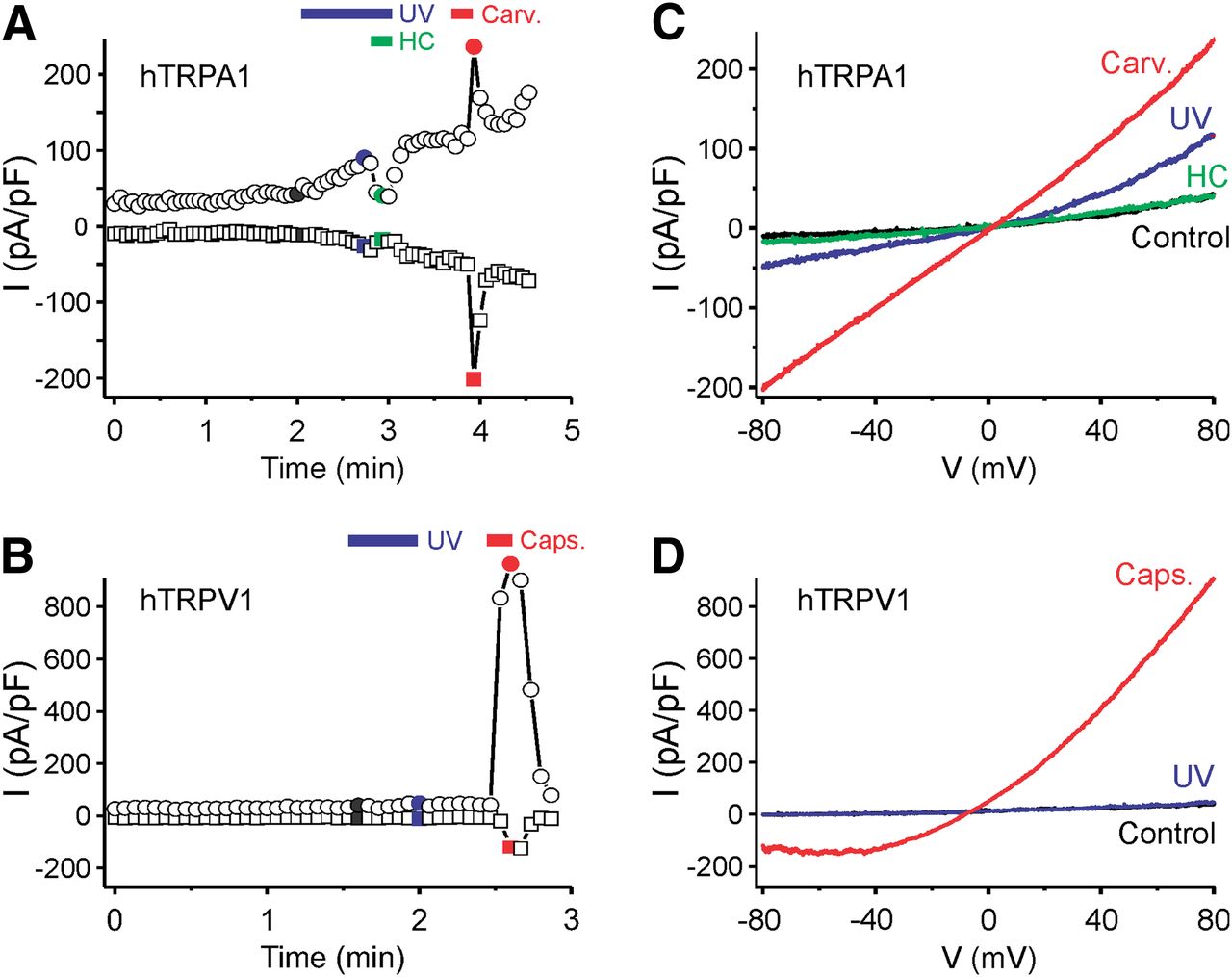
Transfected cells were also exposed to UV light using the 30 W mercury lamp of the inverted microscope and a 410 nm short-pass filter. In whole-cell voltage-clamp mode and calcium-free conditions, UV stimulation caused membrane currents in hTRPA1-expressing cells (Fig. 3A) but not in untransfected HEK293t cells. Voltage ramps induced slightly outwardly rectifying currents with a reversal potential close to 0 mV (Fig. 3B). UV illumination for >30 s frequently resulted in seal destabilization. The TRPA1 agonist carvacrol (100 μm) caused similar but stronger currents, and the UV-evoked membrane currents were completely inhibited by the TRPA1 antagonist HC-030031 (10 μm). The same UV stimulus failed to elicit membrane currents in hTRPV1-expressing cells, in agreement with the low level of light-induced activity observed in calcium imaging experiments (Fig. 3C,D).
