

Abstract
Spinal reflex circuit development requires the precise regulation of axon trajectories, synaptic specificity, and synapse formation. Of these three crucial steps, the molecular mechanisms underlying synapse formation between group Ia proprioceptive sensory neurons and motor neurons is the least understood. Here, we show that the Rho GTPase Cdc42 controls synapse formation in monosynaptic sensory–motor connections in presynaptic, but not postsynaptic, neurons. In mice lacking Cdc42 in presynaptic sensory neurons, proprioceptive sensory axons appropriately reach the ventral spinal cord, but significantly fewer synapses are formed with motor neurons compared with wild-type mice. Concordantly, electrophysiological analyses show diminished EPSP amplitudes in monosynaptic sensory–motor circuits in these mutants. Temporally targeted deletion of Cdc42 in sensory neurons after sensory–motor circuit establishment reveals that Cdc42 does not affect synaptic transmission. Furthermore, addition of the synaptic organizers, neuroligins, induces presynaptic differentiation of wild-type, but not Cdc42-deficient, proprioceptive sensory neurons in vitro. Together, our findings demonstrate that Cdc42 in presynaptic neurons is required for synapse formation in monosynaptic sensory–motor circuits.
SIGNIFICANCE STATEMENT Group Ia proprioceptive sensory neurons form direct synapses with motor neurons, but the molecular mechanisms underlying synapse formation in these monosynaptic sensory–motor connections are unknown. We show that deleting Cdc42 in sensory neurons does not affect proprioceptive sensory axon targeting because axons reach the ventral spinal cord appropriately, but these neurons form significantly fewer presynaptic terminals on motor neurons. Electrophysiological analysis further shows that EPSPs are decreased in these mice. Finally, we demonstrate that Cdc42 is involved in neuroligin-dependent presynaptic differentiation of proprioceptive sensory neurons in vitro. These data suggest that Cdc42 in presynaptic sensory neurons is essential for proper synapse formation in the development of monosynaptic sensory–motor circuits.
Introduction
During nervous system development, important sequential steps such as axon guidance, selective target recognition, and synapse formation must be regulated properly for the appropriate formation of neural circuits. Monosynaptic sensory–motor reflex arcs provide us with an elegant model system with which to understand the molecular mechanisms underlying each step of neural circuit assembly. In these circuits, group Ia proprioceptive sensory neurons, the cell bodies of which are located in the DRGs, project axons centrally to the ventral spinal cord, where they make direct contacts and form mature synapses with particular subsets of motor neuron pools (Brown, 1981). Previous studies have revealed how transcription factors, ligand–receptor interactions, and intracellular kinases control proprioceptive axon trajectories within the spinal cord (Arber et al., 2000; Inoue et al., 2002; Levanon et al., 2002; Patel et al., 2003; Chen et al., 2006; Yoshida et al., 2006; Wang et al., 2007; Leslie et al., 2011; Lilley et al., 2013). More recently, studies have been undertaken to explore the cellular and molecular mechanisms underlying synaptic specificities between group Ia afferents and motor neurons (Pecho-Vrieseling et al., 2009; Sürmeli et al., 2011; Fukuhara et al., 2013). A motor-neuron-independent, dorsoventral tier-targeting system (Sürmeli et al., 2011) and motor-neuron-dependent molecules such as semaphorin 3E (Pecho-Vrieseling et al., 2009; Fukuhara et al., 2013) have been proposed to regulate the exquisite motor pool specificity achieved in the development of monosynaptic sensory–motor circuits. However, in contrast, the molecular mechanisms underlying the final formation of synapses within these connections remain obscure.
Synaptogenesis requires rearrangement of the actin cytoskeleton in both presynaptic and postsynaptic regions. The small Rho GTPases are essential regulators of cytoskeletal reorganization (Heasman and Ridley, 2008; Hall and Lalli, 2010; Hall, 2012). In particular, the small Rho GTPase Cdc42 has been shown to play critical roles in synapse formation in postsynaptic neurons in a variety of systems. For example, Cdc42 influences the dendritic morphologies of various types of neurons, including cortical neurons (Irie and Yamaguchi, 2002; Scott et al., 2003; Murakoshi et al., 2011; Rosário et al., 2012), and loss of Cdc42 in mice causes impaired postsynaptic structural plasticity of dendritic spines in the hippocampus (Kim et al., 2014). Compared with its synaptogenic roles in postsynaptic neurons, the functions of Cdc42 in presynaptic neurons have been largely unexplored. A few studies have been performed using a dominant-negative form of Cdc42, which showed that Cdc42 activation initiates reorganization of the presynaptic network in Aplysia (Udo et al., 2005) and that activity-induced presynaptic maturation in cultured neurons is mediated by Cdc42 signaling through actin (Shen et al., 2006). However, genetic evidence for a presynaptic role for Cdc42 in synapse formation has yet to be documented.
In this study, we show that, during sensory–motor circuit development, Cdc42 expression is detected in both sensory and motor neurons contemporaneously with the period of synapse formation. Motor-neuron-specific deletion of Cdc42 in mice does not yield any obvious defects in sensory–motor synapses. In contrast, both anatomical and electrophysiological analyses demonstrate impairments in monosynaptic sensory–motor connectivity in sensory-neuron-specific Cdc42 mutant mice. Furthermore, the synaptic organizers, neuroligins (NLs), which can induce presynaptic differentiation in proprioceptive sensory neurons in vitro, require Cdc42 for the differentiation process. Therefore, our findings strongly suggest that Cdc42 in presynaptic sensory neurons plays a significant yet restricted role in affecting synaptogenesis in monosynaptic sensory–motor connections without adversely affecting synaptic specificity or synaptic transmission.
Materials and Methods
Mice.
The following mouse lines were used for this study: Cdc42-floxed (Yang et al., 2006), Advilln-Cre (Hasegawa et al., 2007), ChAT-Cre (Rossi et al., 2011), Pv-Cre (Hippenmeyer et al., 2005), and stop-floxed EGFP (Nakamura et al., 2006). Mouse handling and procedures were approved by the Institutional Animal Care and Use Committee at the Cincinnati Children's Hospital Research Foundation. Cdc42flox/flox and Cdc42flox/+;Cre mice were used as littermate controls. Both sexes were used for this study.
Tissue preparation.
Spinal cords, DRGs, and their surrounding tissues were fixed with 4% paraformaldehyde (PFA)/phosphate buffer (PB) for 2 h for immunohistochemistory or overnight for in situ hybridizations. Afterward, they were embedded in agarose gels and sectioned at 150–200 μm using a vibratome or cryoprotected in 30% sucrose, embedded in OCT compound, and sectioned with a cryostat at 10–16 μm.
In situ hybridizations.
