

Abstract
Brain mechanisms compensating for cerebral lesions may mitigate the progression of chronic neurodegenerative disorders such as Alzheimer's disease (AD). Mild cognitive impairment (MCI), which often precedes AD, is characterized by neuronal loss in the entorhinal cortex (EC). This loss leads to a hippocampal disconnection syndrome that drives clinical progression. The concomitant sprouting of cholinergic terminals in the hippocampus has been proposed to compensate for reduced EC glutamatergic input. However, in absence of direct experimental evidence, the compensatory nature of the cholinergic sprouting and its putative mechanisms remain elusive. Transgenic mice expressing the human APOE4 allele, the main genetic risk factor for sporadic MCI/AD, display impaired cholinergic sprouting after EC lesion. Using these mice as a tool to manipulate cholinergic sprouting in a disease-relevant way, we showed that this sprouting was necessary and sufficient for the acute compensation of EC lesion-induced spatial memory deficit before a slower glutamatergic reinnervation took place. We also found that partial EC lesion generates abnormal hyperactivity in EC/dentate networks. Dentate hyperactivity was abolished by optogenetic stimulation of cholinergic fibers. Therefore, control of dentate hyperactivity by cholinergic sprouting may be involved in functional compensation after entorhinal lesion. Our results also suggest that dentate hyperactivity in MCI patients may be directly related to EC neuronal loss. Impaired sprouting during the MCI stage may contribute to the faster cognitive decline reported in APOE4 carriers. Beyond the amyloid contribution, the potential role of both cholinergic sprouting and dentate hyperactivity in AD symptomatogenesis should be considered in designing new therapeutic approaches.
SIGNIFICANCE STATEMENT Currently, curative treatment trials for Alzheimer's disease (AD) have failed. The endogenous ability of the brain to cope with neuronal loss probably represents one of the most promising therapeutic targets, but the underlying mechanisms are still unclear. Here, we show that the mammalian brain is able to manage several deleterious consequences of the loss of entorhinal neurons on hippocampal activity and cognitive performance through a fast cholinergic sprouting followed by a slower glutamatergic reinnervation. The cholinergic sprouting is gender dependent and highly sensitive to the genetic risk factor APOE4. Our findings highlight the specific impact of early loss of entorhinal input on hippocampal hyperactivity and cognitive deficits characterizing early stages of AD, especially in APOE4 carriers.
- Alzheimer's disease
- APOE4
- cholinergic sprouting
- entorhinal cortex
- hippocampal disconnection
- spatial memory
Introduction
Curative treatments trials for Alzheimer's disease (AD) have failed so far, probably because interventions against Aβ accumulation must target stages earlier than mild to moderate AD (Selkoe, 2012). However, even with the best currently available biomarkers, the perspective of blindly treating billions of people at risk several years or decades before a possible onset raises ethical and cost issues. Therefore, an effective symptomatic treatment at the earliest detectable stage of the disease is urgently needed. Understanding how the brain manages to compensate for early AD progression may be particularly relevant, but remains poorly studied.
During the earliest stages of the disease, including mild cognitive impairment (MCI) (Petersen et al., 2001), almost 50% of entorhinal cortex (EC) neurons are lost (Gómez-Isla et al., 1996; Kordower et al., 2001). The consecutive loss of glutamatergic inputs to the hippocampus (Hyman et al., 1984, 1986) is the best correlate for memory impairment onset and progression to dementia (Sze et al., 1997; Scheff et al., 2006). Nevertheless, clinical trajectories often vary from abrupt AD conversion to long-lasting MCI (Petersen et al., 2001) and even reversion to normal cognition (Koepsell and Monsell, 2012), suggesting that some mechanisms compensate for early hippocampal disconnection.
Experimental EC lesions are known to be associated with the subsequent sprouting of cholinergic fibers within deafferented regions of the hippocampus in rodents (Lynch et al., 1972). A similar sprouting has been reported in MCI/AD patients (Geddes et al., 1985; DeKosky et al., 2002). Interestingly, this cholinergic sprouting depends on astrocyte-secreted apolipoprotein E (ApoE), the main cholesterol and lipid transport protein for neurons that is required for de novo membrane synthesis and necessary for the sprouting of terminals (Poirier et al., 1993; Pfrieger, 2010).
Despite the lack of direct experimental evidence, hippocampal cholinergic sprouting has been proposed to compensate for the loss of EC inputs (Mufson et al., 2012). However, cholinergic sprouting has never been demonstrated as being necessary for behavioral recovery after EC lesions. Moreover, putative mechanisms underlying a cholinergic compensation for the consequences of a loss of EC glutamatergic inputs remains undetermined. Some studies even denied the existence of a cholinergic sprouting (Phinney et al., 2004). One difficulty in disambiguating the physiological role of cholinergic sprouting is the difficulty to manipulate sprouting without altering normal cholinergic functions.
Possession of the APOE4 allele coding for the ε4 isoform of ApoE is the strongest genetic risk factor for sporadic AD (Roses, 1996), mostly through a negative influence on the early MCI/AD stages characterized by the hippocampal disconnection syndrome (Barabash et al., 2009). Compared with the neutral APOE3 allele, APOE4 precipitates the conversion to AD (Xu et al., 2013), accelerates spatial memory impairment onset (Laczó et al., 2011), and reduces spontaneous reversion rates (Koepsell and Monsell, 2012), a pattern that matches well with impaired brain compensation. Cholinergic sprouting may be particularly impaired in APOE4 patients because they respond less to acetylcholinesterase inhibitors (Farlow et al., 1996). Accordingly, APOE4-transgenic mice have impaired cholinergic sprouting after EC lesions (Blain et al., 2006) and spatial memory deficits reminiscent of those characterizing APOE4-positive MCI patients (Bott et al., 2013). Therefore, APOE4-transgenic mice represent a tool to manipulate the cholinergic sprouting in a disease-relevant way.
To determine the contribution of hippocampal cholinergic sprouting to the recovery from partial bilateral entorhinal lesions in APOE-transgenic mice, we followed the temporal evolution of performance in a spatial navigation task in relation to hippocampal synaptic reorganizations. Because estrogen facilitates sprouting responses (Stone et al., 1998), both males and females were tested. Finally, using optogenetics, putative mechanisms underlying the cholinergic sprouting compensation were explored (ChAT-ChR2 mice).
Materials and Methods
Animals.
A total of 255 male and female transgenic hAPP-YAC/APOE3-tr (APOE3) and hAPP-YaC/APOE4-tr (APOE4) mice (C57BL/6J background) were used at the age of 11 months at the time of surgery (204 mice were included in the study after exclusion of those failing to meet lesion criteria described below). The mouse lines were generated by Taconic Farms as described previously (Bott et al., 2013). These lines express physiological levels of human APOE3 or APOE4 instead of the murine APOE. They also harbor one supplementary copy of normal human APP (nonmutated) in addition to murine APP. To reduce the number of animals, wild-type littermates were not included because we focused on APOE4 and APOE3 genotype comparisons relevant for humans. Male ChAT-ChR2-transgenic mice (CD1 genetic background, 10 backcross generations) were provided by Prof. Feng Guoping (Neuroscience McGovern Institute for Brain Research, Massachusetts Institute of Technology). These mice express channelrhodopsin 2 under the control of the ChAT promoter (specifically in cholinergic neurons) and were 11 months old at the time of surgery. In accordance with the European Union laws for animal studies, all procedures were approved by the Institutional Ethical Committee (authorization number: AL/15/22/02/13 for APOE experiments and AL/58/65/02/13 for ChAT experiments). Animals were maintained with ad libitum access to food and water under standard a 12/12 light/dark cycle (lights on at 7:00 A.M.).