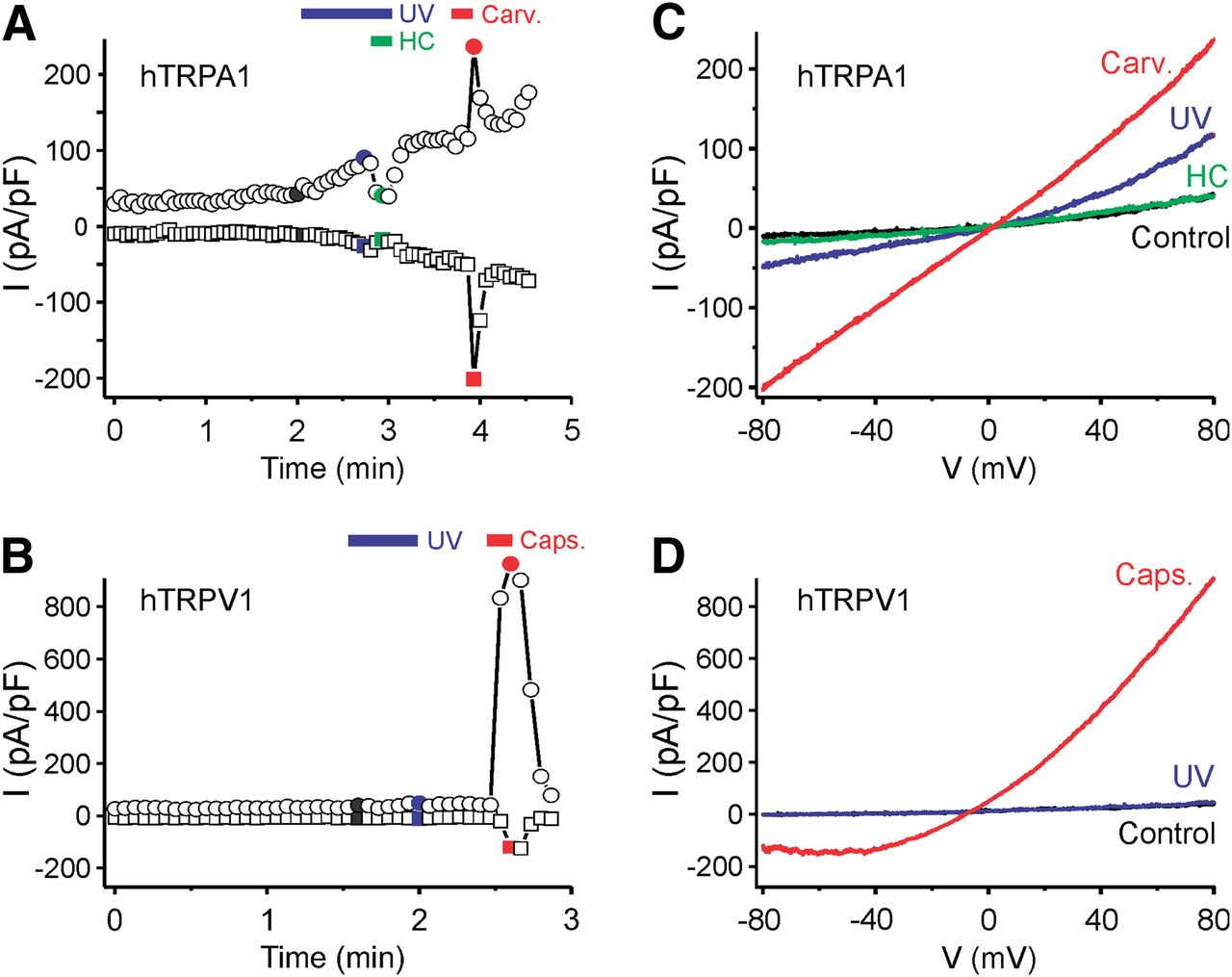
Exposure to UV light induces membrane currents in hTRPA1- but not in hTRPV1-expressing cells. A, Example of a whole-cell patch-clamp recording from a hTRPA1-expressing HEK293t cell stimulated by UV light (<410 nm) and carvacrol (Carv., 100 μm). Following UV stimulation, the TRPA1 antagonist HC-030031 (HC, 10 μm) was applied, leading to a complete inhibition of the current. B, Example of a whole-cell patch-clamp recording from a hTRPV1-expressing HEK293t cell stimulated by UV light (blue bar) and capsaicin (Caps., 1 μm). Illumination with UV light for 30 s did not evoke a TRPV1-mediated current in this cell. A, B, Plot of currents at −80 mV (squares) and 80 mV (circles) extracted from voltage ramps. C, D, Ramp currents recorded at the times indicated with the corresponding color in A and B.
In DRG neurons from TRPA1−/− mice, 390 nm-illumination induced approximately one-third of the calcium increase observed in C57BL/6 neurons (p < 0.001, n = 194 for C57BL/6 and n = 158 for TRPA1−/−; Fig. 4A). In the 194 C57BL/6 DRG neurons, 41 (21%) responded to light exposure with an increase in [Ca2+]i. Of these 41 neurons, 34 (83%) were also activated by carvacrol. Only 7 of 116 (6%) carvacrol-insensitive neurons were activated by the 390 nm stimulus. This indicates a strong coexpression of the sensitivities to light and carvacrol in cultured DRG neurons from C57BL/6 mice (Fig. 4B). Carvacrol (100 μm) activated 40% and capsaicin (1 μm) 61% of all DRG neurons tested. In contrast, in TRPA1−/− mice, the sensitivity to both UV light and carvacrol was almost completely abolished. Only 1 of the 158 neurons from TRPA1−/− DRGs responded to the 390 nm stimulus, and only 7 neurons (4%) were activated by carvacrol, whereas 92 (58%) were activated by capsaicin (Fig. 4B). These results demonstrate that the neuronal responsiveness to illumination at 390 nm is primarily mediated by TRPA1.

UV illumination generates ROS and is almost exclusively mediated by TRPA1 in DRG neurons. A, Neuronal responses to UV light, carvacrol, capsaicin, and KCl in DRG neurons of WT (n = 194) and TRPA1−/− animals (n = 158). Data are mean ± SEM. B, Venn diagram of WT and TRPA1−/− DRG neuron populations in percentage that responded to UV light (390 nm, 4 min), carvacrol (100 μm, 30 s), or capsaicin (1 μm, 20 s). C, D, hTRPA1-expressing HEK293t cells (C) and cultured DRG neurons (D) loaded with the fluorescence ROS indicators were illuminated with 390 nm light as indicated by the boxes (90 s cumulative illumination for hTRPA1-expressing cells and 240 s for DRG neurons). The indicators were excited at 480 nm using a different filter set (hydroethidine HE, black, n = 121 and n = 234; dihydrorhodamine 123 DHR, red, n = 128 and n = 135; H2DCF, blue, n = 100 and n = 175 for HEK293-hTRPA1 and DRG neurons). Note the substantial increase in HE and H2DCF fluorescence but not DHR fluorescence upon UV illumination in both hTRPA1-expressing cells and DRG neurons.