Digoxigenin (DIG)-labeled RNA probes were synthesized using a DIG labeling kit (Roche). Template DNA for the Cdc42 RNA probe was cloned by PCR using the following primers with the underlined sequence showing the T7 promotor: 5′-CTGCTATGAACGCATCTCCA-3′, 5′- GCGCTAATACGACTCACTATAGGGG CACACCCCAAAAGGAGAGAA-3′. In situ hybridizations were performed according to standard protocols (Schaeren-Wiemers and Gerfin-Moser, 1993; Yoshida et al., 2006). For double labeling with in situ hybridization and immunohistochemistory, sections were stained with primary then Alexa Fluor 568-conjugated secondary antibodies and digital images were recorded using a fluorescence microscope (Zeiss Axio Imager). In situ hybridizations were then performed on the same sections. All images were combined using Adobe Photoshop software.
Immunohistochemistry.
Cryosections or vibratome sections were stained with different combinations of the following primary and secondary antibodies: rabbit anti-parvalbumin (anti-Pv; Swant, catalog #PV25); guinea pig anti-vesicular glutamate transporter 1 (anti-vGlut1, Millipore, catalog #AB5905); rabbit anti-vGlut1 (Synaptic Systems, catalog #135303); goat anti-ChAT (Millipore, catalog #AB144); and Alexa Fluor 488-, Cy3-, and Cy5-labeled secondary antibodies (Jackson ImmunoResearch). Images were scanned using a Zeiss LSM510 or a Nikon A1R confocal microscope. Quantification of vGlut1+ presynaptic terminals and Pv+ axons were performed using the spot and surface tools, respectively, in the IMARIS software package (Bitplane) (Leslie et al., 2011; Fukuhara et al., 2013).
Electron microscopy.
Postembedding electron microscopy was performed as described previously (Alvarez et al., 2004). Briefly, spinal cords were fixed with 4% PFA/0.25% glutaraldehyde/PB for 4 h and then sectioned in 50 μm slices using a vibratome. Sections were stained with anti-vGlut1 antibodies, biotinylated secondary antibodies (Jackson ImmunoResearch), and streptavidin 1.4 nm Nanogold probes (Nanoprobes, catalog #2016). The signal was further enhanced using the GoldEnhance kit (Nanoprobes, #2113). Sections were then embedded into resin and ultrathin sectioned (70–90 nm). Ultrathin sections were stained with uranyl acetate and lead citrate. Images were taken with an H-7600 transmission electron microscope (Hitachi).
Extracellular and intracellular recordings.
Dissection of spinal cords and recordings were performed as described previously (Mears and Frank, 1997; Mentis et al., 2011; Fukuhara et al., 2013). Briefly, spinal cords from postnatal day 5 (P5)–P7 newborn pups were removed and hemisectioned in oxygenated (95% O2/5% CO2) artificial CSF (aCSF) containing the following (in mm): NaCl (127), KCl (1.9), KH2PO4 (1.2), CaCl2 (2), MgSO4 (1), NaHCO3 (26), and d-glucose (20.5). For extracellular recordings, spinal cords were placed in a recording chamber containing recirculating oxygenated aCSF after dissection. Using tightly fitting glass pipettes, dorsal roots were stimulated with square pulses of 0.2 ms duration (0.3–0.6 mA, S88X, SIU-C; Grass Technologies). Extracellular potentials were recorded from L3, L4, or L5 ventral roots using tightly fitting glass pipettes. Signals were amplified (P55; Astro-Med) and digitized (Digidata 1440A, Clampex 10; Molecular Devices) for offline analysis. Traces presented are averages of 20 individual stimuli applied at 0.1 Hz.
Intracellular recordings were performed in a recording chamber. Obturator and quadriceps nerves were stimulated (10 mA, S88X, SIU-C; Grass Technologies) via tightly fitting glass pipettes. Intracellular potentials were recorded using glass micropipettes (90–180 MΩ) filled with 2 m potassium acetate with 0.5% fast green and 300 mm lidocaine N-ethyl bromide (Sigma-Aldrich). Traces presented were averages of 20–60 stimulation trials at 1 Hz frequency (MultiClamp 700B, Digidata 1440A, Clampex 10; Molecular Devices). Obturator or quadriceps motor neurons were identified by antidromic activation. Recordings were accepted only from neurons in which the resting membrane potentials were <−40 mV (Mears and Frank, 1997).
Labeling motor neurons.
To label motor neuron cell bodies and dendrites retrogradely, spinal cords were hemisected and incubated in recirculating oxygenated aCSF. L5 ventral nerves (for Fig. 2) or obturator and quadriceps nerves (for Fig. 7) were placed in glass pipettes filled with rhodamine-conjugated dextran (R-Dex) (D3308; Life Technologies) or FITC-conjugated dextran (D3306) and incubated for 24 h. To analyze synapse numbers on rectus femoris motor neurons, R-Dex was injected into the rectus femoris muscle at P4 and mice were killed at P7 (for Fig. 4).
In vitro culture experiments.
For DRG cultures, slides were coated with 0.01% poly-l-lysine (Sigma-Aldrich) and 3.3% Matrigel (Corning). Two DRGs from embryonic day 13.5 (E13.5) mouse embryos were cultured on coated slides and incubated with Neurobasal medium (Invitrogen) with B27 and N2 supplements (Invitrogen), l-gultamine (2 mm; Invitrogen), penicillin–streptomycin (100 U/ml; Invitrogen), and recombinant human NT-3 (20 ng/ml; R&D Systems). Half of the volume of the culture medium was changed every 3 d. 293T cells (1 × 104) transfected with GFP, HA-Neurexin1β (Addgene, catalog #59409) HA-Neuroligin2 (Addgene, catalog #15259), or HA-Neuroligin3 (Addgene, catalog #59318) (Chih et al., 2004; Chih et al., 2006) were added to the cultured DRGs after 10–11 d of incubation. After 2 d of coculture, DRGs were fixed with 2% PFA/PB for 20 min and immunostained with antibodies against HA (3F10; Roche), vGlut1, Pv, and Tuj1 (Covance). Images were taken by Nikon A1R confocal microscopy and analyzed by ImageJ software.
Statistical analysis.
Data were analyzed and reported as mean ± SD.
Results
Expression of Cdc42 in sensory and motor neurons
To determine whether Cdc42 is involved in the establishment of sensory–motor reflex circuits, we examined the expression profiles of Cdc42 in sensory neurons in the DRG and motor neurons in the developing spinal cord of wild-type mice at E15.5 and P0. Monosynaptic sensory–motor connections are generally formed at ∼E17 in mice (Mears and Frank, 1997). Cdc42 was broadly expressed in the spinal cord at E15.5 and P0 (Fig. 1A,C). Cdc42 was also expressed by most of the DRG neurons at E15.5 and P0 (Fig. 1B,D). These data show that the timing of Cdc42 expression in sensory and motor neurons coincides with sensory–motor circuit establishment.

