

Abstract
The etiology and pathophysiology underlying opioid tolerance and dependence are still unknown. Because mu opioid receptor (MOR) plays an essential role in opioid action, many vulnerability-related studies have focused on single nucleotide polymorphisms of MOR, particularly on A118G. In this study, we found that a single-point mutation at the MOR T394 phosphorylation site could be another important susceptive factor in the development of opioid tolerance and dependence in mice. T394A mutation, in which a threonine at 394 was replaced by an alanine, did not alter agonist binding to MOR and opioid analgesia, but resulted in loss of etorphine-induced MOR internalization in spinal dorsal horn neurons and opioid analgesic tolerance induced by either morphine or etorphine. In addition, this mutation also caused an increase in intravenous heroin self-administration and in nucleus accumbens dopamine response to heroin. These findings suggest that T394 phosphorylation following MOR activation causes MOR internalization and desensitization, which subsequently contributes to the development of tolerance in both opioid analgesia and opioid reward. Accordingly, T394A mutation blocks opioid tolerance and leads to an increase in brain dopamine response to opioids and in opioid-taking behavior. Thus, the T394 may serve as a new drug target for modulating opioid tolerance and the development of opioid abuse and addiction.
SIGNIFICANCE STATEMENT The mechanisms underlying opioid tolerance and susceptibility to opioid addiction remain unclear. The present studies demonstrate that a single-point mutation at the T394 phosphorylation site in the C-terminal of mu opioid receptor (MOR) results in loss of opioid tolerance and enhanced vulnerability to heroin self-administration. These findings suggest that modulation of the MOR-T394 phosphorylation or dephosphorylation status may have therapeutic potential in management of pain, opioid tolerance, and opioid abuse and addiction. Accordingly, MOR-T394 mutation or polymorphisms could be a risk factor in developing opioid abuse and addiction and therefore be used as a new biomarker in prediction and prevention of opioid abuse and addiction.
Introduction
Opioids are potent analgesics and addictive drugs. The central effects of opioids are mediated by activation of brain μ, δ, and κ opioid receptors, particularly the mu opioid receptor (MOR) (Matthes et al., 1996; Law et al., 2000). The implications for opioid analgesics in humans are limited because of the concerns of opioid tolerance, dependence, and addiction. However, the cellular and molecular mechanisms underlying the development of opioid tolerance and dependence remain unclear. Human studies suggest that single nucleotide polymorphisms (SNPs) in the MOR gene are associated with vulnerability to opioid abuse and addiction. Of the various SNPs reported in MOR translated regions, rs1799971 (A118G), which results in Asn40Asp variant in MOR protein, has been the most extensively studied in association with opioids or alcohol use and addiction (Bond et al., 1998; Heilig et al., 2011; Haerian and Haerian, 2013; Reed et al., 2014). However, the related findings in humans are controversial and inconclusive (Bergen et al., 1997; Bond et al., 1998; Franke et al., 2001; Nikolov et al., 2011; Haerian and Haerian, 2013). The findings in animal model of human A118G mutation (using A112G mutation mice) are also controversial (Mague et al., 2009; Zhang et al., 2015).
In addition to A118G glycosylation site mutation, other studies suggest that MOR mutations at several MOR phosphorylation sites may also play an important role in the development of opioid tolerance and dependence (Yu et al., 1997; Deng et al., 2000; Williams et al., 2013).There are ∼20 phosphorylation sites identified in the intracellular regions of MOR that may contribute to receptor desensitization and endocytosis (Connor et al., 2004; Koch and Höllt, 2008). For example, activation of MOR by morphine, DAMGO, or fentanyl dose-dependently phosphorylates the Ser375 (S375) residue in the C-terminal of MOR, and site-directed S375A mutation (in which serine 375 is replaced by alanine) produces a dose-dependent increase in opioid analgesia (Grecksch et al., 2011). In contrast to S375A mutation, the T394A phosphorylation site mutation, in which a threonine at 394 is replaced by an alanine, not only abolishes agonist-induced MOR phosphorylation, but also diminished DAMGO-induced MOR desensitization in cultured Chinese hamster ovary (CHO) cells (Pak et al., 1997; Deng et al., 2000). These findings suggest that the S375 and T394 phosphorylation sites play an important role in the development of opioid tolerance. However, in vivo evidence is lacking on the role of T394A mutation in opioid analgesia and tolerance, and little is known as to whether T394A mutation alters opioid abuse potential.
To address these issues, we first developed a T394A mutant mouse strain in the present study. We then evaluated the effects of T394A mutation on MOR binding property, the analgesic tolerance of morphine or etorphine, MOR internalization, and heroin self-administration. Given an important role of nucleus accumbens (NAc) dopamine (DA) in opioid reward and addiction (Wise, 1996; Xi and Stein, 2002), we used in vivo microdialysis with high-performance liquid chromatography to measure NAc DA response to heroin in both wild-type (WT) and T394A mutant mice. We found that T394A mutation causes loss of acute opioid tolerance in antinociception and a significant increase in vulnerability to heroin self-administration behavior.
Materials and Methods
Animals.
Male WT and the MOR-T394A mutant mice with C57BL/6J genetic backgrounds were generated at inGenious Targeting Laboratory, as illustrated in Figure 1, and were bred at the University of Maryland, Baltimore from MOR-T394A breeding pairs purchased from the The Jackson Laboratory. The MOR T394A mutant mice were generated at inGenious Targeting Laboratory, as illustrated in Figure 1. MOR T394A knock-in (KI) mice were backcrossed with C57/BL for >6 generations, and KI mice and WT littermates for testing were produced by heterozygous breeding. The genotype of all progeny was confirmed by PCR analysis of DNA extracted from tail biopsies using the following primers: forward: 5′-GTGATCAACATAGAAGGTGACCCTTC-3′; reverse: 5′- CGCAGCTAGGGAGGAAGATGTCTTC-3′. The PCR product for the WT is 306 bp and the Neo deletion (with remaining set of loxP-FRT sites) is 480 bp.