Short wavelength light is able to generate ROS, including singlet oxygen, superoxide anion radical, and hydrogen peroxide, in the presence of endogenous photosensitizers, such as the ubiquitous PpIX. To investigate whether the activation of TRPA1 by UVA light occurs via ROS, we monitored the production of cellular ROS by 390 nm illumination in hTRPA1-expressing cells and DRG neurons. For this purpose, the cells were loaded with the fluorescent ROS indicators hydroethidine, dihydrorhodamine 123, and dichlorodihydrofluorescein diacetate. While dihydrorhodamine 123 yielded no response in either cell type, hydroethidine and in particular dichlorodihydrofluorescein diacetate displayed large increases in fluorescence upon exposure of both HEK293t cells (Fig. 4C) and DRG neurons (Fig. 4D) to the same UVA stimulus as used for hTRPA1 activation, indicating that substantial production of ROS occurs as a result of illumination.
ALA induces photosensitization of cells expressing TRPA1 and TRPV1
ALA is a precursor in the biochemical pathway leading to heme formation (Ajioka et al., 2006) and is used in PDT for the treatment of a variety of skin conditions (Warren et al., 2009). We investigated photosensitivity of TRPA1- and TRPV1-expressing HEK293t cells after prolonged pretreatment with ALA. Cells were incubated in the dark with ALA (0.2–1 mm, 30–240 min in DMEM), after which the medium was removed and cells were loaded with fura-2 for 30 min and washed for 10 min before calcium microfluorimetry. After this treatment, the brief alternating illumination at 340 and 380 nm (1 Hz, 10 ms each) used to excite fura-2 during calcium imaging was sufficient to evoke an increase in [Ca2+]i in HEK293t cells expressing hTRPA1, and, to a lesser extent, also in cells expressing hTRPV1 (Fig. 5A,B), but not in untransfected HEK293t cells. This light-induced increase in [Ca2+]i required extracellular calcium in both hTRPA1- and hTRPV1-expressing HEK293t cells (Fig. 5C,D) and was abolished by the application of selective antagonists HC-030031(10 μm, Fig. 5E) and BCTC (1 μm, Fig. 5F).

Pretreatment with ALA dramatically increases the photosensitivity of hTRPA1 and hTRPV1. HEK293t cells expressing hTRPA1 (A), hTRPV1 (B), and untransfected control cells were preincubated with ALA (300 μm) for 3 h in the dark at 37°C. The 340/380 nm illumination (10 ms each, 1 Hz) increased intracellular calcium in cells expressing hTRPA1 and hTRPV1 but not in control HEK293t cells. hTRPA1 (C) and hTRPV1 (D) expressing cells preincubated with ALA (300 μm, 3 h) were imaged in the presence or absence of extracellular calcium. The calcium increase by the 340/380 nm illumination was abolished in calcium-free conditions, but large calcium transients occurred upon returning to a calcium-containing extracellular solution. Simply removing extracellular calcium in untreated hTRPA1 or hTRPV1-expressing cells only produced small calcium transients upon returning to a calcium-containing extracellular solution. E, F, Cells were preincubated with ALA (300 μm, 3 h) and imaged in the presence or absence of the TRPA1 antagonist HC-030031 (10 μm, HC, E) and TRPV1 antagonist BCTC (1 μm, F). Calcium transients upon removal of HC-030031 were larger in ALA-pretreated compared with untreated hTRPA1-expressing cells. In contrast, washout of BCTC did not result in calcium transients in hTRPV1-expressing cells. Data are mean ± SEM. G, Example of a whole-cell patch-clamp recording from an hTRPA1-expressing cell pretreated with ALA (300 μm, 3 h). Stimulation by blue light (460–490 nm) induced a current that was inhibited by HC-030031 (10 μm). H, Example of a whole-cell patch-clamp recording from an hTRPV1-expressing cell pretreated with ALA (300 μm, 3 h). The current induced by stimulation with blue light was inhibited by BCTC (1 μm). I, J, Ramp currents recorded at the times indicated with corresponding color in G and H. A–J, Carvacrol 100 μm (Carv.); and capsaicin 300 nm (Caps.).
Light-evoked membrane currents were recorded by whole-cell patch clamp in the voltage-clamp mode after pretreatment of TRPA1- and TRPV1-expressing cells with ALA (300 μm, 3 h). The cells were submitted to 400 ms voltage ramps from −100 to 100 mV every 4 s and were illuminated by a mercury lamp and a 460–490 nm bandpass filter for blue light stimulation. Although illumination with blue light for 1 min did not induce any change in the currents recorded in untreated cells (both n = 5 cells expressing hTRPA1 or hTRPV1), after pretreatment with ALA the same stimulus generated obvious membrane currents in cells expressing hTRPA1 or hTRPV1 (both n = 5; Fig. 5G,H). These currents were outwardly rectifying, had a reversal potential close to 0 mV, and were inhibited by application of the respective antagonists HC-030031 and BCTC (Fig. 5G–J).
Exogenous ALA enters the heme biosynthesis pathway and is converted to porphobilinogen by the enzyme ALA-dehydratase. To demonstrate the involvement of this pathway, we investigated whether succinyl acetone, an inhibitor of ALA-dehydratase (Gibson et al., 2001), can prevent the ALA-induced photosensitization. Succinyl acetone (1 μm, 4 h) alone did not alter the response of hTRPA1-expressing HEK293t cells to the brief UV pulses for fura-2 excitation. However, cells pretreated with succinyl acetone (1 μm, 1 h) and then exposed to ALA (300 μm) and succinyl acetone (3 h) displayed substantially reduced photosensitivity compared with cells exposed to ALA only (Fig. 6A). This indicates that a reduced conversion of ALA decreases photosensitization. The final step in the heme pathway is the enzymatic conversion of PpIX to heme through the insertion of ferrous iron, occurring in the mitochondria. The responsible enzyme, ferrochelatase, can be inhibited by iron-chelating agents, such as DEF (Fukuhara et al., 2013). We hypothesized that inhibiting ferrochelatase, thus mimicking erythropoetic porphyria, would increase photosensitivity due to an accumulation of the endogenous photosensitizer PpIX. This was confirmed by exposing hTRPA1-expressing HEK293t cells to DEF (500 μm), which resulted in a exposure-dependent augmentation of the response to the brief UV pulses used to excite fura-2 and to an additive effect to ALA (200 μm; Fig. 6B–D). Together, these results confirm that exogenous ALA leads to increased photosensitivity in hTRPA1-expressing cells by enhancing PpIX production.