Cdc42 expression in the spinal cord and DRGs. (A–D) Expression of Cdc42 mRNA in the developing spinal cords (A, C) and DRGs (B, D) of E15.5 A, B, and P0 (C,D) wild-type mice. Scale bars: 100 μm (A–D).
Loss of Cdc42 in motor neurons does not affect monosynaptic sensory–motor connections
Because Cdc42 has been shown to have postsynaptic roles in other regions of the CNS (Irie and Yamaguchi, 2002; Scott et al., 2003; Murakoshi et al., 2011; Rosário et al., 2012), we first examined a role for Cdc42 in motor neurons within the sensory–motor reflex arc. We generated motor-neuron-specific Cdc42 mutant mice (Cdc42-ChAT-cKO) by crossing mice that have Cdc42-floxed alleles in which loxP sites flank exon 2 of Cdc42 (Yang et al., 2006) with choline acetyltransferase (ChAT)-Cre mice in which Cre expression is detected in motor neurons starting at E12.5 (Fig. 2A,B) (Rossi et al., 2011; Katayama et al., 2012). We confirmed that the resulting mutants (Cdc42-ChAT-cKO mice) were indeed deficient in Cdc42 in ChAT+ motor neurons (Fig. 2C–F) and were not only viable (surviving until adulthood), but were fertile as well (data not shown). To assess the positioning and dendritic morphologies of motor neurons in Cdc42-ChAT-cKO mice, motor neurons were labeled by retrograde transport of R-Dex from the L5 ventral root. No obvious defects were observed (Fig. 2G,I,J,L).

Loss of Cdc42 in motor neurons does not affect monosynaptic sensory–motor connections. A, B, GFP expression by motor neurons in the spinal cord of a ChAT-Cre;floxed-GFP mouse at P7. B, Enlarged view of GFP expression in the ventral spinal cord. C–F, In situ hybridizations for Cdc42 (E, F) with immunostaining for motor neurons using anti-ChAT antibody (red; C, D) in control (C, E) and mutant mice lacking Cdc42 in motor neurons (Cdc42-ChAT-cKO; D, F) at P7. Arrows indicate ChAT+ motor neurons. G–L, Motor neurons labeled with R-Dex in L5 ventral nerves (magenta) and vGlut1+ proprioceptive axon terminals (green) in P7 control (G–I) and Cdc42-ChAT-cKO (J–L) mice. There were no obvious defects in numbers of vGlut1+ sensory terminals or the morphology of motor neurons in Cdc42-ChAT-cKO mice. M, Schematic drawing of the extracellular recording experiment for monosynaptic EPSP amplitudes. Sensory neurons (blue) were stimulated from dorsal nerves and EPSPs of motor neurons (red) were recorded from ventral nerves. N, Amplitudes of monosynaptic (Mono) EPSPs at L3, L4, or L5 ventral roots in P5–P7 control (n = 6) and Cdc42-ChAT-cKO mice (n = 6). Traces presented are averages of 20 individual stimuli applied at 0.1 Hz. There were no significant differences between control and Cdc42-ChAT-cKO mice (Student's t test). Scale bars: A, B, L, 100 μm; D, 10 μm.
Next, we examined the monosynaptic sensory–motor connections in Cdc42-ChAT-cKO mice. We first visualized the presynaptic regions of proprioceptive afferents in the ventral spinal cord by immunostaining for vGlut1, a marker for proprioceptive synapses in the ventral spinal cord (Oliveira et al., 2003; Alvarez et al., 2004). The density of vGlut1+ proprioceptive synapses in the ventral horn of P7 Cdc42-ChAT-cKO mice appeared to be similar to that observed in littermate control animals (Fig. 2H,I,K,L). The functional connectivity of these sensory–motor circuits was then examined by measuring the amplitudes of monosynaptic EPSPs recorded extracellularly from lumbar ventral roots (L3, L4, or L5) after stimulation of the corresponding dorsal roots (Fig. 2M). The shortest latencies were interpreted to be monosynaptic sensory–motor connections, as was reported previously (Arber et al., 2000; Wang et al., 2007; Mentis et al., 2011). We found no significant differences in the amplitudes of monosynaptic EPSPs between littermate control and Cdc42-ChAT-cKO mice at P5–P7 (Fig. 2N; control L3: 0.797 ± 0.135 mV; mutant L3: 0.898 ± 0.092 mV; control 4: 0.938 ± 0.123 mV; mutant L4: 1.073 ± 0.177 mV; control L5: 1.836 ± 0.380 mV; mutant L5: 1.616 ± 0.286 mV; n = 6). Therefore, unlike studies of other circuits showing that Cdc42 functions primarily in postsynaptic neurons (Irie and Yamaguchi, 2002; Scott et al., 2003; Murakoshi et al., 2011; Rosário et al., 2012), in monosynaptic sensory–motor connections, Cdc42 expression by postsynaptic neurons (motor neurons) does not appear to influence circuit development.
Loss of Cdc42 in sensory neurons affects monosynaptic sensory–motor connections
To determine whether Cdc42 expressed by proprioceptive sensory neurons plays a presynaptic role in the establishment of monosynaptic sensory–motor circuits, we generated sensory-neuron-specific Cdc42 mutant mice (Cdc42-Adv-cKO) by crossing Cdc42-floxed mice with Advillin-Cre mice in which Cre is specifically expressed by DRG sensory neurons before E12.5 (Fig. 3A,B; Hasegawa et al., 2007). In P7 Cdc42-Adv-cKO mice, Cdc42 was confirmed to be absent in proprioceptive sensory neurons in the DRGs, which were identified by the proprioceptive sensory neuron marker Pv (Fig. 3C–F; Honda, 1995; Arber et al., 2000). Overall numbers of proprioceptive sensory neurons in the DRGs remained similar to those of littermate controls at P6–P7 (Fig. 3G–K; control L2: 147 ± 15.3; mutant L2: 137 ± 2.6; control L3: 268.8 ± 45.5; mutant L3: 266 ± 15.4; n = 5). ChAT+ motor neuron numbers were also unaltered in P6–P7 Cdc42-Adv-cKO mice (Fig. 3L–N; control: 9.1 ± 1.1, mutant: 9.2 ± 0.5, n = 5). This suggests that Cdc42 expression in presynaptic proprioceptive sensory neurons is not essential for cell survival or differentiation of the sensory neurons themselves or for the survival of the postsynaptic motor neurons. Similar to littermate controls, Pv+ proprioceptive afferents in P11 Cdc42-Adv-cKO mice projected appropriately into the ventral spinal cord (Fig. 3O,P). Finally, because group Ia proprioceptive sensory neurons also extend axons peripherally to terminate in the muscle spindles located in intrafusal muscle fibers (Maier, 1997), we also examined the vGlut1+ peripheral endings of proprioceptive sensory afferents (Wu et al., 2004; Pang et al., 2006). The vGlut1+ group Ia proprioceptive sensory terminals were similarly detected in the rectus femoris muscles of both littermate controls and P7 Cdc42-Adv-cKO mice (Fig. 3Q,R), indicating that Cdc42 is not required for the formation of peripheral terminals of group Ia proprioceptive sensory afferents. Together, these results suggest that Cdc42 does not affect central axonal trajectories and peripheral terminals of proprioceptive sensory neurons.