All mice used in the present experiments were matched for age (8–14 weeks) and weight (25–35 g). Animals were tested between 12 and 20 weeks of age. Mice were housed 3–5 per cage (for the hotplate test) or housed individually (for the heroin self-administration and microdialysis experiments) in a climate-controlled room under a 12 h light/dark cycle (lights on at 7:00 P.M., lights off at 7:00 A.M.). Food and water were available ad libitum throughout the experiment. All experimental procedures were conducted in accordance with the Guide for the care and use of laboratory animals of the U.S. National Research Council, and were approved by the Animal Care and Use Committee of University of Maryland School of Pharmacy (for the breeding, analgesia, and tolerance experiments) and the National Institute on Drug Abuse of the U.S. National Institutes of Health (for the heroin self-administration, locomotor behavior, and microdialysis experiments).
Drugs.
Morphine sulfate, heroin, etorphine, and cocaine were provided by the National Institute on Drug Abuse drug supply program. All drugs were dissolved into 0.9% NaCl and administered subcutaneously (morphine, etorphine) or intravenously (heroin, cocaine).
Hotplate test.
Pain reflexes in response to a thermal stimulus were measured in male mice using a hotplate device from Socrel (model DS37, Ugo Basile) as described previously (Guang et al., 2004). Briefly, mice were placed on the hotplate (55 ± 0.4°C). The response was defined by the animal forepaw licking; 30 s was the cutoff time to avoid tissue damage. Baseline was established 1 d before the drug testing day by performing several trials 2 min apart until steady latency is obtained. Data were reported as mean ± SE of the maximum possible effect (% MPE), which was determined by using the following calculation: 100% × [(drug response time − basal response time)/(30 s − basal response time)] = % MPE.
Opioid tolerance.
Tolerance to the antinociceptive effect of morphine in the hotplate test was conducted on a 2 d paradigm. On day 1, mice were administered subcutaneously with cumulative doses of morphine dissolved into 0.9% NaCl at doses of 0, 1, 5, 7.5, 10, 15, 20, 25, and 100 mg/kg. Hotplate response was assessed at peak time of 30 min after each dose of the drug. ED50 was calculated from dose–response curve. On day 2, mice were injected with saline (10 ml/kg), and hotplate test was performed to assess basal pain threshold. Mice were then treated with morphine at the ED50 dose, and antinociceptive response was measured 30 min after morphine injection.
For the etorphine tolerance experiment, mice were administered subcutaneously with cumulative doses of etorphine dissolved into 0.9% NaCl at doses of 0, 1, 3, 5, 7.5, 10, and 15 μg/kg etorphine. Antinociceptive effect of etorphine in the hotplate test was assessed at peak time of 15 min after injection for dose–response effect. Three hours following the last etorphine injection, mice were injected with saline (10 ml/kg) first and hotplate response was performed to ensure the analgesic response has returned to the basal level. Mice were then treated with an ED50 dose of etorphine. Antinociceptive response was measured 15 min after etorphine injection.
[3H]-DAMGO binding assays.
Brain membranes were incubated with [3H]-DAMGO ranging from 0.1 to 10 nm, free radioligand was filtered, and the bound radioactivity was measured by scintillation counter. Specific binding was calculated by total binding minus nonspecific binding determined in the presence of 10 μm naloxone.
MOR internalization Fifteen minutes following etorphine (10 μg/kg, s.c.) injection, animals were subjected to the hotplate test during which they exhibited a maximum response at the cutoff time (30 s). Immediately after the antinociception test, mice were anesthetized with ketamine/xylazine (90 mg/kg and 10 mg/kg, i.p., respectively) and perfused intracardially with 50 ml of PBS (10.1 mm Na2HPO4, 1.9 mm KH2PO4, 137 mm NaCl, and 2.7 mm KCl, pH 7.4) 1 m, followed by 20 ml of PFA 4% in PBS 0.1 m. Spinal cords were dissected, postfixed overnight at 4°C in the same fixative, and transferred to a solution of 30% sucrose in PBS 0.1 m at 4°C for cryoprotection until the tissues sank in the solution. The lumbar segments of spinal cord were frozen and kept at −80°C until sectioning.
Immunofluorescence staining of the μ receptor.
The cross sections of lumbar segments of the spinal cord were cut at 30 μm using a cryostat (Leica CM3050S) maintained at −20°C. Sections were washed with PBS for 3 × 5 min and blocked with 5% normal goat serum and 0.2% Triton X-100 in PBS for 1 h at room temperature. The sections were incubated with anti-MOR antibody (1:10,000) overnight at 4°C and then washed with PBS for 3 × 5 min. MOR antiserum was generated in the rabbit against the synthetic peptide corresponding to the 16 amino acid peptide (383–398) of the C-terminal domain of the MOR, which was conjugated to keyhole limpet hemocyanin. Antiserum was purified with antigen affinity chromatography (Cheng et al., 1996; Huang et al., 2008). This stretch of sequence is identical among the rat, mouse, and human MOR. Subsequently, the sections were incubated with secondary antibody AlexaFluor-488-conjugated goat anti-rabbit (1:1000, Invitrogen) for 90 min at room temperature. The sections were washed again for 3 × 5 min and then mounted with anti-fade mounting medium with DAPI (Invitrogen). The sections were examined with a fluorescence microscope (Nikon ECLIPSE TE300) and a confocal microscope (Leica TCS SP5) with a 60× objective to examine the MOR distribution pattern.
Drug self-administration.