Modulation of photosensitivity by ALA and the activated population of sensory neurons. A, hTRPA1-expressing HEK293t cells were pretreated with the ALA-dehydratase inhibitor succinyl acetone (SA, 1 μm, 4 h), ALA (300 μm, 3 h), or both (SA 4 h + ALA 3 h). Pretreatment with SA led to a diminished ALA-induced photosensitization of hTRPA1. B, hTRPA1-expressing cells pretreated with ferrochelatase inhibitor DEF (500 μm) were sensitized to the 340/380 nm illumination (10 ms each, 1 Hz) compared with untreated hTRPA1-expressing cells. C, D, hTRPA1-expressing cells were pretreated with ALA (200 μm, C) or with both ALA and DEF (500 μm, D) for 30, 60, or 120 min. E, ALA-induced photosensitization was reduced in DRGs from TRPA1 or TRPV1 knock-out mice. DRG neurons from C57BL/6 (n = 310), TRPA1−/− (n = 297), TRPV1−/− (n = 295), and TRPA1−/−/TRPV1−/− (n = 399) were preincubated with ALA (600 μm, 3 h). Bar chart represents the ratio increase by 340/380 nm illumination (10 ms each; 1 Hz). F, Venn diagram of the neuronal populations activated by 340/380 nm light, carvacrol-sensitive neurons, and capsaicin-sensitive neurons. The number reflects the respective percentage of neurons. Encompassing circle represents the total neuronal population for each genotype. A–E, Data are mean ± SEM. *p < 0.01. A–F, Carvacrol 50 μm (Carv.); and capsaicin 1 μm (Caps.).
DRG neurons pretreated with ALA (600 μm, 3 h) and loaded with fura-2 were illuminated by 340 and 380 nm (10 ms each, sampling rate 1 Hz). This illumination activated 30% (93 of 310) of WT neurons; substantially fewer responses occurred in both TRPA1−/− (18%, 53 of 297; p < 0.001, χ2 test) and TRPV1−/− mice (19%, 56 of 295; p = 0.002; Fig. 6E). The average amplitude of the increase in [Ca2+]i induced in DRG neurons by 340/380 nm illumination was reduced after genetic ablation of either TRPA1 (to 33%, p < 0.001, t test independent samples) or TRPV1 (to 46%, p < 0.001). In DRG neurons cultured from WT mice, light sensitivity was strongly correlated with the responsiveness to carvacrol and capsaicin: 61% of light-sensitive neurons responded to carvacrol and 83% to capsaicin; >90% of light-sensitive neurons were activated by either carvacrol or capsaicin or both. Moreover, of WT DRG neurons that responded to carvacrol, capsaicin, or both, 48% (84 of 174) were also activated by UV light. In contrast, only 7% (9 of 136) of carvacrol-insensitive and capsaicin-insensitive neurons were activated by UV light. In TRPV1−/− DRGs, 84% of photosensitive neurons also responded to carvacrol; and in TRPA1−/−, 81% of photosensitive neurons also responded to capsaicin (Fig. 6F). The ALA-induced light sensitivity was dependent on TRPA1 and TRPV1, as it was almost entirely abolished in DRGs from TRPA1−/−/TRPV1−/− mice (only 3 of 399 neurons were light-activated).
PpIX-induced photosensitization is mediated by TRPA1 and TRPV1
Exposure to exogenous ALA leads to accumulation of the photosensitizer PpIX through the heme pathway within the cells. We investigated whether PpIX is able to photosensitize cells expressing recombinant hTRPA1 and hTRPV1 by acutely exposing the cells to PpIX. In the presence of PpIX, the brief UV pulses used to excite fura-2 induced a concentration-dependent rise in [Ca2+]i in both hTRPA1- and hTRPV1-expressing HEK293t cells, whereby higher PpIX concentration increased the slope of the calcium rise rather than the finally reached [Ca2+]i (Fig. 7A,B). PpIX was highly potent; therefore, the concentrations were further reduced to 0.1 and 1.0 nm; PpIX was first applied in the dark, then in the presence of the 340/380 nm fura-2 excitation and finally during continuous illumination with 360 nm UV light, each for 1 min. In hTRPA1-expressing cells, 1 nm PpIX caused a small calcium increase in the dark. At both 0.1 and 1 nm, PpIX acted as a concentration-dependent photosensitizer enhancing hTRPA1 activation by 360 nm light (Fig. 7C). A similar photosensitization to 360 nm light was recorded in hTRPV1-expressing cells (Fig. 7D). Specific involvement of hTRPA1 in the photosensitizing action of PpIX was demonstrated by complete inhibition for as long as HC-030031 (10 μm) was present (Fig. 7E). To investigate whether intracellular PpIX is sufficient to photosensitize cells expressing hTRPA1, we only pretreated the cells for 10 min with PpIX (10–1000 nm), relying on the fact that the compound is lipophilic and membrane-permeable. The subsequent 340/380 nm fura-2 excitation in the absence of extracellular PpIX resulted in concentration-dependent light-evoked calcium transients (Fig. 7F). However, only the highest PpIX concentration tested (1 μm) reached the effect magnitude of the coapplication of PpIX with the UV light stimuli. This suggests that also extracellular PpIX, generating membrane-permeable singlet oxygen upon illumination, may contribute to hTRPA1 activation by oxidation (Snyder et al., 2005).