Experimental design.
To investigate synaptic reorganization in relation to behavioral performance 30, 70, and 170 d postlesion (dpl) (see Fig. 1), APOE-transgenic mice were pseudorandomly assigned to 24 experimental groups (3 delays * 2 sex * 2 genotypes * 2 treatments). ChAT-ChR2 mice underwent the same procedure as APOE mice: 5 sham and 5 lesioned mice were used for behavioral and histological experiments, 9 sham and 14 lesioned mice were used for medial septum/diagonal band of Broca (MSDB) cholinergic neuron stimulations coupled with intrahippocampal recordings, and 10 sham and 19 lesioned mice (5 at 7 dpl and 14 at 30 dpl) were used for local stimulation of cholinergic terminals in the dorsal hippocampus together with perforant path electrical stimulations. Experimenters were blinded to genotype and treatments.
EC lesions.
To perform partial bilateral EC lesions, deeply anesthetized mice (complete loss of tail and paw-pinch retraction reflexes; sodium pentobarbital, 70 mg/kg, i.p., Ceva Santé Animale) received stereotaxic microinjections of NMDA (120 mm) diluted in PBS (pH 7.4; 0.1 μl/min). To control pain, anesthetized mice received 0.05 ml of Xylocaïn (Xylovet, France; 21 mg/ml) subcutaneously before scalp incision. Coordinates were taken from bregma as follows: site 1 (0.075 μl): A = −4.1 mm; L = ±4.3 mm; 0.5 mm above the bottom of the skull; site 2 (0.1 μl): A = −4.7 mm; L = ±3.5 mm; 0.5 mm above the bottom of the skull; and site 3 (0.05 μl): A = −4.7 mm; L = ±3.5 mm; 1 mm above the bottom of the skull. After surgery, mice received a 5 mg/kg nonsteroidal anti-inflammatory treatment (Meloxicam; Boehringer Ingelheim) and were carefully monitored for 1 week (for the complete procedure, see Bott et al., 2013).
Barnes maze.
The maze was a 1-m-diameter circular platform with 12 regularly spaced holes (4 cm from the edge of the platform) raised 1 m above the floor and brightly illuminated (900 lux) to motivate escape through a single target hole connected to the mouse home cage. All trials were recorded with a video-tracking system (ANY-maze 4.3; Ugo Basile). Mice were trained during 5 acquisition days (3 daily trials 15 min apart; 180 s cutoff) to escape the platform through the target hole. To assess spatial memory performance, a probe trial (2 min, all holes closed) was conducted 24 h after the last acquisition session. Between each trial, the platform was cleaned with 70% ethanol and pseudorandomly rotated to avoid olfactory-based strategy (for more detailed methodology, see Bott et al., 2013).
To evaluate learning, the number of errors (visit to nontarget holes) and latency to the first access to the target hole were recorded. Because mice from the C57BL/6J background are known to prefer nonspatial strategies in this task, each acquisition trial was classified into one of three search strategies adapted from Harrison et al. (2006): spatial (direct visit to the target or an adjacent hole), serial (at least 2 successive visits before reaching the target), and mixed (remaining trials) strategies. Memory performances during the probe trial were evaluated by comparing the distance run in each of the four quadrants of the Barnes maze. Because the total distance run differed between groups, performances were normalized by dividing individual distance run in each quadrant by the total distance run.
Perfusion and tissue preparation.
Twenty-four hours after the probe trial, mice were killed by an overdose of sodium pentobarbital (150 mg/kg, i.p.) and received a 4% paraformaldehyde intracardiac perfusion. Brains were further postfixed during 4 h before being cryoprotected for 48 h in a 20% sucrose solution and finally frozen in isopentane (−35°C). Brains were cut into 20-μm-thick sections. Coronal slices were serially taken (1/4) from the dorsal hippocampus for immunohistochemistry and horizontal slices were taken serially for EC lesion delimitation.
Lesion measurements.
Lesions were delimited on a cresyl violet staining. Lesion percentage was calculated for each slice and then averaged by hemisphere. The absence of significant lesions [<30% medial EC (MEC)], unilateral lesions (>20% of difference between hemispheres), and lesions extending to adjacent structures (>30% in subiculum, perirhinal, and postrhinal cortices) led to mouse exclusion.
Immunohistochemistry.
VGLuT1 immunoreactivity was used to evaluate changes in cortical and intrahippocampal glutamatergic terminal densities (Fremeau et al., 2004) and VAChT for cholinergic terminal densities. Immunofluorescent staining was done using primary antibodies against VAChT (rabbit polyclonal; 1:1000; Synaptic Systems catalog #139 103 RRID:AB_887864) and VGluT1 (guinea pig polyclonal; 1:1000; Synaptic Systems catalog #135 304, RRID:AB_887878) and secondary antibodies (Alexa Fluor 488 anti-guinea-pig; 1:1000 and Alexa Fluor 555 anti-rabbit; 1:1000) from Invitrogen. Slices were incubated with 5% normal donkey serum diluted in PBS (0.1% of Triton ×100) for 2 h at room temperature and then incubated at room temperature for 18 h with primary antibodies diluted in PBS (0.1% of Triton X-100). After 3 washes in PBS, sections were incubated for 2 h with secondary antibodies. After three washes in PBS, sections were mounted with a DAPI slide mounting kit (DAPI Fluoromount-G; SouthernBiotech) to facilitate anatomical delimitations. Each immunohistochemical staining was done in one run for all mice included in the study.
Image analysis.
Images were taken with a 20× magnification objective lens mounted on a Leica bright-field microscope, with all settings kept identical for all sections of each type of staining. Image analysis was performed with ImageJ (RRID:SCR_003070). Synaptic densities were measured in all synaptic layers of the dorsal DG, CA3, CA2, and CA1 (for each layer, three measurements per section in 100 μm2 sampling box on four sequential sections spaced by 120 μm: these 12 measurements for each synaptic layer for each mouse were averaged to give one value for each layer per mouse; see Fig. 1). Cholinergic density was estimated from binearized pictures as a percentage of area covered by the staining after thresholding. The threshold was set to quantify only synaptic terminals: threshold level was increased until all fibers disappeared from thresholded images. This approach was selected as more appropriate to reveal changes in the sparse VAChT signal than relative fluorescent density. Glutamatergic density was assessed through the intensity of fluorescence after subtraction of background fluorescence taken from the surrounding corpus callosum. This relative fluorescence approach was selected for assessing VGluT1 changes because VGluT1-positive terminal dense packing and the absence of fiber staining precludes a thresholding approach similar to that used for cholinergic terminals. Synaptic cholinergic and glutamatergic data were expressed as a ratio of the respective sham density (for each delay, genotype, and sex) to extract and compare specifically lesion-related synaptic reorganizations throughout groups. Density of staining was first measured for all mice. Then, for each genotype, sex, and delay, density of all sham and lesioned mice was divided by the averaged density calculated in the corresponding sham group. The presumable main origin of glutamatergic innervation for each layer was indicated in our graphs based on Van Strien et al. (2009).