Deletion of Cdc42 in presynaptic proprioceptive sensory neurons does not affect axon projections. A, B, GFP expression in the spinal cord (A) and DRGs (B) of an Adv-Cre;floxed-GFP mouse at P7. C–F, In situ hybridizations for Cdc42 (E, F) with immunostaining using anti-Pv antibody to identify proprioceptive sensory neurons (red) in P7 control (C, E) and mutant mice lacking Cdc42 in Pv+ proprioceptive sensory neurons (Cdc42-Adv-cKO) (D, F). Arrows indicate Pv+ neurons. G–J, Pv (red) and GFP (green) staining in DRGs of P7 control (G, H) and Cdc42-Adv-cKO mice (I, J). Control and Cdc42-Adv-cKO mice have a GFP reporter transgene (floxed-GFP) that permits visual verification of Cre-mediated recombination. K, Quantification of Pv+ sensory neurons in the DRGs of control (n = 5) and Cdc42-Adv-cKO mutant mice (n = 5) at P6. No significant differences were observed (Student's t test). L, M ChAT staining showing motor neurons in the ventral spinal cords of control (L) and Cdc42-Adv-cKO (M) mice at P6. N, Quantification of ChAT+ motor neurons per section in P6 control and Cdc42-Adv-cKO mice (n = 5 in each genotypes). No significant differences were observed. O, P, Pv staining in the ventral spinal cords of control (O) and Cdc42-Adv-cKO (P) mice at P11. There were no obvious defects on Pv+ axon projection in Cdc42-Adv-cKO mice. Q, R, vGlut1+ peripheral terminals in the rectus femoris muscle in P7 control (Q) and Cdc42-Adv-cKO (R) mice. Muscle spindles seem to be normally formed and maintained. Scale bars: A, J, M, P, R, 100 μm; D, 10 μm.
We then examined the presynaptic terminals of proprioceptive sensory afferents in the ventral spinal cord during early postnatal stages. Interestingly, in Cdc42-Adv-cKO mice, the proportion of vGlut1+ presynaptic terminals in the ventral spinal cord was significantly reduced (30–40%) in both P0 and P11 mice compared with littermate controls (Fig. 4A–E; control P0: 100 ± 10.7%; mutant P0: 68 ± 5.5%, p = 0.0062; control P11: 100 ± 9.6%; mutant P11: 64 ± 4.7%, p = 0.0047; Student's t test; n = 4). We then looked specifically at the vGlut1+ presynaptic terminals on motor neurons by injecting R-Dex into the rectus femoris muscle and tracing the dye back to their corresponding motor neurons (Fig. 4F–I). In P7 Cdc42-Adv-cKO mice, the vGlut1+ proprioceptive synapses on R-Dex+ motor neurons were significantly reduced in number (Fig. 4F–J; control: 55.7 ± 6.3; mutant 16.8 ± 11.5; p = 0.0000050, Student's t test; n = 5). This is unlikely due to fewer projections or diminished branching in proprioceptive sensory axons because Pv+ proprioceptive sensory axon density in the ventral spinal cord was similar in Cdc42-Adv-cKO and control mice (Fig. 4H,I,K; control: 100 ± 35%; mutant: 80 ± 35; n = 5). Therefore, Cdc42 in proprioceptive sensory neurons plays an important role in the presynaptic differentiation of proprioceptive sensory afferents, which is likely required for proper synaptogenesis.
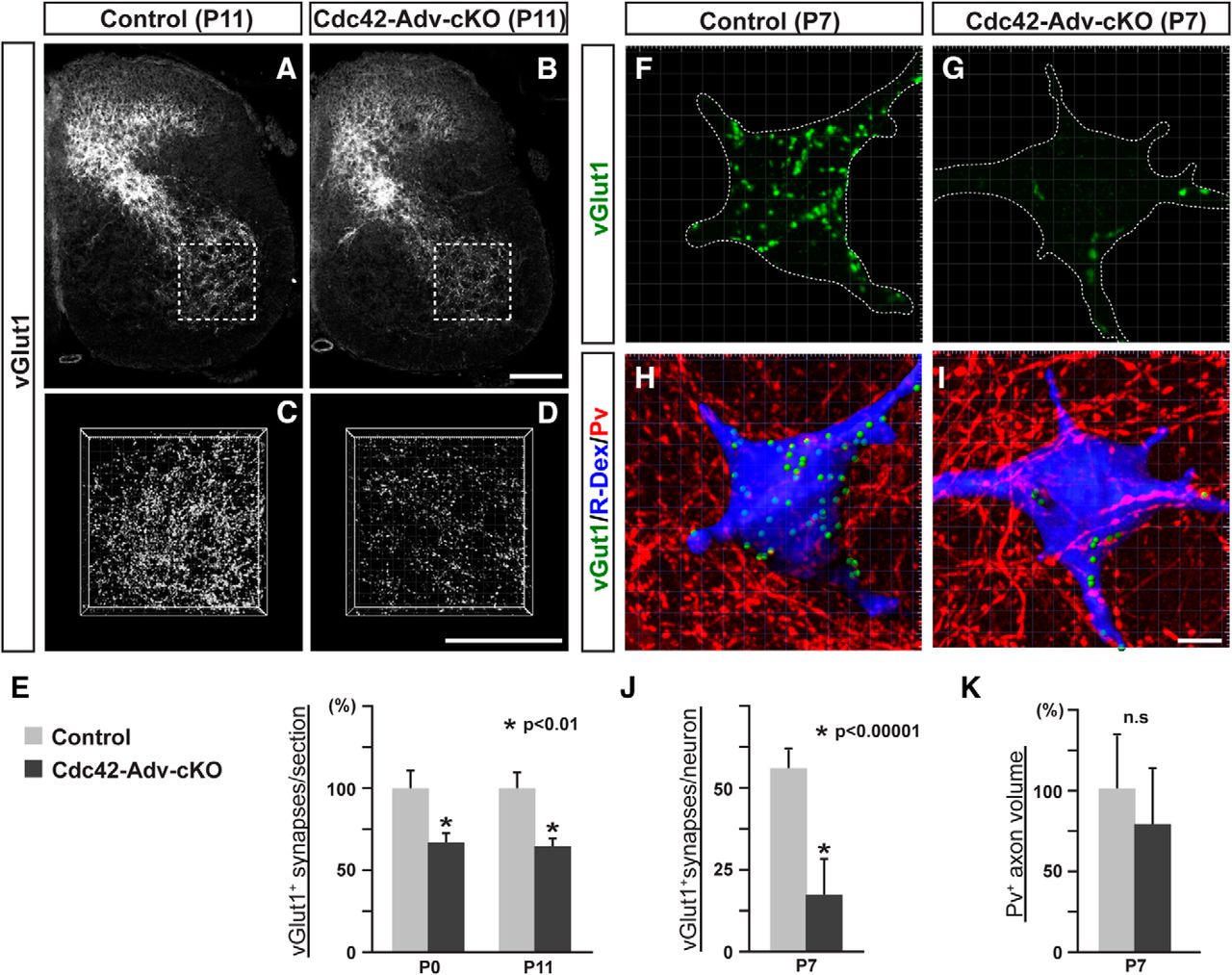
Deletion of Cdc42 in presynaptic proprioceptive sensory neurons affects synapse formation. A–D, vGlut1+ proprioceptive central terminals in the spinal cords of control (A, C) and Cdc42-Adv-cKO (B, D) mice at P11. C, D, 3D views of vGlut1+ presynaptic terminals in the ventral spinal cord at P11. E, Quantification of vGlut1+ synapses in control and Cdc42-Adv-cKO mice (n = 4 for each genotype, Student's t test, *p < 0.01) showing significant reductions in vGlut1+ proprioceptive terminals at both P0 and P11. F–I, vGlut1 staining (green), R-Dex (blue), and Pv (red) on rectus femoris motor neurons in control (F, H) and Cdc42-Adv-cKO (G, I) mice at P7. H, I, 3D reconstructions of synapses (vGlut1; green), motor neurons (R-Dex; blue), and proprioceptive sensory axons (P; red). J, Quantification of vGlut1+ synapses per rectus femoris motor neuron (n = 5 for each genotype) showing a decrease in vGlut1+ proprioceptive terminals (*p < 0.00001, Student's t test). K, Comparison of Pv+ proprioceptive axon volume per section (n = 5 for each genotype). There was no significant difference (n.s, Student's t test). Scale bars: B, D, 100 μm; H, 10 μm.
To determine whether corresponding functional deficits are observed in monosynaptic sensory–motor circuits in Cdc42-Adv-cKO mice, we performed extracellular recordings to measure EPSP amplitudes of monosynaptic sensory–motor connections from L3, L4, and L5 ventral roots (Fig. 5A–C). Monosynaptic EPSPs were observed in P6 Cdc42-Adv-cKO mutants; however, the amplitudes of these EPSPs were markedly reduced (40–60%) compared with littermate controls (Fig. 5A–C; control L3: 0.872 ± 0.135 mV; control L4: 1.053 ± 0.120; control L5: 1.132 ± 0.016 mV; mutant L3: 0.356 ± 0.045 mV, p = 0.025; mutant L4: 0.415 ± 0.098 mV, p = 0.0022; mutant L5: 0.674 ± 0.104 mV, p = 0.0072; Student's t test; n = 10 controls; n = 7 mutants). Therefore, reduced synapse numbers in the sensory–motor circuits of Cdc42-Adv-cKO mice affect electrophysiological responses in these circuits by way of reduced EPSP amplitudes.