PpIX activates hTRPA1 and hTRPV1. A, In hTRPA1-expressing HEK293t cells PpIX (PpIX, 5 min) evoked a concentration-dependent increase in [Ca2+]i. B, Similarly, hTRPV1-expressing cells challenged with PpIX showed concentration-dependent calcium increases. C, In hTRPA1-expressing cells, PpIX increased the UV response compared with untreated hTRPA1-expressing cells or HEK293t cells. When the 340/380 nm illumination (10 ms each; 1 Hz) within the first minute of PpIX application was omitted (gray box), a direct effect on intracellular calcium was observed for PpIX 1 nm, but not for PpIX 0.1 nm. D, hTRPV1-expressing cells exposed to PpIX increased calcium upon illumination more than untreated hTRPV1-expressing cells or HEK293t cells. E, hTRPA1-expressing cells were stimulated with PpIX (20 nm) in the absence or in the presence of TRPA1 antagonist HC-030031 (10 μm, HC). F, In hTRPA1-expressing cells preincubated for 10 min with PpIX and washed, the 340/380 nm illumination (10 ms each, 1 Hz) caused a concentration-dependent increase in [Ca2+]i. Data are mean ± SEM.
To scrutinize PpIX-induced photosensitization, whole-cell membrane currents were recorded. Application of PpIX (20 nm, 2 min) in the dark did not produce any change in membrane currents in HEK293t cells expressing hTRPA1 or hTRPV1 cells (both n = 5). Similarly, without PpIX applications, illumination with blue light (460–490 nm bandpass filter) did not elicit any change in whole-cell currents in these cells (both n = 5 for hTRPA1 and hTRPV1). However, blue light illumination in the presence of preapplied PpIX evoked large outwardly rectifying currents with a reversal potential close to 0 mV both in hTRPA1-expressing cells (n = 6; Fig. 8A,B) and in hTRPV1-expressing cells (n = 5; Fig. 8C,D).

Blue light (460–490 nm) evokes hTRPA1- and hTRPV1-dependent membrane currents in the presence of PpIX. Example of a whole-cell patch-clamp recording from an (A) hTRPA1- or (B) hTRPV1-expressing HEK293t cell stimulated with PpIX (20 nm) and then blue light (60 s) in the presence of PpIX, followed by carvacrol (100 μm). Currents at −80 mV (squares) and 80 mV (circles) of voltage ramps at regular intervals were recorded. C, D, Ramp currents recorded at the times indicated by the corresponding color in A and B, respectively. E, F, PpIX activates a subpopulation of mouse DRG neurons; the effect is substantially reduced in DRGs from TRPA1−/− and TRPV1−/− mice and abolished in TRPA1−/−/TRPV1−/− mice. E, Compared with DRG neurons from WT (n = 278 cells) stimulated by PpIX (PpIX, 100 μm), the responses were substantially reduced in TRPA1−/− (n = 330), TRPV1−/− neurons (n = 209), and TRPA1−/−/TRPV1−/− (n = 266). Data are mean ± SEM. Inset, Comparison of the response amplitudes to PpIX during 340/380 illumination. *p < 0.001. F, Venn diagram of neuronal populations sensitive to PpIX, carvacrol, and capsaicin. The number reflects the respective percentage of neurons; the encompassing circle represents the total neuronal population. A–F, Carvacrol 50 μm (Carv.); and capsaicin 1 μm (Caps.).
In C57BL/6 DRG neurons exposed to PpIX (100 nm, 4 min) the fura-2 illumination induced substantial calcium influx (n = 278). Compared with the WT, the PpIX-induced light response was only 15% in TRPA1−/− neurons (n = 330, p < 0.001, t test independent samples), and 24% in TRPV1−/− neurons (n = 209, p < 0.001; Fig. 8E). Illuminated PpIX activated 37% of the WT neurons (103 of 278), and 63% of these PpIX-sensitive neurons responded to carvacrol or capsaicin (65 neurons; Fig. 8F). Within all carvacrol- or capsaicin-sensitive neurons, 44% (65 of 148) responded to light under PpIX; the fraction of carvacrol- and capsaicin-insensitive neurons that were sensitized to light by PpIX was lower (29%; 38 of 130, p < 0.011, χ2 test). Compared with C57BL/6 DRG neurons, the fraction of PpIX light-sensitive cells was strongly reduced in TRPA1−/− neurons to 6% (18 of 330 neurons, p < 0.001, χ2 test), as well as in DRG neurons from TRPV1−/− mice (7%, 14 of 209, p < 0.001). Finally, in TRPA1−/−/TRPV1−/− neurons, photosensitization by PpIX was almost completely abolished (1 of 266).
To ensure that the blue-light activation of hTRPA1 is mediated by excitation of PpIX producing an activator of the channel, most likely singlet oxygen, we used a cell-free model using purified full-length hTRPA1 protein integrated into the artificial lipid bilayer for voltage-clamp recording of single-channel activity (Moparthi et al., 2014). The artificial membrane was illuminated by a 405 nm laser with a power density of 0.45 mW/mm2, comparable with the monochromator illumination of HEK293t cells and DRG neurons described above. This stimulation evoked single-channel currents in the presence of PpIX (1 μm) at positive and negative holding potential (Fig. 9A,B). The Po values were 0.30 ± 0.03 at 60 mV (n = 7) and 0.41 ± 0.08 at −60 mV (n = 3). The Gs values were 21 ± 2 pS at 60 mV (n = 8) and 15 ± 1 pS at −60 mV (n = 4). Without hTRPA1, no currents were elicited by the combination of 405 nm light and PpIX (n = 4; Fig. 9B). The unselective TRP channel pore blocker ruthenium red (40 μm) abolished the hTRPA1 activity (n = 3; Fig. 9C). In 2 of 4 separate experiments exposing hTRPA1 to only violet light, short initial (10 s) and minimal TRPA1 activity at 60 mV was observed (Po values were 0.04 and 0.07). TRPA1 activity was negligible in the presence of PpIX alone (Po value was 0.02) and only in 1 of 3 separate experiments at 60 mV. Thus, substantial hTRPA1 single-channel activity in this reductionistic model was only accomplished with violet light irradiation in the presence of PpIX.

Purified hTRPA1 is activated by blue light in the presence of PpIX. A, Purified hTRPA1 was inserted into planar lipid bilayers. In the presence of protoporphyrin (PpIX) at 1 μm, exposure to 405 nm light triggered TRPA1 single-channel currents. B, Neither light nor PpIX alone evoked channel activity, whereas their subsequent combination induced distinct single-channel openings at the steady-state test potentials of 60 mV and −60 mV. C, Bilayers without hTRPA1 did not respond to light in the presence of PpIX. D, The nonselective TRP channel blocker ruthenium red (40 μm) abolished TRPA1 activity. Representative traces and the corresponding amplitude histograms are shown. c, Closed channel state.