Standard intravenous drug self-administration methods were used to assess the rewarding effects of heroin or cocaine in WT and T394A mutant mice. Intravenous catheterization surgery and the self-administration procedures are the same as described previously (Xi et al., 2011; Song et al., 2012). Briefly, one group of WT or T394A mutant mice was initially trained for oral sucrose (5% solution) self-administration for 10 d to compare the effects of a nondrug reinforce and then followed by three different doses (in a sequence of 15, 30, 60 μg/kg/infusion) of heroin and cocaine. After stable sucrose-taking behavior was achieved, animals were prepared for intravenous catheterization surgery. After 5–7 d of recovery from surgery, each mouse was placed into a test chamber (Med Associates) and allowed to lever press for intravenous heroin (15, 30, or 60 μg/kg/infusion) or cocaine (0.5 mg/kg/infusion). Another group of WT or T394A mutant mice was trained for heroin (beginning from 60 μg/kg/infusion to 30 μg/kg/infusion) or cocaine (from 1 mg/kg/infusion to 0.5 mg/kg/infusion) without initial oral sucrose self-administration. Each lever press led to a delivery of 0.015 ml of the drug solution over 4.2 s under an fixed-ratio 1 reinforcement schedule. Each session lasted 3 h or until the animal received the maximally allowed 50 drug infusions to prevent drug overdose. Daily drug self-administration continued until stable day-to-day operant behavior was established with a steady behavioral response pattern for at least 3 consecutive days. The total numbers of infusions, active lever presses, and inactive lever presses for sucrose, heroin, and cocaine were compared between WT and T394A mice.
Progressive ratio (PR) heroin self-administration.
Additional groups of animals were initially trained for heroin self-administration (30 μg/kg/infusion) under fixed-ratio 1 reinforcement as outlined above. After stable heroin self-administration was established, animals were switched to PR reinforcement, under which the work requirement (lever presses) to receive an infusion was progressively raised within each test session (Xi et al., 2011). The PR breakpoint was defined as the maximal number of lever presses completed for the last heroin infusion before a 1 h period during which no infusions were obtained by the animal. Animals were allowed to continue daily sessions of heroin self-administration under PR reinforcement conditions for 5–10 d until day-to-day variability in breakpoint fell within 1–2 ratio increments for 3 consecutive days. The total numbers of heroin infusions and the breakpoint levels under PR reinforcement were compared between the two strains of mice.
In vivo microdialysis.
In vivo microdialysis procedures were as reported previously (Xi et al., 2011; Song et al., 2012). Briefly, mice were anesthetized with sodium pentobarbital, and guide cannulae (20 gauge, Plastics One) were surgically implanted into the NAc (anteroposterior 1.4 mm, mediolateral ±1.5 mm, dorsoventral −3.8 mm, 8° from vertical). The guide cannulae were fixed to the skull with 2–3 stainless steel jeweler's screws (Small Parts) and dental acrylic. After 7 d of recovery from surgery, microdialysis probes were inserted into the NAc 12 h before the onset of microdialysis. Microdialysis samples were collected every 20 min into 10 μl 0.5 m perchloric acid to prevent DA degradation. After collection, samples were frozen at −80°C. Dialysate DA was measured using high-pressure liquid chromatography with electrochemical and fluorometric detection, as reported previously (Xi et al., 2011). After the completion of microdialysis experiments, mice were anesthetized with a high dose of pentobarbital (>100 mg/kg, i.p.) and perfused transcardially with 0.9% saline followed by 10% formalin. Brains were removed and placed in 10% formalin for histological verification of microdialysis probe locations in mouse brain.
Locomotor activity.
The locomotor-stimulating response to heroin in WT and T394A mutant mice was recorded in a locomotor detection chamber (Accuscan Instruments). Before drug administration, each animal was placed in the locomotor chamber for 3 d (4 h per day) for environmental habituation. On each test day, mice were placed in the chamber for 1 h of habituation, and then removed and given either saline or one dose of heroin (0, 0.5, 1.0 mg/kg, i.p.). Additional groups of WT and T394A mutant mice were used to compare the effects of T394A mutation on cocaine-induced increase in locomotion. Animals were then placed back into the locomotor chambers to record locomotor activity for 4 h. After each test, animals received an additional 3–5 d of locomotor habituation before testing the next dose of drug. The order of testing for the various doses of heroin was counterbalanced. Data were collected in 10 min intervals. Total distance was used to compare locomotor response to heroin between two strains of mice.
Data analysis.
Data are presented as mean ± SEM. Two-way ANOVAs were used to compare the results of behavioral and microdialysis experiments. When appropriate, Student's t test was used for comparisons between two groups.
Results
Generation and characterization of T394A mutant mice
The targeting vector for creating T394A mutant mice contains a 10.1 kb DNA sequence, including one point mutation in exon 4 of MOR and the floxed Neo cassette. After confirmation by restriction analysis and by sequencing at each modification step, the targeting vector was first transfected to C57BL/6 × 129/SvEv hybrid embryonic stem cells, and then microinjected into C57BL/6 blastocysts. Resulting chimeras with a high percentage of agouti coat color were mated to WT C57BL/6 mice to generate F1 heterozygous offspring. The F1 heterozygous mice were identified for targeted integration and confirmed with the introduced point mutation, and then mated with flippase (FLP) recombinase-expressing mice to generate F2 heterozygous mice and subsequent homozygous breeding (Fig. 1). The homozygous T394A mutant mice were viable and fertile and had no gross abnormalities. The T394A mutation has no effect on basal MOR protein expression. Receptor binding assays showed that T394A mutant mice did not alter receptor binding to the MOR agonist [3H]-DAMGO (Fig. 2).