Electrophysiological analysis of monosynaptic sensory–motor connections in Cdc42-Adv-cKO mice. A, B, Recording traces from L5 ventral nerves of control (A) and Cdc42-Adv-cKO (B) mice at P6. Traces presented are averages of 20 individual stimuli applied at 0.1 Hz. Arrows indicate peak amplitudes of monosynaptic EPSPs. C, Average peak amplitudes of monosynaptic EPSPs at L3, L4, and L5 ventral nerves of P6 control (n = 10) and Cdc42-Adv-cKO mice (n = 7). EPSP amplitudes were significantly decreased in mutant mice (*p < 0.05, Student's t test).
To determine whether morphological defects are present at presynaptic sites in Cdc42-Adv-cKO mice, we performed immunoelectron microscopy to examine the ultrastructures of vGlut1+ proprioceptive sensory terminals in the ventral spinal cord of P30 mice. We analyzed vesicle sizes in vGlut1+ proprioceptive sensory terminals, but no obvious defects were observed (Fig. 6A–E; control: 43.7 ± 6.83 nm; mutant: 43.8 ± 6.62 nm; n = 5), suggesting that, although reduced in number, the presynaptic structures of the synapses that do form in Cdc42-Adv-cKO mice are not altered. Together, these data indicate that Cdc42 in presynaptic sensory neurons promotes the formation of the proper complement of monosynaptic connections in sensory–motor circuit development.

Ultrastructure of sensory–motor synapses. A–D, vGlut1 immunoelectron microscopy (EM) in P30 control (A, B) and mutant mice lacking Cdc42 in proprioceptive sensory neurons (Cdc42-Adv-cKO; C, D). B, D, Enlarged views from the dashed box regions of A and C, respectively. E, Quantification of synaptic vesicle sizes of control (n = 5) and Cdc42-Adv-cKO mice (n = 5). There were no significant size differences (t test). Scale bars in D, 100 nm.
Deletion of Cdc42 in sensory neurons does not affect synaptic specificity of monosynaptic sensory–motor circuits
Next we investigated whether Cdc42 regulates synaptic specificity in addition to synapse formation because Cdc42 may be a downstream intracellular molecule of various signaling pathways that affect synaptic specificity. We focused on obturator and quadriceps sensory–motor reflex arcs because previous studies have shown that Ia afferents conveying sensory information from either the obturator or quadriceps nerves do not form synaptic contacts with motor neurons supplying the opposite muscles (Mears and Frank, 1997). This selectivity in sensory–motor connections is particularly striking given the close proximity of motor pools supplying the obturator and quadriceps nerves in the spinal cord (Fig. 7A). We performed intracellular recordings from obturator and quadriceps motor neurons after obturator and quadriceps sensory nerve stimulation, respectively, in P5–P7 mice (Fig. 7B). Short-latency inputs with a variance in onset latency of <0.2 after repeated trials (20–60 trials at 1 Hz) were identified as monosynaptic responses (Doyle and Andresen, 2001; Rose and Metherate, 2005; Vrieseling and Arber, 2006; Fukuhara et al., 2013). In control mice, the sets of latencies from homonymous connections (obturator-to-obturator and quadriceps-to-quadriceps) ranged between 4.0 and 5.8 ms and 4.1 and 5.5 ms, respectively (gray bins in Fig. 7G,H). In P5–P7 Cdc42-Adv-cKO mice, the amplitudes of homonymous monosynaptic EPSPs were markedly reduced (30–40%) for both obturator and quadriceps motor neurons compared with control mice (Fig. 7C–F,I,J; control Ob recording: 5.324 ± 2.342 mV, n = 9; Cdc42-Adv-cKO Ob recording: 1.689 ± 1.872 mV, n = 10, p = 0.013; control Q recording: 7.432 ± 2.643 mV, n = 16; Cdc42-Adv-cKO Q recording: 3.417 ± 2.490 mV, n = 14, p = 0.0020, Student's t test). This finding was consistent with the reduced EPSP amplitudes obtained in extracellular recordings from these mutants (Fig. 5A–C). Finally, to find out whether Cdc42 is involved in determining the synaptic specificity of monosynaptic sensory–motor connections, we recorded from obturator motor neurons after quadriceps stimulation and from quadriceps motor neurons after obturator stimulation. We did not find any monosynaptic connections in these sensory–motor circuits in either littermate control or Cdc42-Adv-cKO mice (Fig. 7C–H). These data suggest that, whereas Cdc42 is involved in synapse formation, it is not required for synaptic specificity in monosynaptic sensory–motor connections.