ALA and PpIX induce photosensitization of neuropeptide release through TRPA1 and TRPV1
Illumination of the C57BL/6 mouse hindpaw skin ex vivo with a cold (white) light source for 10 min did not induce an increase of CGRP release. Also, in the absence of light, PpIX (5 μm) for 10 min caused no increase of CGRP release. However, in skin flaps preincubated with PpIX (5 μm) in darkness for 30 min, the same light stimulation caused a 36 ± 4 pg/ml increase in CGRP release compared with the period before illumination (p = 0.001, n = 12, t test dependent samples; Fig. 10A). This photosensitization by PpIX was substantially decreased in TRPA1−/− and TRPV1−/− mice (ANOVA, F(3,38) = 6.2, p = 0.002 and p = 0.013, HSD post hoc tests; Fig. 10B). Compared with the large difference between WT and TRPA1−/−, less reduction of PpIX photosensitization was observed in the presence of the TRPA1 antagonist A-967079 (2 μm) and in TRPV1−/−/TRPV1−/− mice (both n = 9).
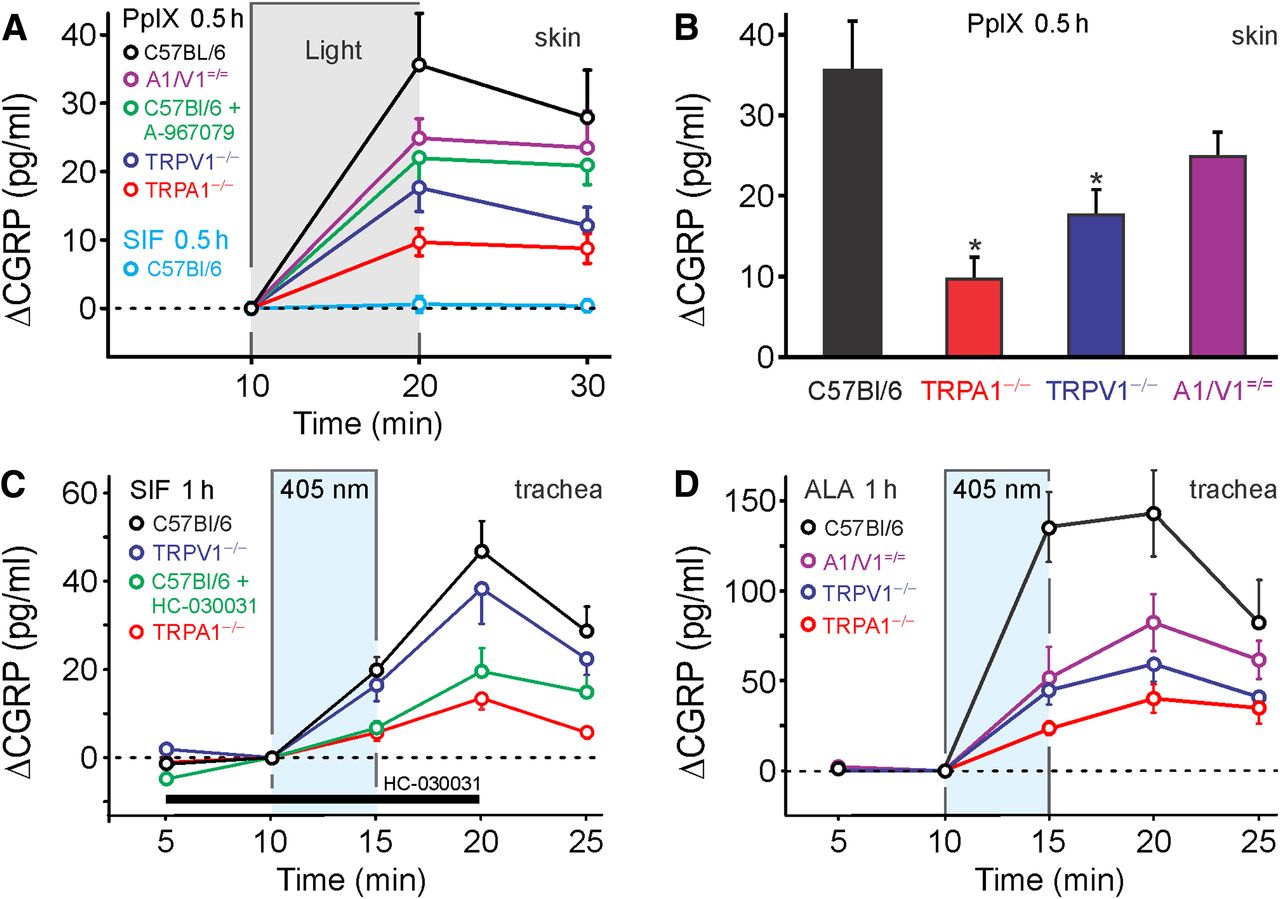
ALA and PpIX photosensitize neuropeptide release by activation of TRPA1 and TRPV1. A, In isolated hindpaw skin, release of the neuropeptide CGRP was measured by EIA as index of neuronal activation. Compared with a period without illumination, an intense broad-spectrum light caused the release of CGRP from skin pretreated with PpIX (PpIX 5 μm for 40 min, n = 10–12). Compared with C57BL/6, CGRP release was substantially reduced in hindpaw skin of TRPA1−/− and TRPV1−/− animals and absent without pretreatment by PpIX (n = 8). B, Comparison of the light-induced increase in CGRP release after PpIX pretreatment in the different genotypes (n = 10–12, p < 0.05 vs C57BL/6). C, The isolated superfused mouse trachea was stimulated by 405 nm laser illumination after 1 h in control solution (SIF). A slight but significant increase in CGRP release was observed in trachea from WT animals (C57BL/6, n = 7); this was reduced in TRPA1−/− animals (n = 4) and in the presence of HC-030031 (n = 4) but not in TRPV1−/− animals (n = 4). D, After 1 h exposure to ALA (100 μm), stimulation by the 405 nm laser caused a substantial increase in CGRP release. Compared with WT mice (n = 6), the light-induced CGRP release was significantly reduced in the trachea of TRPA1−/− (n = 5), TRPV1−/− (n = 4), and TRPA1−/−/TRPV1−/− (n = 4) animals. Data in the trachea are given as increase of CGRP (ΔCGRP) over baseline.