Optogenetic and electrophysiology.
Mice under urethane (1.5 g/kg; Sigma-Aldrich) and ketamine (50 mg/kg; Imalgen; Merial)/xylazine (10 mg/kg; Rompun; Bayer) terminal anesthesia were placed in a stereotaxic apparatus after the complete loss of tail- and paw-pinch retraction reflexes. Coordinates were calculated from bregma (dorsal hippocampus recordings: AP −1.94 mm; ML ±0.11 mm; P −0.12 mm from dura; MSDB optical fiber: AP +0.078 mm; ML −0.015 mm; P-3.9 mm from dura; perforant path stimulation: AP-0.43 mm; ML ±0.24 mm; P −0.15 from dura). Optogenetic stimulations were done with an LED stimulation light (Prizmatix) at 470 nm through a 250 μm optic fiber (10 ms pulses at 0.2, 5, or 20 Hz; 15–20 mW output power). Recordings were done with a linear 16-channel silicon probe (Neuronexus, A1x16-2 mm-50-177) connected to an AlphaLab recording system (Alpha-Omega). Raw signal was amplified (200×), filtered between 0 and 9 kHz, and digitalized at 22 kHz. Silicon probes, optical fiber, and stimulating electrodes were painted with 2% Dil solution (Sigma-Aldrich) for localization.
Analyses were performed using custom-made scripts in MATLAB (MathWorks, RRID:SCR_001622). When present, slow drift and electrical noise were removed using the Chronux signal processing toolbox (Bokil et al., 2010; RRID:SCR_005547). LFP data were down-sampled to 2200 Hz and filtered between 0.1 and 500 Hz. Spectral analysis were carried with the Chronux toolbox with a time-frequency product of three and five tapers. Time-frequency analyses were done on 4 s window moved across the data in 1 s increments. Due to the high occurrence of dentate LFP spikes in lesioned mice, power in the theta band (3–9 Hz in anesthetized mice) was measured as peak power, a measure removing contamination caused by high-amplitude events.
Urethane anesthesia is characterized by alternating theta and non-theta states (Pagliardini et al., 2013). Only periods associated with theta (theta/delta power ratio >5) were analyzed. Electrophysiological changes induced by optogenetic cholinergic stimulations were extracted from the comparison between 60 s of baseline with 60 s of stimulation followed by 60 s of recovery. Scopolamine hydrobromide (Sigma-Aldrich; 3 mg/kg, i.p., dissolved in saline) was used to assess muscarinic cholinergic receptor involvement.
Dentate LFP spikes (Bragin et al., 1995) were detected as high-amplitude events on wide-band LFP (>5SD above mean of LFP power from 1 to 500 Hz) recorded in hilar channels. Current-source-density (CSD) calculation was used to compare the sink-source profile associated with spontaneous dentate LFP spikes with the activity elicited by perforant path electrical stimulations. The CSD value for a given time point t was calculated as follows:
 where LFP(n, t) is the LFP recorded at the electrode n, LFP(n + 1, t) and LFP(n − 1, t) are the LFP from electrodes above and below, respectively, and Δd is the spacing (in millimeters) between sites.
where LFP(n, t) is the LFP recorded at the electrode n, LFP(n + 1, t) and LFP(n − 1, t) are the LFP from electrodes above and below, respectively, and Δd is the spacing (in millimeters) between sites.
Statistical analysis.
Analyses were done with Statistica version 10 (StatSoft). Data are expressed as the mean ± SEM calculated across animals except for unit firing, in which the sampling unit was the cell. Factorial ANOVA (factor genotype, lesion, delay, and sex) and ANOVA with repeated measures (days and holes for the behavior; synaptic layers for immunohistochemistry) were performed. For histological data, any interaction in which the lesion factor is not an interaction per se (i.e., a different effect for each modality of the lesion factor) because sham group means were all forced (normalized) to 1. The interaction simply expresses a difference between the lesioned groups and their corresponding sham groups. In case of significant interaction among factors, multiple comparisons among groups were performed using Fisher's LSD post hoc test (after factorial ANOVA for comparison between numerous groups) or Newman–Keuls post hoc test (after ANOVA with repeated measures). Comparison to chance level in the Barnes maze was done using a one-sample Student's t test. The threshold for statistical significance was set at p < 0.05. The experimenter was blinded to the treatment groups until the end of statistical analyses.
Results
To determine the role of hippocampal cholinergic sprouting in the recovery from partial hippocampal disconnection induced by partial bilateral entorhinal lesions, we monitored the behavioral recovery of APOE-transgenic mice in a Barnes maze task as well as cholinergic and glutamatergic plastic reorganizations in main hippocampal subfields (Fig. 1). In addition, because estrogen facilitates sprouting responses (Stone et al., 1998), both males and females were tested to determine whether APOE4's inhibitory effect on cholinergic sprouting could be overpassed to facilitate functional recovery in females. Finally, using an optogenetic approach, we also explored the putative mechanism underlying the cholinergic sprouting compensation on hippocampal networks in mice expressing light-activable opsin in cholinergic neurons (ChAT-ChR2 mice).

Experimental timeline and hippocampal regions of interest. A, Experimental timeline for hAPP/APOE and ChAT-ChR2 mice experiments. B, Photomicrograph of a DAPI-stained dorsal hippocampus slice showing the different regions of interest for histological measurements. oML, outer part of the molecular layer; iML, inner part of the molecular layer; HIL, hilus; OR, stratum oriens; LUC, stratum lucidium; RAD, stratum radiatum; LM, stratum lacunosum moleculare; iLM, 2, inner part of the stratum lacunosum-moleculare and 1,3, outer part of the stratum lacunosum moleculare.
Impaired recovery and compromised sprouting response in male APOE4 mice
Lesion magnitude was similar among all male lesioned groups (Fig. 2A; genotype*delay interaction: F(2,46) = 0.4215; p = 0.658; genotype effect F(1,46) = 1.7638, p = 0.1907; delay effect: F(2,46) = 0.2403, p = 0.7874), suggesting that APOE4 mice were not more sensitive to NMDA excitotoxicity. Lesions were quite specific to the MEC, with minimal impact on surrounding regions such as lateral EC (LEC), subiculum, and perirhinal cortex (region effect: F(7,322) = 44.1709; p < 0.00001).

EC lesion and acquisition performance in the Barnes maze for male hAPP/APOE mice. A, Photomicrograph of representative cresyl violet stainings from a sham (top) and a lesioned (bottom) mouse. MEC and LEC are delineated with dashed yellow lines and the lesion area is delineated with a dashed red line. Scale bar, 600 μm. Left bar graph shows that NMDA microinjections induced partial lesions of similar magnitude in all groups. MEC was clearly the most lesioned region, whereas LEC, perirhinal cortex, and subiculum were much less affected ($p < 0.05 MEC vs other regions; LSD post hoc analysis). B, During the acquisition phase of the Barnes maze task throughout all postlesion delays, all groups improved their performance in a similar fashion, although without relying much on a spatial strategy (inserts represent the proportion of trials with spatial strategy; *p < 0.05 sham vs lesioned; **p < 0.01 sham vs lesioned; two-sample unpaired t test).