Intracellular recordings from quadriceps and obturator motor neurons. A, Retrograde labeling of obturator (blue) and quadriceps (red) motor neurons with FITC and R-Dex, respectively, in the spinal cord of a P6 wild-type mouse. B, Schematic drawing of the intracellular recording experiment for sensory–motor connectivity. Obturator (Ob, blue) and quadriceps (Q, red) motor neurons (MNs) were identified by antidromic responses from obturator and quadriceps sensory nerve stimulation (Ob stim. and Q stim.), respectively, at P5–P7. C–F, Obturator (C, D) and quadriceps (E, F) motor neuron recordings in control (C, E) and mutant mice lacking Cdc42 in proprioceptive sensory neurons (Cdc42-Adv-cKO; D, F), respectively. Blue and red traces show the results of obturator and quadriceps sensory nerve stimulations (Ob stim. and Q stim.), respectively. Traces presented are averages of 20–60 stimulation trials at 1 Hz. Gray bins indicate ranges of monosynaptic responses (G, H). G, H, Quantification of the shortest EPSP onset latencies from recordings of individual quadriceps motor neurons with obturator stimulation (G) and obturator motor neurons with quadriceps stimulation (H). Monosynaptic ranges (gray bins) were defined by homonymous connections (recordings of obturator motor neurons with obturator nerve stimulation or recordings of quadriceps motor neurons with quadriceps nerve stimulation) in P5–P7 control and Cdc42-Adv-cKO mice. No aberrant connections were observed in Cdc42-Adv-cKO mice. I, J, Dot plots of the peak amplitudes of homonymous monosynaptic EPSPs from obturator (I) and quadriceps (J) motor neurons of P5–P7 control and mutant mice in which Cdc42 had been deleted before (Cdc42-Adv-cKO) or after sensory–motor circuit formation (Cdc42-Pv-cKO). Amplitudes of monosynaptic EPSPs were significantly decreased in Cdc42-Adv-cKO mice, but not Cdc42-Pv-cKO mice. *p < 0.02 (Student's t test; I), *p < 0.01 (Student's t test; J).
Cdc42 in proprioceptive sensory neurons is not required for synaptic transmission
To determine whether Cdc42 in proprioceptive sensory neurons regulates synaptic transmission as well as synaptogenesis, we used Pv-Cre mice (Hippenmeyer et al., 2005) to delete Cdc42 in proprioceptive sensory neurons after monosynaptic sensory–motor connections had formed. In Pv-Cre mice, Cre-mediated recombination in the DRGs is only observed from P0 (Fig. 8A,B; Lilley et al., 2014). Because sensory–motor connections are usually formed by E17 (Mears and Frank, 1997), these mutants (Cdc42-Pv-cKO) permitted us to examine the effects of Cdc42 deletion in properly formed sensory–motor circuits (Fig. 8C–F). First, we examined the vGlut1+ presynaptic terminals of proprioceptive sensory neurons in the ventral spinal cords of P11 mice, but no defects were observed (Fig. 8G–I; control: 100 ± 13.4%; mutant: 117 ± 13.2%, n = 5). We then analyzed the homonymous monosynaptic EPSP amplitudes from obturator and quadriceps motor neurons after obturator and quadriceps nerve stimulation, respectively, but, again, the Cdc42-Pv-cKO mutants (P5–P7) did not differ significantly from controls (Fig. 7I,J, Ob recording: 4.646 ± 3.769 mV, n = 22; Q recording: 6.034 ± 3.413 mV, n = 30). This suggests that the connectivity of sensory–motor circuits is unaffected in the mutants. These data strongly suggest that Cdc42 in proprioceptive sensory neurons is not likely required for synaptic transmission.

Axonal projections and presynaptic terminals of proprioceptive sensory neurons in the spinal cords of Cdc42-Pv-cKO mice. A, B, GFP expression in the spinal cord of a Pv-Cre; floxed-GFP mouse at P7. B, Enlarged view of the ventral spinal cord from A. C–F, In situ hybridizations for Cdc42 (D, F) with immunostaining for proprioceptive sensory neurons using anti-Pv antibody (red; C, E) in P7 control (C, D) and mutant mice lacking Cdc42 in proprioceptive sensory neurons from postnatal stages only (Cdc42-Pv-cKO; E, F). Arrows in C–F show Pv+ neurons. G, H, vGlut1+ presynaptic terminals of proprioceptive sensory neurons in control (G) and Cdc42-Pv-cKO (H) mice at P11. I, Comparison of vGlut1+ presynaptic terminals in the ventral spinal cord (n = 5 for each genotype). There were no significant differences between P11 control and Cdc42-Pv-cKO mice (Student's t test). Scale bars: A, B, H, 100 μm; E, 10 μm.