In addition, the mouse trachea preparation was used because the sensory nerve endings come close to the very surface of the respiratory epithelium and because the preparation can be superfused with a thin layer of SIF not impeding light penetration. Exposure of the mouse trachea to 405 nm violet laser light (0.45 mW/mm2) evoked a small but significant increase in CGRP release (19 ± 4 pg/ml, n = 5; Fig. 10C), which continued rising during the first wash-out period. This constitutive light response in the SIF-preincubated trachea was almost abolished by the antagonist HC030031 (50 μm) as well as in TRPA1−/− animals, while it was unchanged in TRPV1−/− mice (p = 0.013 and p = 0.008 compared with control, n = 4–7, HSD post hoc tests). The ubiquitous presence of PpIX in all cells might be sufficient to provide a genuine blue light sensitivity mediated by TRPA1, in agreement with our psychophysical finding that the same blue light caused pain in human skin.
After ALA (100 μm, 1 h) preincubation, the 405 nm laser stimulation increased CGRP release sevenfold (135 ± 20 pg/ml, n = 6; Fig. 10D). Compared with their WT littermates, the 405 nm-stimulated tracheal CGRP release after ALA pretreatment was only 17% in TRPA1−/−, 33% in TRPV1−/−, and 38% in TRPA1−/−/TRPV1−/− (ANOVA, F(3,15) = 12.3, p < 0.001, HSD post hoc tests; Fig. 10D). In the presence of both antagonists HC-030031 (50 μm) and BCTC (10 μm), the photosensitization by ALA was decreased to 37% (data not shown). Intriguingly, neither of the two TRP channel antagonists alone was able to significantly reduce the blue light response of the photosensitized trachea, only the combination was effective, while either of the two knock-out strains showed a marked reduction of the light sensitivity. The mere pharmacological inhibition of the one channel seems to be overcome by the activation of the other channel, as long as both proteins are present. If one of the channel proteins is missing, a putative synergism between the two TRP channels is lacking and light activation falls to the same low level as achieved by the combination of inhibitors.
Discussion
Porphyrias comprise a group of rare, but severe and highly disabling, diseases (Kauppinen, 2005). The cause of the disease is known: a nonfunctional enzyme in the ubiquitous heme biosynthetic pathway causes an accumulation, in particular in bone marrow and liver cells, of a biochemical intermediate, such as PpIX, which spills into the blood and extracellular fluids. This leads to either visceral pain symptoms (in acute porphyrias) or, in the case of porphyrin accumulation, cutaneous light hypersensitivity, the most disabling symptom of which is intense burning pain occurring within minutes of exposure to light, particularly in erythropoietic porphyria (Foran and Abel, 2003; Sassa, 2006; Horner et al., 2013; Tintle et al., 2014). The mechanism causing this pain was unexplained. In the most severely affected patients, the disease manifests during childhood, can be life-threatening due to liver failure, and eventually necessitates blood transfusions and even bone marrow transplantation. Similarly, pain is the main adverse effect of PDT, which uses exogenous ALA or analogs to enhance production of PpIX in the target tissue, followed by prolonged illumination with visible red light (Halldin et al., 2011, 2013). However, all living cells contain and require PpIX as a part of the vitally important heme pathway. Thus, illumination of normal skin with the optimal wavelength (405 nm) to excite PpIX and generate singlet oxygen should also be able to evoke pain. This was the case in all of our human volunteers, and it occurred within 60 s. The double-blind study with subepidermal injections of ALA and vehicle revealed a dramatic augmentation of the blue light-induced pain due to photosensitization. Even then, the pain immediately subsided when the stimulating laser was switched off.
Our results identify the thermo-TRP channels TRPA1 and TRPV1 as key targets in the treatment of pain and inflammation evoked by light exposure in patients with cutaneous porphyrias or treated with PDT. Activation of TRPA1 by UVA light and the increased light sensitivity of the channel in presence of certain photosenzitizer molecules (hypericin and acridine orange) has been reported, as well as evidence for a role for ROS in mediating these effects (Hill and Schaefer, 2009). We found that constitutive TRPA1 photosensitivity occurs over a wide range of wavelengths, spanning both UVA and blue visible light, with highest efficiency in the range of 400–430 nm. The increase in intracellular calcium concentration evoked in TRPA1-expressing HEK293t cells by illumination with 390 nm light is due to calcium entry, not release from intracellular stores, and is completely abolished in the presence of the TRPA1 antagonist HC-030031. Several cysteine residues from the cytoplasmic N terminus domain of TRPA1 (C621, C641, C665, and particularly C633 and C651) appear to be critical for the light activation of the channel. Our previous work indicated the oxidative generation of N-terminal disulfide bonds between some of these cysteines; upon exposure to methylglyoxal, disulfide bonds between cysteines at positions 621/633 and positions 641/651 were observed (Eberhardt et al., 2012), while nitroxyl induced bonds between cysteines at positions 621/633 and positions 651/665 (Eberhardt et al., 2014). In both cases, the previously unsuspected amino acids 633 and 651 appear to be involved. Interestingly, sensitivity to light was abolished when cysteines 633 and 651 were replaced by serine, underlining the importance of these juxtamembrane cysteines for the sensitivity to electrophilic agonists and for oxidation of hTRPA1. The mechanism responsible for hTRPV1 activation by oxidative agents also involves covalent modification of intracellular cysteines at positions 158, 387, 391, and 767 (Chuang and Lin, 2009).