Male hAPP/APOE mice were trained in the Barnes maze at three different postlesion delays (dpl): 24 d (probe trial at 30 dpl), 64 d (probe trial at 70 dpl), and 164 d (probe trial at 170 dpl). Along the five acquisition days, all groups similarly reduced the time to reach the target hole (Fig. 2B: day effect: F(4,380) = 378.821, p < 0.00001), suggesting comparable motivation to escape the maze. However, mice did not rely preferentially on a spatial-based strategy, as documented frequently in C57BL/6J mice (O'Leary and Brown, 2012).
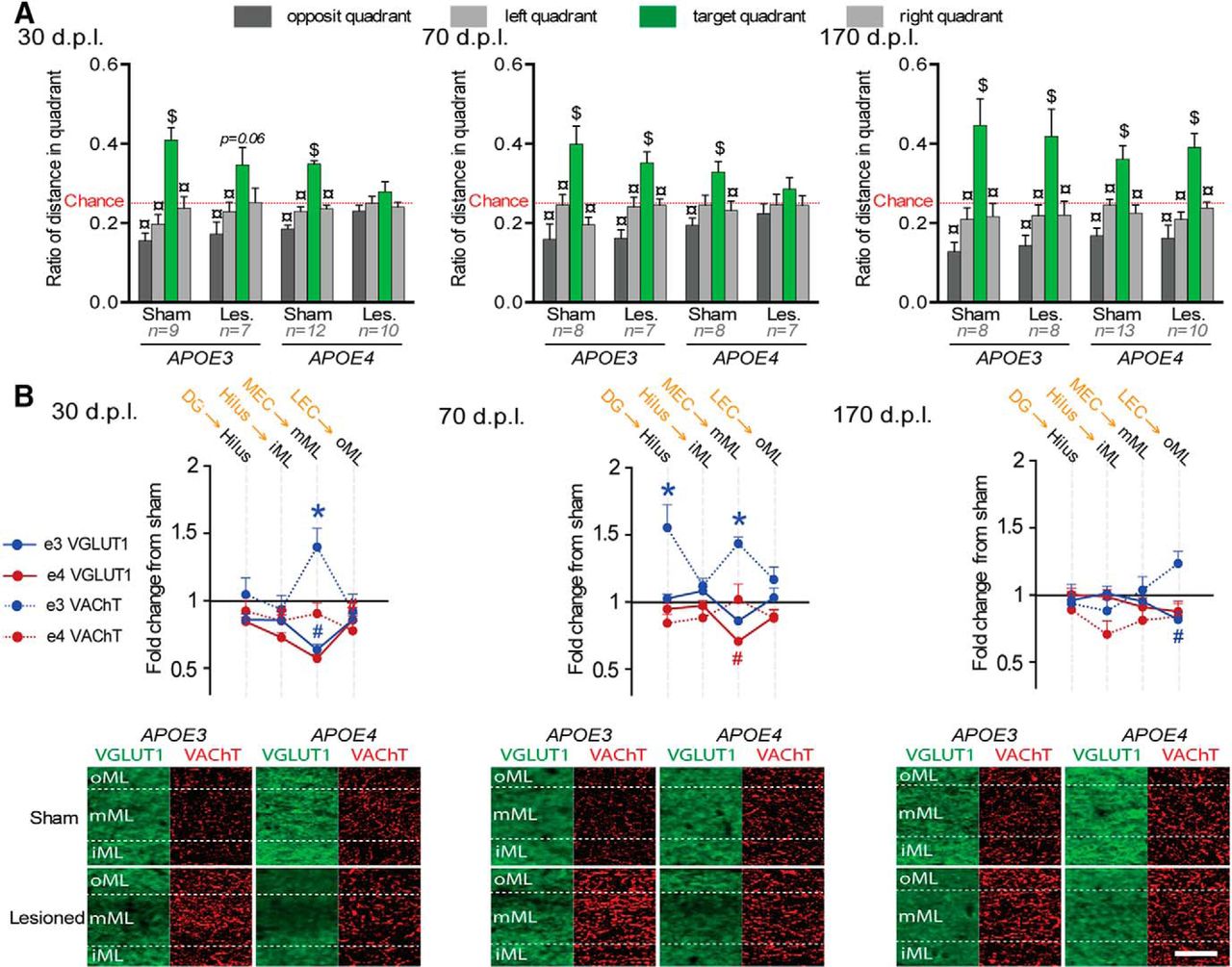
The probe trial allowed us to evaluate the mouse's knowledge of the target hole location independently of its preferred strategy. Accordingly, this trial was used to assess spatial memory performances that were influenced independently by postlesion delay (Fig. 3A, delay*quadrant interaction: F(6,285) = 2.49; p = 0.02) and genotype (genotype*quadrant interaction: F(3,285) = 5.68; p = 0.0008). However, all sham mice visited the target quadrant more than the other quadrants and more than the chance level, suggesting a successful recall of the target location (Fig. 3A). Lesioned APOE3 mice were slightly impaired, but only at 30 dpl (Fig. 3A). In contrast, lesioned APOE4 mice were deeply impaired at both 30 and 70 dpl before recovering at 170 dpl (Fig. 3A). Therefore, lesioned APOE4-mice displayed a slower behavioral recovery than lesioned APOE3 mice after partial EC lesion.

Delayed behavioral recovery and impaired cholinergic sprouting in lesioned male APOE4 mice. A, During the Barnes maze probe trial, lesioned APOE4 mice were impaired at 30 and 70 dpl, whereas lesioned APOE3 mice were only mildly affected at 30 dpl. $Distance in target quadrant differs from chance level (p < 0.05; t test); ¤differs from target quadrant (p < 0.05; Fisher's LSD). B, Top, Lesioned APOE4 mice (red) showed a broader glutamatergic loss at 30 dpl and a delayed glutamatergic reinnervation at 70 dpl. Only lesioned APOE3 mice (blue) displayed a cholinergic sprouting at 30 and 70 dpl. The presumed main origin of VGLuT1-positive inputs is indicated in orange above the corresponding hippocampal layer in black. *VAChT density differs from sham level (p < 0.05, Fisher's LSD); #VGLuT1 immunoreactivity differs from sham level (p < 0.05, Fisher's LSD). Bottom, Examples of VGLUT1 (green) and VAChT stainings (red) taken from the molecular layer of the DG. Scale bar, 50 μm.
The extent of glutamatergic (VGluT1) and cholinergic (VAChT) presynaptic reorganizations were measured for each postlesion delay (Fig. 3B, Table 1, Table 2, respectively). Lesion-induced glutamatergic changes were layer and delay dependent (lesion*layers*delay interaction: F(30,1005) = 2.117; p = 0.00046). In lesioned APOE3 mice, glutamatergic input loss was only significant at 30 dpl and was limited to layers receiving MEC inputs (Fig. 3B). In contrast, at 30 dpl, lesioned APOE4 mice exhibited a significant loss of glutamatergic input in most hippocampal layers (Fig. 3B, Table 1), suggesting perturbation of hippocampal glutamatergic networks broader than just in layers targeted directly by EC inputs. Furthermore, the persistence of glutamatergic loss in lesioned APOE4 mice at 70 dpl in layers receiving MEC inputs (Fig. 3B, Table 1) indicated a slower glutamatergic reinnervation.