NL-induced presynaptic differentiation of proprioceptive sensory neurons is impaired in the absence of Cdc42
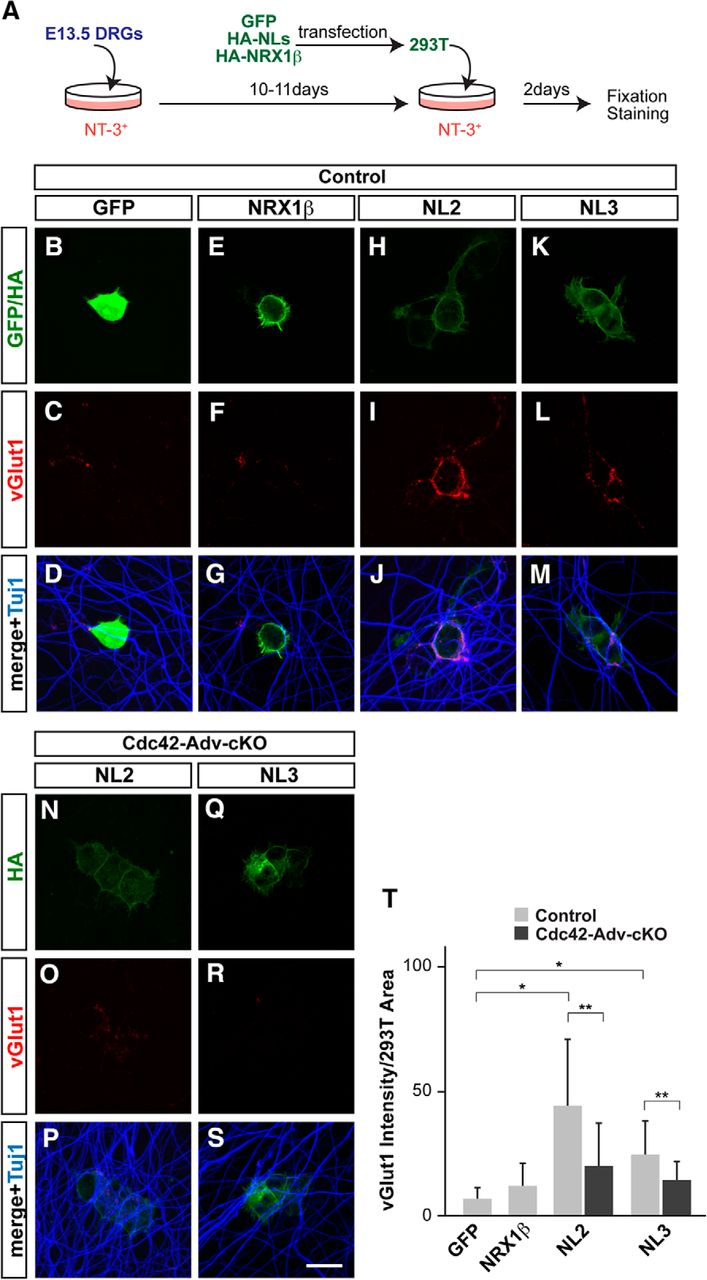
To elucidate the molecular mechanisms underlying Cdc42-mediated synapse formation, we focused on NLs that function as synaptic organizers in various types of neurons (Scheiffele et al., 2000; Südhof, 2008; Shen and Scheiffele, 2010; Siddiqui and Craig, 2011; Bemben et al., 2015). The NL family consists of NL1–NL4 in mammals, with NL2 and NL3 being strongly expressed by motor neurons (Zelano et al., 2007; Berg et al., 2013; Bemben et al., 2015). In addition, in Aplysia, NL and its receptor, neurexin (NRX), are located on motor and sensory neurons, respectively, and they regulate synaptic strength and remodeling of sensory–motor connections (Choi et al., 2011). To determine whether NLs act as synaptic organizers in developing monosynaptic sensory–motor circuits in mice, we examined vGlut1+ presynaptic terminals using cocultures of DRGs dissected from E13.5 wild-type mice and 293T cells expressing GFP, NRX1β, NL2, and NL3 in the presence of NT-3, which is essential for proprioceptive sensory neuron survival (Fig. 9A; Hohn et al., 1990; Hory-Lee et al., 1993). After 2 d in culture, DRGs in coculture with NL2- or NL3-expressing 293T cells showed numerous vGlut1+ presynaptic terminals (vGlut1 intensity; NL2: 44.0 ± 26.9, p = 0.0000022; NL3: 24.8 ± 13.2, p = 0.00060; Student's t test; n = 15), whereas these terminals were rarely detected when cocultured with GFP- or NRX1β-expressing 293T cells (GFP: 6.9 ± 4.3; NRX1β: 12.0 ± 9.1; n = 15; Fig. 9B–M,T). Interestingly, in the absence of Cdc42, axon development, as highlighted by the β-tubulin marker Tuj1, appeared to be normal, but significantly fewer vGlut1+ sensory terminals developed in the NL cocultures (NL2: 19.6 ± 17.5, p = 0.021; NL3: 14.2 ± 7.6, p = 0.024; Student's t test; n = 15; Fig. 9N–S,T). These results suggest that NL2 and NL3 can induce the development of presynaptic terminals in proprioceptive sensory neurons in vitro and, furthermore, that Cdc42 plays a role in this process.

Cdc42 is involved in NL-induced synaptogenesis of proprioceptive sensory neurons. A, Schematic drawing of the in vitro proprioceptive sensory neuron culture. DRGs from E13.5 mouse embryos were cultured with NT-3-supplemented media. After 10–11 d, 293T cells expressing GFP, HA-NLs, or HA-NRX1β were added and cocultured for 2 d. DRGs were then fixed for immunostaining. B–S, Control (B–M) and Cdc42-deficient DRGs (N–S) were cocultured with 293T cells expressing GFP (B–D), HA-NRX1β (E–G), HA-NL2 (H–J, N–P), and HA-NL3 (K–M, Q–S). Cells were immunostained with antibodies against GFP or HA (green; B, E, H, K, N, Q), vGlut1 for presynaptic structures on proprioceptive sensory neuron (red; C, F, I, L, O, R), or Tuj1 for axons of proprioceptive sensory neurons (blue; D, G, J, M, P, S). T, Quantification of total vGlut1 integrated intensity staining associated with surface area of 293T cells (n = 15 in each experiment). NLs, but not NRX1β, induced vGlut1+ presynaptic structures and loss of Cdc42 affected NL-dependent presynaptic structure formation. *p < 0.001, **p < 0.05 (Student's t test). Scale bar in S, 10 μm.
Discussion
Monosynaptic sensory–motor reflex circuits have been studied extensively using anatomical, electrophysiological, molecular, and mouse genetic approaches (Brown, 1981; Ladle et al., 2007; Arber, 2012). Despite this, very little is known about the molecular mechanisms underlying synapse formation in these circuits. Although muscle-spindle-derived factors such as NT-3 have been suggested to strengthen monosynaptic sensory–motor connectivity, they are not necessary for initial synapse formation (Chen et al., 2002; Hippenmeyer et al., 2002; Shneider et al., 2009; Mentis et al., 2010). Other studies have identified many ligand–receptor pairs that control synaptogenesis in the mammalian CNS (Waites et al., 2005; Shen and Scheiffele, 2010; Dabrowski and Umemori, 2011; Siddiqui and Craig, 2011; Terauchi and Umemori, 2012); however, synaptic organizers have never been implicated in the development of monosynaptic sensory–motor reflex arcs. Difficulties in identifying molecules that control sensory–motor synaptogenesis directly may arise from these regulators being numerous and possibly overlapping in function in vivo. Therefore, disruption of a single extracellular signaling pathway might not be sufficient to interrupt overall synapse formation in monosynaptic sensory–motor circuits. To circumvent this logistical problem, we decided to focus on a common intracellular molecule functioning downstream of various extracellular signaling cascades that control synaptogenesis. This molecule was the broadly functioning small Rho GTPase Cdc42. Cdc42 has been shown to have critical roles in a variety of cellular processes, including regulating actin cytoskeleton rearrangements that are necessary for synapse formation (Govek et al., 2005; Murakoshi and Yasuda, 2012).