Generation of T394A KI mice. A, Construct of the targeting vector. The region was designed such that the long homology arm (LA) extended ∼8.0 kb from the 5′ end to exon 4. The floxed Neo cassette was inserted downstream of exon 4, including one point mutation (A–G) (marked by red star) generated by PCR mutagenesis. The short homology arm (SA) extended 2.1 kb downstream of the floxed Neo cassette. The targeting vector was confirmed by restriction analysis after each modification step and by sequencing with primers designed to read from the selection cassette into the LA and the SA. B, Secondary confirmation of positive clones identified by PCR. Clones 121, 232, 233, 251, and 281 were confirmed by Southern blot as correctly targeted and recommended for blastocyst injection; B6, 129, and hybrid were positive controls from the DNA of WT C57BL/6, 129/SvEv, and BA1 (C57BL/6 × 129/SvEv) mice, respectively. C, Genotyping of F1 mice. The F1 heterozygous mice 14906, 13054, and 13056 were identified for targeted integration and confirmed with the introduced point mutation. The expanded ES clone (121), which was used as a positive control, is denoted by the “+” in the gel photos. D, Genotyping illustration and result for Neo deletion of MOR KI mice (OPIO) after flippase (FLP) mating. Primer set (NDEL1 and NDEL2) was used to screen F2 mice for deletion of the Neo cassette. The PCR product for the WT is 306 bp, and the Neo deletion (with remaining set of loxP-FRT sites) is 480 bp. The presence of the Neo cassette could not be detected from this PCR screening. F1 heterozygous mouse 13056 and WT DNA were both used as negative controls, and are denoted by “−” in the gel photo. The PCR products were run on a 2% agarose gel with a 100 bp ladder as a reference. Primers for PCR screening: NDEL1, 5′-GTGATCAACATAGAAGGTGACCCTTC-3′; NDEL2, 5′-CGCAGCTAGGGAGGAAGATGTCTTC-3′.

Effects of T394A mutation on agonist binding to MOR, illustrating that T394A mutation did not alter [3H]-DMAGO binding to the MOR in brain membrane. No differences in Bmax and EC50 were found between WT and T394A mutant.
T394A mutation blocks acute opioid tolerance
The analgesic effects of morphine and etorphine were assessed in WT and T394A mutant mice using the hotplate test. Figure 3A shows the time course of the analgesic effects after a single morphine injection (10 mg/kg, s.c.) in WT mice. As peak antinociception was observed 30 min after morphine treatment, we used the 30 min time-point to compare the dose effects of morphine between WT and T394A mutant mice. Figure 3B shows the dose–response curve of morphine analgesia using an escalating morphine dose regimen (1, 5, 7.5, 10, 15, 20, 25, and 100 mg/kg, s.c.). There was no difference in morphine-induced analgesia between the two genotypes of mice (F(1,98) = 0.046, not significant). To determine whether T394A mutation alters morphine-induced analgesic tolerance, mice were challenged with morphine at the ED50 dose (15 mg/kg) 24 h after receiving an accumulative morphine dose of 100 mg/kg. We found that morphine-induced analgesia was significantly reduced in WT, but not in T394A mutant mice (two-way ANOVA, genotype main effect, F(1,14) = 17.47, p < 0.05; morphine treatment main effect, F(1,32) = 4.59, p < 0.05; genotype × treatment interaction, F(1,32) = 8.26, p < 0.05) (Fig. 3C).

Effects of T394A mutation on opioid analgesia and tolerance in antinociception to morphine or etorphine. A, Time course of the analgesic effect of morphine (10 mg/kg, s.c., n = 3) in WT. B, Cumulative dose–response curve of the antinociception produced by morphine (1, 5, 7.5, 10, 15, 20, 25, and 100 mg/kg, s.c.) in both WT and T394A mutant mice (n = 8 each strain). C, Acute tolerance to morphine (produced by the ED50 dose of 15 mg/kg, s.c.) assessed 24 h after the dose–response experiment (B) in both WT (n = 9) and T394A mutant (n = 10) mice. D, Time course of the analgesic effect of etorphine (10 μg/kg) in WT mice (n = 6). E, Cumulative dose–response curve of etorphine (1, 3, 5, 7.5, 10, and 15 μg/kg, s.c.)-induced antinociception in WT (n = 10) and T394A mutant (n = 13) mice. F, Acute tolerance to etorphine (produced by the ED50 dose of 6 μg/kg, s.c.), assessed 3 h after the dose–response experiment (E) in both WT (n = 7) and T394A mutant (n = 6) mice. #p < 0.05, compared with the Injection 1-induced analgesia. ##p < 0.01, compared with the Injection 1-induced analgesia. ***p < 0.001, compared with WT mice.
To determine whether this lack of acute tolerance to morphine in T394A mutant mice can be generalized to other opioids, we measured the analgesic effects of etorphine, a highly potent MOR agonist, using a similar experimental procedure. Figure 3D shows the time course of the analgesic effect after a single dose etorphine injection (10 μg/kg, s.c.). Peak effect was observed 15 min after drug treatment. Figure 3E shows the dose–response analgesic effects of etorphine after an escalating dose regimen (1, 3, 5, 7.5, 10, and 15 μg/kg, s.c.) measured at 15 min after each dose administration. There were no significant differences observed in etorphine-produced analgesia between the two strains of mice (Fig. 3E; F(1,123) = 0.024, not significant). However, when given etorphine injection again 3 h after the completion of the above dose–response test, etorphine (at the ED50 dose of 6 μg/kg, s.c.) produced analgesic tolerance in WT mice, but not in T394A mutant mice (Fig. 3F; two-way ANOVA, for the genotype main effect: F(1,14) = 17.47, p < 0.05; etorphine treatment main effect: F(1,32) = 4.59, p < 0.05; genotype × treatment interaction: F(1,32) = 8.26, p < 0.05).