Changes in VGLUT1 immunoreactivity in male APOE3 and APOE4 mice
Changes in VACHT immunoreactivity in male APOE3 and APOE4 mice
Postlesion delay and genotype also influenced layer-specific cholinergic changes (Fig. 3B, Table 2, layer*delay*genotype*lesion interaction: F(30,1005) = 1.505; p = 0.04). At 30 dpl, lesioned APOE3 displayed a cholinergic sprouting specific to the deafferented DG middle part of the molecular layer (mML) (Fig. 3B, Table 2). At 70 dpl, this cholinergic sprouting expanded to stratum oriens in CA fields (Table 2), layers that are not primarily targeted by entorhinal inputs. Finally, this transient cholinergic sprouting returned to baseline at 170 dpl (Table 2). Lesioned APOE4 mice showed no evidence of cholinergic sprouting whatever the layer and delay (Fig. 3B, Table 2), suggesting a complete deficit of cholinergic sprouting. In contrast, glutamatergic reinnervation was effective, albeit slower than in lesioned APOE3 mice.
Neither glutamatergic or cholinergic changes were restricted to DG. However, among all histological measures, cholinergic sprouting in DG mML appeared sufficient to compensate for EC lesion, as suggested by the minimal memory impairment of lesioned APOE3 mice at 30 dpl (Fig. 3, Table 1, Table 2). Nevertheless, when all postlesion delays were taken into account, it appeared likely that both the cholinergic sprouting and the faster glutamatergic reinnervation expressed by lesioned APOE3 mice contributed to their faster behavioral recovery compared with lesioned APOE4 mice.
Preserved behavioral recovery and cholinergic sprouting in female APOE4 mice
To determine whether sex hormones reverse APOE4-related impairments, the same experiment was replicated in female hAPP/APOE mice. Lesion magnitude was similar in APOE3 and APOE4 female mice for all delays (Fig. 4A; genotype*delay interaction: F(2,33) = 1.21; p = 0.1962). Lesions were quite specific to the MEC (region effect: F(7,231) = 67.1464; p < 0.00001) and similar in their extent to those of males (gender effect: F(1,79) = 0.0002; p = 0.9875). During the Barnes maze acquisition phase, similarly to males, females from all groups reduced their latency to the target, whatever the genotype or lesion status (Fig. 4B: day effect: F(4,340) = 378.437, p < 0.00001). This confirms that all groups had similar motivation to escape the device. Like males, females mainly relied on nonspatial strategies.

EC lesion and Barnes maze acquisition performance of female hAPP/APOE mice. A, Photomicrograph of representative cresyl violet stainings from a sham (top) and a lesioned (bottom) mouse. MEC and LEC are delineated with dashed yellow lines and the lesion area is delineated with a dashed red line. Scale bar, 600 μm. Left bar graph shows that NMDA microinjections induced partial lesion of similar magnitude in all groups. MEC was clearly the most lesioned region, whereas LEC, perirhinal cortex, and subiculum were much less affected ($p < 0.05 MEC vs other regions; LSD post hoc analysis). B, In the acquisition phase of the Barnes maze task, all groups improved their performance in a similar fashion, although without relying much on a spatial strategy (see inserts; *p < 0.05 sham vs lesioned; two-sample unpaired t test).
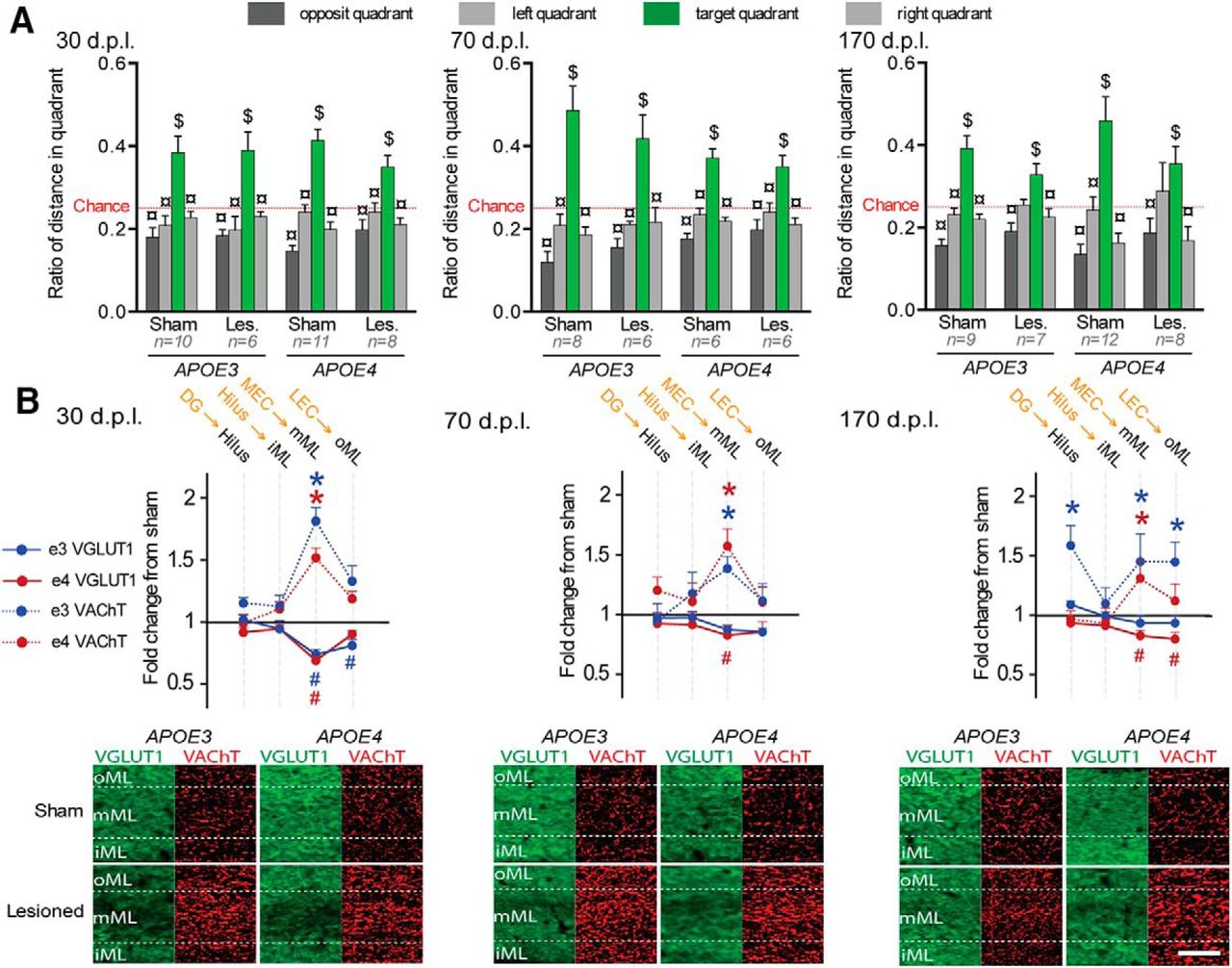
During the probe trial (Fig. 5A), the preference for the target quadrant was only influenced by the lesion status (lesion*quadrant interaction: F(3,255) = 2.86; p = 0.037). Among lesioned mice, preference for the target quadrant was influenced by gender as a function of genotype (quadrant*gender*genotype interaction: F(3,234) = 2.26; p = 0.038). Contrary to males, lesioned females from both genotypes displayed a significant preference for the target quadrant at all delays (Fig. 5A), suggesting intact spatial memory whatever the genotype and postlesion delay. Therefore, lesioned APOE4 female mice exhibited an efficient recovery similar to that of lesioned APOE3 mice from both sexes, but in contrast to the poor recovery of lesioned APOE4 males.