Because Cdc42 could potentially function in both presynaptic and postsynaptic neurons, we deleted it selectively in either motor or proprioceptive sensory neurons to examine its effects on synaptogenesis within the framework of the spinal reflex arc. Previous studies have shown that Cdc42 functions in postsynaptic neurons to influence dendritic growth of hippocampal and cortical neurons in the mouse (Irie and Yamaguchi, 2002; Scott et al., 2003; Murakoshi et al., 2011; Rosário et al., 2012), but in our system, we did not observe any defects in dendritic length or positioning in postsynaptic motor neurons in mice lacking Cdc42 in motor neurons (Cdc42-ChAT-cKO mutants; Fig. 2). Moreover, our anatomical and electrophysiological analyses did not reveal any defects in sensory–motor synapses in these mutants. These data suggest that the postsynaptic structure and function of sensory–motor synapses in Cdc42-ChAT-cKO mice are not affected. Therefore, even though Cdc42 is expressed in wild-type motor neurons, it may not have an important synaptogenic role in monosynaptic sensory–motor connections. However, it is also possible that other closely related Rho GTPases such as TC10/RhoQ, TCL/RhoJ, Chp/RhoV, or Wrch1/RhoU (Heasman and Ridley, 2008) may compensate for the missing activity of Cdc42 in these mutants. This will be further explored in follow-up studies.
Presynaptic deletion of Cdc42 in sensory neurons (Cdc42-Adv-cKO mice) similarly did not cause obvious defects in neuronal survival or axonal projections (Fig. 3). However, anatomical and electrophysiological analyses showed significant impairments in sensory–motor connectivity (Figs. 4, 5, 7). In Cdc42-Adv-cKO mutants, vGlut1+ proprioceptive synapses were reduced by ∼30–40% compared with controls and the extent of this reduction seemed to correspond appropriately with the decreased peak amplitudes observed in our monosynaptic EPSP recordings. Interestingly, structural analyses of the existing vGlut1+ proprioceptive synapses in Cdc42-Adv-cKO mice by electron microscopy did not reveal any obvious structural abnormalities (Fig. 6). These data indicate that, although the numbers of monosynaptic sensory–motor connections are reduced in Cdc42-Adv-cKO mice, the remaining synapses appear to be functional. Together, our findings suggest that Cdc42 regulates the initiation of synapse formation in subsets of monosynaptic sensory–motor connections.
How does Cdc42 function in synapse formation in monosynaptic sensory–motor connections? We theorized a few possible mechanisms. First, Cdc42-Adv-cKO mice may have impaired development of peripheral terminals of group Ia proprioceptive axons, leading to compromised sensory–motor connectivity. However, in these mutants, the muscle spindles appeared to be properly innervated by proprioceptive afferents (Fig. 3Q,R), making this scenario unlikely. Second, Cdc42 may regulate axonal projections or axon branching of proprioceptive sensory neurons. Again, we did not find such morphological defects in Cdc42-Adv-cKO mice (Figs. 3, 4), so this too seems doubtful. Third, Cdc42 may regulate synaptic transmission including vesicular trafficking events such as neurotransmitter release. However, deletion of Cdc42 in proprioceptive sensory neurons after sensory–motor connections had formed (Cdc42-Pv-cKO mice) did not show impairments in sensory–motor connections (Figs. 7I,J, 8G–I). Therefore, our genetic evidence strongly suggests that Cdc42 regulates synapse formation in monosynaptic sensory–motor connections and we predict that it functions by affecting cytoskeletal rearrangements.
Finally, we found that Cdc42 is involved in NL-dependent synapse formation of proprioceptive sensory neurons in vitro. Because NL2 and NL3 are expressed by motor neurons (Zelano et al., 2007; Berg et al., 2013; Bemben et al., 2015), we examined their effects on synapse formation in vitro and found that both NL2 and NL3 induced vGlut1+ excitatory presynaptic structures in proprioceptive sensory neurons (Fig. 9). Other studies have shown that NL2 is almost exclusively localized at inhibitory synapses, with only weak localization of NL2 at excitatory synapses (Levinson et al., 2005; Dolique et al., 2013). In contrast, NL3 is found equally at excitatory and inhibitory synapses (Budreck and Scheiffele, 2007). Moreover, overexpression of NL2 and NL3 increased both excitatory and inhibitory terminal density in vitro (Chih et al., 2005; Takahashi et al., 2011), although NL2−/− mice show defects in only inhibitory synapses (Chubykin et al., 2007). Therefore, NL2 and/or NL3 may participate in synapse formation in sensory–motor connections in vivo. Future analyses using NL2 and NL3 mutant mice will address this question.
One unresolved issue is why the reduction of vGlut1+ proprioceptive sensory terminals and monosynaptic EPSP amplitudes in Cdc42-Adv-cKO mice are only partial reductions. It may be that Cdc42 transduces the intracellular signaling cascade of only a portion of the extracellular signaling pathways regulating synpaptogenesis, with other pathways being responsible for inducing the remaining 60–70% of sensory–motor synapses. Alternatively, other Rho GTPases in proprioceptive sensory neurons may function redundantly with Cdc42 to control sensory–motor synapse formation. Future studies will explore these possibilities.
In summary, our studies provide two novel findings using mouse genetics. First, we present the first evidence of molecular controls for synapse formation in monosynaptic sensory–motor circuits and, second, we show a novel role for Cdc42 in presynaptic neurons during a critical stage in sensory–motor circuit development. Future studies involving Cdc42 will identify the precise mechanisms and signaling cascades in presynaptic neurons that lead to appropriate synapse formation in monosynaptic sensory–motor circuits in vivo.
Footnotes
This work was supported by a the National Institute of Neurological Disorders and Stroke–National Institutes of Health (Grant NS093002 to Y.Y.). We thank F. Wang (Duke University Medical Center) and S. Arber (University of Basel) for providing Advillin-Cre and Pv-Cre mice, respectively; F. Alvarez (Emory University), M. Baccei (University of Cincinnati), Xiaoyi Cheng (Cincinnati Children's Hospital Medical Center), and M. Kofron (Cincinnati Children's Hospital Medical Center) for help with electron microscopy analyses, electrophysiological analyses, sharing unpublished materials, and confocal microscope analyses, respectively; and the members of the Yoshida and Crone laboratories for helpful discussions regarding these experiments.
The authors declare no competing financial interests.
- Correspondence should be addressed to Yutaka Yoshida, Division of Developmental Biology, Cincinnati Children's Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45229. yutaka.yoshida{at}cchmc.org





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}