T394A mutation attenuates etorphine-induced MOR internalization
To determine whether the loss of opioid tolerance observed above in T394A mutant mice is related to MOR internalization, we examined the effect of the T394A mutation on etorphine-induced MOR internalization using immunohistochemistry. Figure 4 shows the MOR distribution pattern in the dorsal horn of the lumbar spinal cord in saline- or etorphine-treated WT and T394A mutant mice. Under low magnification, MOR was seen in soma (bright green dots) and neuronal processes (bright green lines) in both saline-treated WT (Fig. 4A) and T394A mice (Fig. 4C). Under higher magnification, MOR appeared primarily on plasma membranes (Fig. 4A1 in WT; Fig. 4C1 in T394A mutants). Strikingly, etorphine (10 μg/kg, s.c.) treatment significantly altered the distribution pattern in WT mice. Bright dots and long lines were no longer evident (Fig. 4B). Under high magnification, the MOR was seen mostly in the intracellular compartment, rather than on plasma membranes. MORs appeared as scattered punctates or clusters in subcellular compartments (Fig. 4B1), suggesting receptor internalization. In contrast to these findings observed in WT mice, etorphine treatment did not alter MOR distribution in T394A mutant mice (Fig. 4D). MOR immunoreactivity was still mostly present on plasma membranes and did not show the same intracellular scattered pattern as seen in etorphine-treated WT mice (Fig. 4D1).

Effects of T394A mutation on etorphine-induced MOR internalization in dorsal horn neurons of the spinal cord. A, MOR immunostaining in WT mice 15 min after saline administration. B, MOR immunostaining in WT mice 15 min after after 15 μg/kg etorphine administration. C, MOR immunostaining in T394A mutant mice 15 min after saline administration. D, MOR immunostaining 15 min after etorphine administration. The right panels show the MOR immunostaining, DAPI-stained nuclei, and merged images in individual neurons under high magnification (60×) in each group of mice. Scale bars: A–D, low magnification (20×), 100 μm; high magnification (60×), 10 μm. The figures represent the results from 4 or 5 mice/group, 3–5 sections/mouse.
T394A mutation increases heroin self-administration
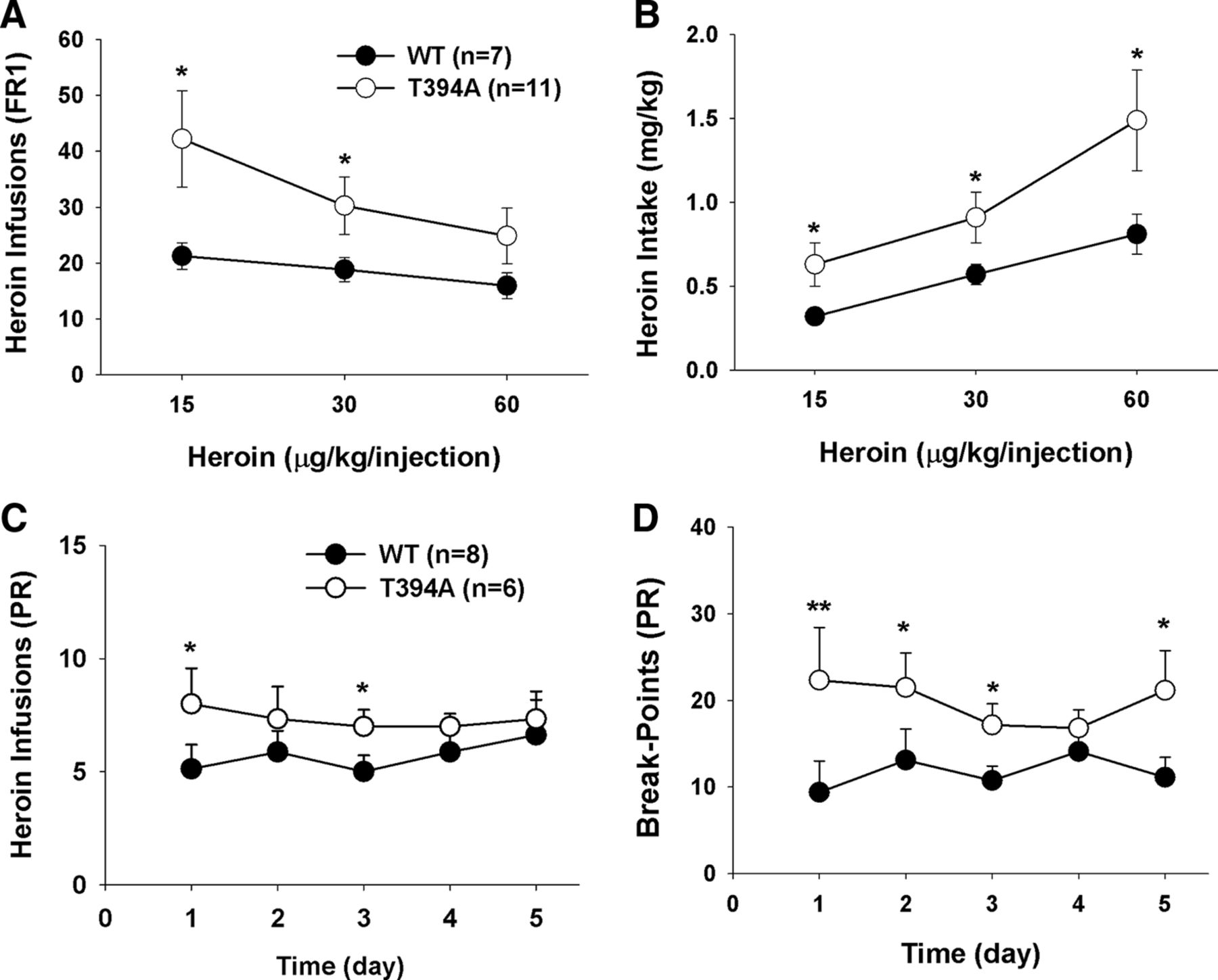
We then investigated whether the T394A mutation alters vulnerability to opioid use and abuse. Figure 5 shows the timeline of the self-administration experiment, illustrating the total numbers of drug/sucrose infusions (Fig. 5A), active lever presses (Fig. 5B), and inactive lever presses (Fig. 5C) over time for different reinforcement contingencies. In this experiment, we first trained animals to orally self-administer a 5% sucrose solution. We did not find a significant difference in acquisition and maintenance of sucrose self-administration between WT and T394A mutant mice (Fig. 5A, left; F(1,21) = 0.19, not significant). Then, sucrose was replaced by heroin in the same groups of mice. We found that T394A mutant mice displayed significantly higher levels of heroin infusions (Fig. 5A, top) and higher levers of active lever presses (Fig. 5B, middle panels) than WT mice. Two-way ANOVA for repeated measures over time revealed a significant genotype main effect in heroin infusions maintained by 15 μg/kg/infusion (Fig. 5A; F(1,21) = 5.38, p < 0.05) and 30 μg/kg/infusion (F(1,18) = 4.49, p < 0.05), but not 60 μg/kg/infusion (F(1,16) = 1.05, not significant) heroin. When heroin was replaced by cocaine, there was no difference in self-administration for cocaine between the two strains of mice (F(1,10) = 0.11, not significant), suggesting that the T394A mutation selectively alters vulnerability to heroin, not to sucrose or cocaine.