Lesioned APOE4 female mice displayed intact behavioral performance associated with a cholinergic sprouting. A, Lesioned females from both genotypes were never dramatically impaired during the Barnes maze probe trial, suggesting intact spatial memory abilities. $Distance in target quadrant differs from chance level (p < 0.05; t test); ¤distance differs from target (p < 0.05; Fisher's LSD). B, Top, Lesioned mice from both genotypes were characterized by a long-lasting cholinergic sprouting, but lesioned APOE4 mice displayed an impaired glutamatergic reinnervation. The presumed main origin of VGLuT1-positive inputs is indicated in orange above the corresponding targeted hippocampal layer in black. *VAChT density differs from sham level (p < 0.05, Fisher's LSD); #VGLuT1 immunoreactivity differs from sham level (p < 0.05, Fisher's LSD); *VAChT density differs from sham level (p < 0.05, Fisher's LSD); #VGLuT1 immunoreactivity differs from sham level (p < 0.05, Fisher's LSD). Bottom, Examples of VGLUT1 (green) and VAChT stainings (red) taken from the molecular layer of the DG. Scale bar, 50 μm.
Lesion-induced glutamatergic changes in female mice (Fig. 5B, Table 3) were layer and genotype specific (layers*genotype*lesion interaction: F(15,900) = 2.303; p = 0.0032). Among lesioned mice, gender influenced glutamatergic changes (Table 4), but differentially as a function of genotype and postlesion delay (layers*gender*delay*genotype interaction: F(15,1170) = 2.546; p < 0.0001). Contrary to males, in lesioned females from both genotypes, VGLUT1 loss was restricted to layers receiving entorhinal inputs at 30 dpl (Fig. 5B, Table 3). Therefore, in contrast to lesioned APOE4 males (Fig. 3B, Table 1), lesioned APOE4 females did not show a broad glutamatergic perturbation (Fig. 5B, Table 3, Table 4). However, glutamatergic loss in lesioned APOE4 females persisted throughout delays (Fig. 5B, Table 3), which suggests reduced long-term glutamatergic reinnervation capabilities compared with APOE4 males.
Changes in VGLUT1 immunoreactivity in female APOE3 and APOE4 mice
VGLUT1 immunoreactivity changes in female and male APOE3 and APOE4 lesioned mice
Lesion-induced cholinergic sprouting in female mice (Fig. 5B, Table 5) was layer specific and independently influenced by the delay (layers*lesion*delay interaction: F(30,900) = 1.607; p = 0.021) and the genotype (layers*lesion*genotype interaction: F(15,900) = 1.746; p = 0.038). When data from male and female lesioned mice are analyzed together (Table 6), lesion-induced cholinergic sprouting was influenced by gender in a genotype-dependent manner (layers*gender*delay*genotype interaction: F(30,1170) = 3.003; p < 0.0001), further suggesting that APOE4-dependent impairment of cholinergic sprouting was reversed in females. In lesioned APOE3 females, at 30 dpl, cholinergic sprouting expanded in all deafferented layers and in CA1 stratum oriens, CA1 radiatum, and all CA2 layers (Table 5). At 70 dpl, this sprouting was restricted to layers receiving MEC inputs in the DG, CA3, and CA2 (Fig. 5B, Table 5). Finally, and despite a complete glutamatergic reinnervation at 170 dpl, the cholinergic sprouting was still maintained in the DG from lesioned APOE3 females (Fig. 5B), particularly in layers receiving EC inputs and in the hilus. In contrast to what was found in lesioned APOE4 males, lesioned APOE4 females displayed a marked cholinergic sprouting (Fig. 5B, Table 5, Table 6). At 30 dpl, this sprouting was present in deafferented layers and in the stratum lucidium of the CA3 region (Fig. 5B, Table 5). A similar pattern was maintained at 70 dpl. At 170 dpl, the cholinergic sprouting of lesioned APOE4 females was maintained only in the DG mML receiving MEC inputs. Finally, their glutamatergic deafferentation persisted and even spread at 170 dpl to all EC-receiving layers of DG, CA2, and CA3 regions (Fig. 5B, Table 5).
Changes in VACHT immunoreactivity in female APOE3 and APOE4 mice
VACHT changes in female and male APOE3 and APOE4 lesioned mice
Despite impaired glutamatergic reinnervation, lesioned APOE4 female mice had intact spatial memory performances (Fig. 5A) associated with long-lasting cholinergic sprouting in the DG (Fig. 5B). This result suggests that the cholinergic sprouting compensated efficiently for the hippocampal glutamatergic disconnection. Therefore, the cholinergic sprouting appears necessary and sufficient to mediate behavioral recovery through the compensation of partial loss of EC glutamatergic inputs to the hippocampus. Furthermore, cholinergic sprouting in the DG may also be sufficient to compensate for EC lesion despite broader glutamatergic loss.
Cholinergic sprouting associated with increased hippocampal cholinergic modulation
Despite the link between cholinergic sprouting and behavioral recovery found in hAPP/APOE mice, we further determined whether the sprouting, which is supposed to reflect the proliferation of cholinergic terminals, was actually associated with increased cholinergic drive to the hippocampus. EC lesions were replicated in male ChAT-ChR2 mice expressing channelrhodopsin-2 in cholinergic neurons, thereby allowing their selective activation through 470 nm light pulses. According to the hAPP/APOE results, we focused on changes in the DG at 7 dpl (before the cholinergic sprouting took place) and at 30 dpl (with sprouting in place).
Lesions were similar at 7 and 30 dpl (Fig. 6A). At 30 dpl, lesioned Chat-ChR2 mice were not impaired in the Barnes maze (Fig. 6B, quadrant effect: F(3,24) = 12.23, p = 0.0004). They showed glutamatergic (Fig. 6C, layers*lesion interaction: F(3,24) = 17.1912; p = 0.0002) and cholinergic (Fig. 6C, layers*lesion interaction: F(3,24) = 17.1912; p = 0.0003) changes similar to those of APOE mice exhibiting cholinergic sprouting (Figs. 3A–C, 5A–C). At 7 dpl, lesioned Chat-ChR2 mice displayed only a glutamatergic disconnection, confirming that the cholinergic sprouting was weak at this delay (Fig. 6C). However, because the Barnes maze protocol already requires 6 d of testing and the cholinergic sprouting is maximal within 10 d (Steward, 1992), the behavioral impact of early cholinergic sprouting could not be evaluated at our minimal 7 dpl delay. Nevertheless, lesional, behavioral, and histological profiles of lesioned ChAT-ChR2 mice at 30 dpl were similar to those of unimpaired lesioned hAPP/APOE mice, confirming in a different strain that cholinergic sprouting acted as a compensatory mechanism to cope with partial EC lesion.