Intracellular singlet oxygen generation by red light irradiation through Type II photosensitizing reactions appears to be the main effect responsible for biological actions during PDT, albeit superoxide anion radical may also be involved to some extent (Henderson and Dougherty, 1992). While exogenous photosensitizers can substantially enhance light-induced responses, native untreated (but not untransfected) cells also demonstrate constitutive photosensitivity, possibly due to intermediates of the ubiquitous heme pathway. Both hTRPA1-expressing cells and native mouse DRG neurons displayed calcium transients in response to illumination with 390 nm light. This activity is mediated via generation of ROS, as antioxidants NAC and DTT reduced the response to light stimulation. Moreover, the same stimulus that was used to evoke TRPA1-mediated calcium entry was sufficient to generate substantial responses from the fluorescent ROS indicators hydroethidine and H2DCF. The precise identification and quantification of the actual ROS generated remain to be addressed. While, for example, increased hydroethidine fluorescence is generally interpreted as being associated with superoxide formation, recent work has shown that most fluorescent ROS indicators available, including those used in this study, do not possess sufficient specificity to allow unequivocal ROS identification (Price and Kessel, 2010; Zielonka and Kalyanaraman, 2010). The fact that neither light per se nor the photosensitizer PpIX alone was able to evoke substantial hTRPA1 activity was emphasized with the purified channel protein reconstituted in artificial lipid bilayers, which showed single-channel activity as soon as PpIX was irradiated by the optimal wavelength (405 nm) to generate singlet oxygen (Rossi et al., 2008).
Using a heterologous expression system, we demonstrate that three procedures (application of ALA, PpIX, or inhibition of PpIX processing) leading to excess PpIX in human cells cause marked photosensitization mediated by two ion channels involved in pain signaling. Similarly, mouse DRG neurons in primary culture are rendered hypersensitive to light by preincubation with ALA as well as by acute application of PpIX. In both cases, photosensitivity is largely restricted to a subpopulation of carvacrol- and/or capsaicin-sensitive neurons, most likely nociceptors. Genetic ablation of TRPA1 and TRPV1 leads to a substantial reduction in the constitutive light sensitivity as well as in the hypersensitivity induced by ALA or PpIX. Interestingly, the fact that the PpIX-induced light sensitivity is reduced from 37% among WT neurons to the same extent by deletion of either TRPA1 (to 6%) or TRPV1 (to 7%) suggests a synergy between the two channels, whereby activation of both TRPA1 and TRPV1 is required for a full-scale development of PpIX-enhanced photosensitivity. The same logical “or” relationship is evident from the trachea results: the photosensitized CGRP release is decreased to a similar degree in either TRPA1 or TRPV1 knock-outs, and the double knock-outs show no additive effect of the gene deletions. A similar interaction with supra-additive effects was recently reported for TRPA1 and TRPV1 expressed in rat vagal pulmonary C-fiber afferents (Lin et al., 2015). Several forms of indirect or direct interaction between both channels have been described and discussed (Fischer and Edwardson, 2014; Fischer et al., 2014). Although photosensitization induced by both ALA and PpIX was almost entirely abolished in DRG neurons from TRPA1−/−/TRPV1−/− mice, we cannot rule out additional mechanisms contributing to this effect. ROS generated through PpIX-mediated photosensitization may activate other neuronal sensors of oxidative stress, such as TRPM2 (Nazıroğlu et al., 2011).
Our results obtained in cellular models were complemented by the investigation of intact nociceptive nerve endings ex vivo in mouse trachea and skin. The constitutive light sensitivity of mouse cultured sensory neurons was recapitulated in the particularly sensitive and translucent trachea preparation that even responds to visible, violet light in the absence of photosensitization and dependent on TRPA1 expression. Pretreatment with ALA or PpIX leads to photosensitization of these nerve endings in both trachea and skin from WT mice, as demonstrated by the increase in CGRP release during illumination. Light-induced CGRP release from tissues of TRPA1−/−/TRPV1−/− mice was markedly diminished compared with the WT. The pharmacological inhibition of one TRP channel seems largely compensated by the activation of the other channel; each of the TRP channels alone was apparently able to provide the calcium influx required for the marked, but still submaximal, CGRP release. However, the complete deficiency of either one of the channel proteins was not compensated, as if both proteins together were required for full responsiveness. Consistently, the TRPA1−/−/TRPV1−/− did not show a stronger, additive, reduction of the light response than the single knock-outs. This was in contrast to the complete abolition of photosensitization in DRG neurons from TRPA1−/−/TRPV1−/− mice and may be explained by contributions from other cell types releasing nociceptor-activating agents in the intact tissue preparations. The described TRPA1-TRPV1 protein-protein interaction (Fischer et al., 2014) may in part be explained by the recently discovered Tmem100, a small transmembrane protein that modulates the TRPA1-TRPV1-complex (Weng et al., 2015). Our results support the conclusion that TRPA1 and TRPV1 have an essential contribution to the pathologically enhanced activity of cutaneous nociceptors during PDT and in light-exposed porphyria patients.
The receptor channels TRPA1 and TRPV1 expressed in pain-signaling nerve endings act as cellular sensors to detect the oxidative stress produced by near UV and visible blue light, which is enhanced in the presence of cellular or exogenous photosensitizers. Selective antagonists for these channels might provide pain relief in patients suffering from cutaneous porphyrias, as well as to increase the tolerance of PDT and thus the therapeutic efficiency of this procedure.
Footnotes
A.B. was supported by the Alexander von Humboldt Foundation. C.N., A.B., M.F., and P.W.R. were supported as members of the Emerging Fields Initiative consortium Medicinal Redox Inorganic Chemistry of the University of Erlangen-Nürnberg. B.N. was supported by DFG Grant NA 970/1–1. P.M.Z. and L.M. were supported by the Swedish Research Council (2014-3801), the Medical Faculty at Lund University, and the Research School of Pharmaceutical Sciences at Lund University. We thank Iwona Izydorczyk, Annette Kuhn, and Susi Haux-Oertel for excellent technical assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Prof. Dr. med. Peter W. Reeh, Institute of Physiology and Pathophysiology, Universitätsstrasse 17, 91054 Erlangen, Germany. peter.reeh{at}fau.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}