Effects of T394A mutation on self-administration of sucrose, heroin, or cocaine in the same groups of mice (within-groups design). A, Total numbers of infusions. B, Total numbers of active lever presses. C, Total numbers of inactive lever presses during the last 5 d of daily 3 h session for initial 5% sucrose, 3 different doses (15, 30, 60 μg/kg/infusion) of heroin, and cocaine (0.5 mg/kg/infusion), illustrating that T394A mutation caused enhanced heroin self-administration under fixed-ratio 1 reinforcement schedules. *p < 0.05, compared with WT mice.
To further determine whether previous sucrose or heroin self-administration alters subsequent heroin or cocaine self-administration, additional groups of mice were trained directly for heroin or cocaine self-administration without initial sucrose self-administration.
Figure 6A shows that T394A mutant mice displayed fast learning for heroin self-administration with significantly higher heroin infusion level (F(1,21) = 7.78, p < 0.01). In contrast, no significant difference was seen in the acquisition for cocaine self-administration between the two groups of mice (Fig. 6B; cocaine infusions: F(1,11) = 0.68, not significant). Figure 6C shows representative original records of heroin self-administration maintained by two different doses of heroin, illustrating evenly distributed patterns of heroin-taking behavior in both strains of mice. T394A mice displayed more burst-like drug-taking and higher numbers of heroin infusions with shorter interinfusion intervals than WT mice as shown in Figure 6A.

Effects of T394A mutation on heroin or cocaine self-administration in mice without initial sucrose self-administration. A, Time course of heroin self-administration, illustrating faster learning and acquisition of heroin self-administration in T394A mutant mice than in WT mice. B, Time course of cocaine self-administration, illustrating no significant difference in the acquisition for cocaine self-administration between two groups of mice. C, Representative heroin self-administration patterns, illustrating increased heroin infusions with shorter interinfusion intervals in T394A mutant mice than in WT mice. *p < 0.05, compared with WT mice (heroin infusions).
Figure 7A shows the dose–response effects of heroin self-administration, indicating that the T394A mutation significantly shifted the heroin self-administration dose–response curve upward (F(1,16) = 4.59, p < 0.05), as assessed by the mean heroin infusions over the last 5 d of heroin self-administration at each dose. Figure 7B shows the heroin intake dose–response curve, calculated by number of heroin infusions × heroin self-administration dose in each mouse, illustrating that the T394A mutation significantly shifted the heroin intake dose–response curve leftward and upward (F(1,16) = 4.86, p < 0.05).

Effects of T394A mutation on multiple dose heroin self-administration and heroin self-administration under PR reinforcement. T394A mutation causes an increase in heroin self-administration (A), heroin intake (B), and motivation for heroin self-administration (C, D). *p < 0.05, compared with WT mice. **p < 0.01, compared with WT mice.
We also observed the effects of the T394A mutation on breakpoint levels for heroin self-administration under PR reinforcement conditions. The PR breakpoint level, which is defined as the maximal work amount (lever presses) performed by an animal to get a heroin infusion, has been shown to be drug dose-dependent and positively correlated with reward strength or motivation for reward-taking behavior. Figure 7C shows that T394A mutant mice display higher levels of heroin infusions than WT mice during the last 5 d of heroin self-administration under PR reinforcement conditions (Fig. 7C; F(1,12) = 3.94, p < 0.05). Figure 7D shows a significant increase in PR breakpoint for heroin self-administration after T394A mutation (Fig. 7D; F(1,12) = 6.19, p < 0.05). These findings suggest that T394A mutation significantly enhances motivation for opioid-taking behavior.
T394A mutation causes a reduction in basal locomotion
To determine whether the above findings in heroin self-administration can be generalized to other actions of heroin, we observed the effects of T394A mutation on basal and heroin-enhanced locomotion. Figure 8 shows that T394A mutant mice displayed a significant reduction in basal levels of locomotion compared with WT mice. Systemic administration of heroin (0.5, 1 mg/kg, i.p.) produced a significant and dose-dependent increase in locomotion in both groups of mice. However, there is no significant difference in peak locomotor response to each dose of heroin observed between WT and T394A mutant mice (Fig. 8B,C, left panels). Because the basal levels of locomotion are different, we then compared heroin-induced changes (% baseline) in locomotion. We found that T394A mutation produced a significantly enhanced locomotor response to heroin (Fig. 8B,C, right panels). Two-way ANOVA for repeated measures over time revealed a statistically significant genotype main effect in locomotion after 1 mg/kg (Fig. 8C, right; F(1,14) = 11.34, p < 0.01), but not after 0.5 mg/kg (Fig. 8B, right; F(1,14) = 0.86, not significant) heroin. We also compared the effects of T394A mutation on cocaine-enhanced locomotor response. We found that T394A mutation did not alter the peak locomotor response to 10 mg/kg cocaine but produced a significant increase in normalized (% baseline) locomotor response to cocaine (Fig. 8D, right; F(1,9) = 13.72, p < 0.01).