Lesioned ChAT-ChR2 mice displayed intact behavioral recovery and cholinergic sprouting. A, Lesioned ChAT-ChR2 mice at both delays displayed similar EC lesion magnitude. B, At 30 dpl, lesioned ChAT-ChR2 mice had intact preference for the target quadrant. $Different from chance level (p < 0.05, t test); ¤different from the target quadrant (p < 0.05, Fisher's LSD). C, At 30 dpl, lesioned ChAT-ChR2 mice showed cholinergic sprouting specifically in deafferented layers. This cholinergic sprouting was not in place at 7 dpl. *VAChT density differs from sham level (p < 0.05, Newman–Keuls); #VGLuT1 immunoreactivity differs from sham level (p < 0.05, Newman–Keuls).
To explore hippocampal functional changes induced by EC lesion and the reactive cholinergic sprouting, LFP was recorded in the dorsal DG (Fig. 7A, top) under urethane/ketamine anesthesia. Despite the loss of glutamatergic terminals, EC lesion did not significantly change the basic oscillatory properties of the DG (Fig. 7B). However, when cholinergic neurons from the MSDB (Fig. 7A, bottom) were stimulated optogenetically at 5 Hz, the theta-band power (3–7 Hz under urethane anesthesia) increased more reliably in 30 dpl lesioned mice than in sham mice (Fig. 7C,D). Increasing optogenetic stimulation frequencies of cholinergic neurons is known to increase their firing rate in association with upregulated theta power in the hippocampus (Vandecasteele et al., 2014). During our MSDB stimulations, theta peak power (Fig. 7E) was not significantly influenced by 0.2 Hz stimulation, but 5 Hz stimulation increased this peak specifically in 30 dpl mice (stimulation*lesion interaction: F(2,18) = 3.6721; p = 0.0459). Under 20 Hz stimulation, both sham and lesioned mice exhibited enhanced theta-peak power (stimulation effect: F(2,18) = 8.3781: p = 0.0026). Local 5 Hz stimulation of cholinergic terminals in the dorsal hippocampus (dHPC) led to similar results in lesioned mice at 30 dpl, but not in 7 dpl mice lacking cholinergic sprouting, which responded similarly to sham control mice (Fig. 7E). The effects of 5 Hz optogenetic stimulations were inhibited by scopolamine administration, suggesting that theta modulation by the cholinergic sprouting is muscarinic dependent even under urethane/ketamine/xylazine anesthesia, a state in which theta oscillations are resistant to anticholinergic drugs (Klausberger et al., 2003). Therefore, compared with sham mice or lesioned mice with no sprouting, the cholinergic sprouting in 30 dpl mice increased the strength of the cholinergic modulation on theta power (Fig. 7E). Accordingly, the cholinergic sprouting is more likely to be a true physiological compensatory response rather than a shrinkage-related artifact as proposed earlier (Phinney et al., 2004).

Cholinergic sprouting enhanced cholinergic drive in dentate mML in ChAT-ChR2 mice. A, Microphotograph showing the recording/local stimulation zone in the dHPC (top) and the stimulating zone in the MSDB (bottom). Scale bar, 500 μm. Red, Dil embedding of silicon probe (top) and optical fiber (bottom); blue, DAPI staining; green, YFP staining. B, Baseline LFP properties: partial EC lesion did not change total power of LFP, relative theta or gamma power, or oscillatory strength. C, Optogenetic stimulation (5 Hz for 0.5 ms) of cholinergic neurons for 30 s in the MSDB induced a marked increase in theta-band power in the DG mML of lesioned mice, but not in sham mice. D, Dentate mML theta-band power increased during optical stimulation in lesioned mice, but not in sham mice. Dashed lines indicate SEM. E, F, Compared with sham mice, the presence of a cholinergic sprouting in 30 dpl mice increased the strength of the modulation of MSDB cholinergic neurons on dentate mML theta-peak power. Local optical stimulations in the dHPC led to similar effects but with a lower magnitude. *p < 0.05, Newman–Keuls; **p < 0.01, Newman–Keuls; ***p < 0.001, Newman–Keuls.
Lesion-related DG hyperactivity is controlled by cholinergic sprouting activity
Because cholinergic septohippocampal projections are functionally nonhomologous to the glutamatergic innervation coming from the EC, the putative mechanisms enabling a cholinergic compensation for the loss of glutamatergic inputs remain enigmatic. We hypothesized that such compensation is most probably due to network modulation rather than to direct functional replacement of entorhinal inputs. To clarify how the cholinergic sprouting compensated for a partial loss of glutamatergic EC inputs in the DG, we first aimed to determine the major changes in DG network activity induced by the partial EC lesion.
Compared with sham animals, lesioned mice displayed LFP with high occurrence of high-amplitude (<1 mV) transient bursts of activity. Bragin et al. (1995) described similar events in normal rodents and characterized them as high-amplitude LFP events reflecting population discharges of dentate neurons in response to synchronous EC input activities. Both lesioned and sham mice displayed these “dentate LFP spikes” in association with high-amplitude CSD sinks similar to those induced by perforant path (pp) electrical stimulations (Fig. 8A). To detect dentate LFP spikes unambiguously, we relied on a CSD method that leaves out passive volume-conducted currents. Single events were characterized by a sink in the DG molecular layer with an amplitude of at least 3 SD's above the background mean sink amplitude. The 7- and 30-dpl lesioned mice both displayed increased occurrence of dentate LFP spikes compared with sham mice based on LFP traces (lesion effect: F(2,26) = 8.731: p = 0.0013; Fig. 8A,B) as well as CSD sink located in the DG molecular layer (lesion effect: F(2,26) = 10.51: p = 0.0005; Fig. 8A,C). Moreover, multiunit firing frequency in the DG granular layer was also higher in 30 dpl mice compared with sham mice (2-tailed unpaired t test: t(25) = 3.697: p = 0.0011; Fig. 8D). Unfortunately, the number of units recorded in 7 dpl mice was not sufficient to determine a baseline firing frequency for this group.

EC lesion-induced dentate hyperactivity is normalized by optogenetic stimulation of the cholinergic sprouting. A, Lesioned mice at 7 and 30 dpl and sham-operated mice showed spontaneous dentate LFP spikes that were visible in raw LFP traces, especially in DG layers (top). These spontaneous events were associated with a strong CSD sink located in the dentate molecular layer. Both CSD and LFP signals associated with spontaneous dentate LFP spikes strongly resembled pp-evoked responses, suggesting that EC inputs contributed to dentate LFP spikes. Arrows indicate dentate LFP spikes or pp stimulations. B, Under baseline conditions, lesioned mice at both 7 and 30 dpl had an increased frequency of dentate LFP spikes that was normalized by local optical stimulation of cholinergic terminals within the dHPC. C, Lesioned mice from both postlesion delays had increased CSD pp-like sink frequency (3SD above mean sink amplitude) in the deafferented DG molecular layer that was also normalized by local optical stimulation. D, Multiunit spontaneous firing frequency was three times higher in lesioned mice than in sham mice at 30 dpl. However, the firing frequency was also normalized to sham level by local optical stimulation. $p < 0.05, $$p < 0.01, $$$p < 0.001, **p < 0.01, ***p < 0.001, each differs from baseline (Newman–Keuls); n indicates the number of animals in each group, except for D, where it refers to the number of cells recorded.