Effects of T394A mutation on heroin- or cocaine-induced increase in locomotion. A–D, Left panels, Original traveled distance counts 1 h before and 2 h after different doses (0.5, 1 mg/kg, s.c) of heroin or cocaine (10 mg/kg, i.p.). Right panels, Heroin- or cocaine-induced changes in locomotion (% baseline). *p < 0.05, compared with WT mice at each time point. **p < 0.01, compared with WT mice at each time point. ***p < 0.001, compared with WT mice at each time point.
T394A mutation enhances DA response to heroin
Given that heroin self-administration and locomotor behavior are, at least in part, brain DA-dependent (Xi et al., 1998; Xi and Stein, 2002), we further observed and compared brain DA response with heroin in both groups of mice. We did not see significant difference in basal levels of extracellular DA in the NAc between two groups of mice. However, T394A mutant mice displayed an overall higher NAc DA response to heroin than WT mice, as assessed by extracellular DA concentration (Fig. 9A) or normalized percentage change of DA over baseline (Fig. 9B). Two-way ANOVA for repeated measures over time did not reveal a statistically significant genotype main effect (Fig. 9A; F(1,11) = 2.76, not significant; Fig. 9B; F(1,11) = 1.89, not significant) but revealed a statistically significant time main effect (Fig. 9A; F(20,220) = 3.38, p < 0.001; Fig. 9B; F(20,220) = 6.55, p < 0.001) and significant genotype × time interaction (Fig. 9A; F(20,220) = 3.29, p < 0.05; Fig. 9B; F(20,220) = 2.98, p < 0.05). Post hoc individual group comparisons revealed statistically significant differences in heroin-induced DA release between WT and T394A mice at several time points after heroin injections (Fig. 9). Figure 9C, D shows the placements of microdialysis probes in the brain, demonstrating that active microdialysis membranes tended to span the length of the core and shell compartments of the NAc.

Effects of T394A mutation on heroin-induced DA release in the NAc. A, Extracellular DA concentrations before and after 0.25 mg/kg and 1 mg/kg heroin injection in WT and T394A mutant mice. B, Heroin-induced changes in NAc DA normalized to baseline before heroin injection (% baseline). C, Representative brain section, showing path of a guide cannula and a microdialysis probe in the brain. D, Reconstruction of the placements of microdialysis probes in the brain, illustrating that active microdialysis membranes tended to span the length of the core and shell compartments of the NAc. *p < 0.05, compared with WT mice.
Discussion
In the present study, we demonstrate that the T394A single-point mutation results in robust changes in opioid actions in vivo, including attenuated opioid tolerance and enhanced vulnerability to heroin self-administration. The reduction in opioid tolerance appears to be related to attenuated MOR internalization as shown in spinal dorsal horn neurons, and that the increase in vulnerability to heroin self-administration appears to be related to enhanced NAc DA response to heroin after T394A mutation. These findings suggest an important role of T394 phosphorylation in the development of opioid tolerance and opioid abuse and dependence.
Opioid analgesia and MOR phosphorylation
It is well documented that opioid analgesia is mediated by activation of MOR-coupled Gi/o proteins (Christie, 2008). Such activation inhibits the intracellular cAMP-PKA signal cascade, causing neuronal inhibition by activation of G-protein-activated inwardly rectifying potassium channels and/or by inactivation of voltage-dependent calcium channels (Ikeda et al., 2002; Zhao et al., 2012; Williams et al., 2013). In addition, recent studies suggest that MOR agonists (such as morphine, DAMGO, or fentanyl) dose-dependently phosphorylate the Ser375 (S375) residue in the C-terminal of MOR, and S375A mutation produces a dose-dependent increase in morphine or fentanyl analgesia (Grecksch et al., 2011), suggesting that MOR phosphorylation plays an important role in mediating opioid analgesia. Congruent with this finding, modification of the downstream signaling pathway of MOR activation (using β-arrestin-2 knock-out mice) caused enhanced morphine-induced analgesia (Bohn et al., 1999, 2002).
In addition to S375, threonine 394 (T394) is another well-characterized phosphorylation site on the MOR in cultured cell lines. In the present study, we developed T394A mutant mice for characterizing the role of T394 in vivo. We found that T394A mutant mice appeared healthy and fertile, and did not display significant changes in mortality, body weight, and general behavior except moderately lower basal locomotion than WT mice. T394A mutation did not alter basal pain threshold and maximal antinociceptive response to morphine or etorphine as assessed by the hotplate test, suggesting that MOR T394 phosphorylation is not involved in opioid analgesia, which is different from S375 phosphorylation that causes an increase in opioid analgesia.
Opioid tolerance and MOR phosphorylation
It is well documented that long-term use of opioids causes the development of opioid tolerance that attenuates opioid-induced analgesia. The molecular mechanisms underlying analgesic tolerance are not fully understood. A well accepted view is that tolerance is associated with receptor phosphorylation, desensitization, and downregulation, with phosphorylation of specific residues in the intracellular domains of MOR being an initial step (Williams et al., 2013). There are ∼20 phosphorylation sites identified in the intracellular regions of MOR that may contribute to receptor desensitization and endocytosis (Connor et al., 2004; Koch and Höllt, 2008). These residues are phosphorylated putatively by G-protein receptor kinase (GRK) (Thr180, Ser355, Ser357, Thr370, Ser370) and/or by non-GRK kinases, such as tyrosine hydroxylase (Tyr106, Tyr166), PKC (Ser363), and calcium-calmodulin kinase (Ser266) (Liu and Anand, 2001; Koch and Höllt, 2008; Williams et al., 2013). However, it is not yet known whether such phosphorylation per se truly desensitizes MORs in vivo, and which amino acid residue phosphorylation is critical to the development of analgesic tolerance in experimental animals. In a previous report using S375A mutant mice, it was found that the S375A mutation selectively abolished analgesic tolerance to DAMGO, but not to morphine or fentanyl (Grecksch et al., 2011), suggesting that S375 phosphorylation is contributing to the development of opioid tolerance; however, its role may depend on distinct MOR agonists.
In the present study, we investigated the role of another phosphorylation site (T394) in the development of opioid tolerance. Previous studies have shown that T394A mutation at the MOR C-terminal leads to a reduction of receptor phosphorylation in transfected CHO cells (Deng et al., 2000), suggesting that T394 phosphorylation may regulate MOR function in vitro (Pak et al., 1997; Wolf et al., 1999; Deng et al., 2000; Wang et al., 2007). In contrast, another group reported opposite results: facilitation in MOR desensitization and faster internalization after T394A mutation (Wolf et al., 1999). The reasons for such conflicting findings are unclear but may be related to the different cell types used (El Kouhen et al., 2001). For example, morphine caused moderate MOR phosphorylation in CHO cells or in cells of rat thalamus, but not in HEK293 cells unless GRK2 was overexpressed, whereas etorphine produced fast and robust MOR phosphorylation in HEK293 cells (Yu et al., 1997; J. Zhang et al., 1998; Deng et al., 2000). These findings suggest that differential MOR phosphorylation may result from distinct agonists and distinct protein kinases in the different cells.
To further address whether the T394 phosphorylation and subsequent receptor internalization are truly important in the development of opioid tolerance in animals, we generated T394A mutation mice in this study. We found that T394A mutation altered neither basal MOR expression in cell membranes, as assessed by the Bmax of [3H]DAMGO receptor binding, nor receptor/ligand binding Kd, but attenuated etorphine-induced MOR internalization and endocytosis as assessed by MOR immunofluorescence staining. Strikingly, T394A mutation also abolished analgesic tolerance to morphine or etorphine as measured by hotplate test 24 h after the last injection of morphine or 3 h after the last injection of etorphine, suggesting that MOR phosphorylation is an initial adaptive process underlying opioid tolerance.
MOR phosphorylation and opioid tolerance in drug reward
We and others have reported that chronic heroin self-administration desensitizes MOR-activated G-proteins (Sim-Selley et al., 2000) and attenuated brain functional response to heroin in rats as assessed by fMRI (Luo et al., 2004; Xi et al., 2004), suggesting opioid tolerance development in opioid reward. However, the molecular mechanisms underlying such MOR desensitization by heroin are unclear. In the present study, we demonstrate that T394A mutation attenuated agonist-induced MOR internalization in spinal dorsal horn neurons and blocked opioid analgesic tolerance, suggesting that a similar MOR phosphorylation mechanism may occur in the mesolimbic DA system, for example, in the VTA GABAergic neurons where MOR is expressed, that leads to tolerance in opioid reward. Accordingly, a reduction in drug reward (tolerance) may lead to the compensatory increase in heroin self-administration or in heroin intake in WT mice. However, the findings in the present study do not appear to support this hypothesis because there is no significant increase in heroin self-administration or heroin intake over time (days) in WT mice during the maintenance phase or after the acquisition of heroin self-administration. This may be related to the experimental conditions in which a short-access (3 h per session) to a very low dose (0.5 mg/kg/infusion) of heroin was used in the present study. This is in contrast to the experimental conditions in which prolonged exposure (30–60 d) or a long-access (6 h per session) to much higher doses of heroin was used to study chronic heroin-induced compulsive drug-taking behavior (Ahmed et al., 2000; Sim-Selley et al., 2000). These findings suggest that the development of opioid tolerance and subsequent behavioral response may largely depend on heroin doses took during the self-administration experiments.
An unexpected finding in this experiment is that T394A mutation produced an increase in heroin self-administration. The simplest interpretation is that T394A mutation causes an increase in membrane MOR availability in the mesolimbic DA system (e.g., in VTA GABAergic interneurons) that leads to an enhanced DA response to heroin via a GABAergic disinhibition hypothesis (Xi and Stein, 2002). Because extracellular DA is positively correlated to a drug's rewarding and addictive effects: the faster the increase in extracellular DA, the higher the drug-induced reward and psychomotor-stimulation (Busto and Sellers, 1986; Volkow et al., 1995), enhanced DA response to heroin may lead to enhanced heroin-taking behavior. We note that the T394A mutation selectively altered heroin, but not cocaine, self-administration. This is related to the fact that heroin and cocaine act on different binding sites in the brain and T394 phosphorylation depends on MOR activation only. In addition, T394A mutation also significantly lowered basal level of locomotion. One possibility is that T394A mutation may increase endogenous opioid tone on MOR due to increased membrane MOR availability, producing sedation-like effects. Finally, T394A mutation did not significantly alter peak locomotor response to heroin. This may be related to the relatively mild DA-enhancing effect in T394A mutant mice.
In conclusion, the present study, for the first time, demonstrates that a single-point mutation at the T394 phosphorylation site blocked acute opioid tolerance and produced enhanced heroin-taking behavior, suggesting an important role of MOR T394 phosphorylation in the development of opioid tolerance and dependence. Accordingly, the MOR T394 phosphorylation site may constitute a new therapeutic target in medication development for the treatment of opioid addiction. Furthermore, T394 mutation or SNP (if identified in humans) may be used as a new biomarker in predicting susceptibility to opioid addiction.
Footnotes
This work was supported by the Intramural Research Program of the National Institute on Drug Abuse and National Institute on Drug Abuse Grants DA11925 and DA018722 to J.B.W. and P30 DA013429 to L.Y.L.-C.
The authors declare no conflict of interest.
- Correspondence should be addressed to either of the following: Dr. Zheng-Xiong Xi, National Institute on Drug Abuse, Intramural Research Program, Baltimore, MD 21224, zxi{at}mail.nih.gov; or Jia Bei Wang, Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, Baltimore, MD 21201. jwang{at}rx.umaryland.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}