The baseline of dentate LFP spike frequency did not differ significantly between 7 and 30 dpl (Fig. 8B). However, the frequency of CSD discharges was higher in the 7 dpl mice devoid of significant sprouting than in the 30 dpl mice displaying a sprouting response (Fig. 8C). This suggests that the cholinergic sprouting may act to inhibit the occurrence of hypersynchronous discharges in EC–DG pathways (CSD sink) and related population discharges in the DG (dentate LFP spikes). To test the relationship between this lesion-induced entorhinal/dentate hyperactivity and cholinergic sprouting, we used local optogenetic stimulation of cholinergic inputs within the dHPC. In lesioned groups, 5 Hz stimulations decreased the frequency of dentate LFP spikes (optogenetic stimulation*lesion interaction: F(4,52) = 4.915; p = 0.0019; Fig. 8B) and spontaneous pp-associated high-amplitude sink frequency (optogenetic stimulation*lesion interaction: F(4,52) = 6.128; p = 0.0004; Fig. 8C) toward sham levels. Moreover, multiunit firing in the DG granular layer was also normalized to sham levels in lesioned mice (optogenetic stimulation*lesion interaction: F(2,50) = 8.541; p = 0.0006; Fig. 8D).
Together, these results suggest that partial entorhinal lesion increases the probability of hypersynchronous discharges in EC–dHPC pathways (CSD sinks recorded in DG molecular layer and similar to pp electrical stimulation), resulting in a dentate hyperactivity characterized by more frequent population hypersynchronous discharges (dentate LFP spikes) and increased multiunit firing. When cholinergic inputs in the dHPC were activated locally, this dentate hyperactivity was normalized to the sham level, suggesting that the local cholinergic sprouting response in the dHPC is able to turn down lesion-induced entorhino-hippocampal hyperactivity toward normal levels.
Discussion
Hippocampal cholinergic sprouting is a classical outcome of EC lesion in rodents (Lynch et al., 1972). Similar responses have been reported in MCI patients (DeKosky et al., 2002). However, the hypothetic compensatory nature of the cholinergic sprouting and related underlying mechanisms remain undetermined.
To manipulate the cholinergic sprouting in a disease-relevant way, bilateral partial EC lesions were induced in mice transgenic for the human APOE4 or APOE3, APOE4 being the stronger genetic risk factor for MCI and sporadic AD (Xu et al., 2013). Our lesions mainly involved MEC but spared LEC. We found that functional recovery of spatial memory was associated with both cholinergic and glutamatergic sprouting across the whole hippocampus.
Cholinergic sprouting was necessary for the maintenance of spatial memory function throughout the duration of the glutamatergic denervation (30 dpl male and female APOE3; 30–170 dpl female APOE4; male ChAT-ChR2 mice). Its absence was associated with dramatic spatial memory impairments (30 and 70 dpl male APOE4). Later, a slower glutamatergic reinnervation also contributed to spatial memory recovery. It often coexisted with cholinergic sprouting (APOE3 male at 70 dpl, APOE3 females at 70 and 170 dpl) and eventually replaced it (APOE3 males at 170 dpl). In the absence of cholinergic sprouting (APOE4 male), functional recovery occurred only after a complete glutamatergic reinnervation, long after the acute phase of the deafferentation (170 dpl). Females from both genotypes had a better behavioral recovery and more extensive and long-lasting cholinergic sprouting, as described previously in rats (Roof et al., 1993; Stone et al., 1998). Interestingly, gender facilitation of this sprouting reaction was strong enough to reverse APOE4-impaired cholinergic sprouting and spatial memory recovery. However, contrary to male APOE4 mice, females never displayed a complete glutamatergic reinnervation, suggesting that sex hormones may modulate APOE4's negative influence on cholinergic sprouting and glutamatergic reinnervation differentially. Nevertheless, in the absence of a complete glutamatergic reinnervation (APOE4 females), cholinergic sprouting was clearly sufficient to maintain spatial memory function. Our finding contrasts with the increased sensitivity to APOE4 reported in female mice (Raber et al., 1998; Bour et al., 2008). However, those studies investigated age-related deficits in older mice (Bour et al., 2008) or mice overexpressing APOE4 (Raber et al., 1998), whereas we focused here on lesion-induced impairments. It is most probable that, in older females, gender facilitation would disappear as a result of altered levels of sex hormones. In conclusion, under partial hippocampal disconnection, cholinergic sprouting appears both required and sufficient for spatial memory, demonstrating its compensatory nature.
The concept of cholinergic compensation of lesion must not be limited to rodents because similar cholinergic-dependent functional recovery has been reported in primates (Croxson et al., 2012). Moreover, cholinergic activity correlates with both cognitive reserve and residual memory extent in patients (Garibotto et al., 2013; Ray et al., 2015). Therefore, cholinergic sprouting in MCI patients (DeKosky et al., 2002) probably denotes a compensatory mechanism allowing adaptation to EC neuronal loss.
Cholinergic sprouting was more consistent in deafferented layers and occurred in most hippocampal subregions. However, cholinergic sprouting in DG molecular layer appears sufficient for spatial memory maintenance despite enduring glutamatergic deafferentation in other hippocampal subregions (30 dpl APOE3 males and 170 dpl APOE4 females). This suggests that cholinergic sprouting in DG is sufficient for functional compensation of EC lesion, at least for spatial memory. Studies using unilateral lesions in rats suggested that DG reinnervation from contralateral EC inputs sustained behavioral recovery (Loesche and Steward, 1977), whereas our study shows recovery after sprouting of cholinergic septohippocampal projections that are nonhomologous to glutamatergic EC inputs. Because our lesions were partial (<50%), it is possible that the increased VGLUT1 staining reflected sprouting of surviving EC terminals, which also played a role in spatial memory maintenance. However, our data suggest that spatial memory maintenance first requires a proper cholinergic sprouting that probably modulates surviving EC inputs before a complete glutamatergic reinnervation occurs.
Unexpectedly, the most prominent activity change found after partial EC lesion was hyperactivity in EC–DG networks (CSD sink in the molecular layer). Spontaneous bursts of activity have been reported in EC neurons (Paré and Llinás, 1995). Therefore, increased occurrence of CSD sink probably reflects synchronous bursts of activity in surviving EC inputs. Increased CSD sink was associated with DG hyperactivity (increased occurrence of dentate LFP spikes and enhanced multiunit firing). During MCI, transient hyperactivity has been described in medial temporal lobe and DG (Dickerson and Sperling, 2008; Yassa et al., 2010). Although initially interpreted as compensatory, these hyperactivities have been linked recently to memory impairments in rodents (Jinde et al., 2012) and MCI (Yassa et al., 2011). Moreover, pharmacological reduction of DG/CA3 hyperactivity in MCI led to cognitive recovery (Bakker et al., 2012, 2015). Studies on transgenic mice overexpressing Aβ suggested that hippocampal hyperactivity might be amyloid dependent (Palop and Mucke, 2010). However, our results suggest that structural factors such as partial EC neuronal loss may also contribute to aberrant hyperactivity. In support of this hypothesis, DG hyperactivity correlates with pp integrity in patients (Yassa et al., 2011). Lesioned mice displayed spontaneous DG hyperactivity despite the presence of cholinergic sprouting (30 dpl ChAT-ChR2 mice). However, this DG hyperactivity was higher before cholinergic sprouting in 7 dpl ChAT-ChR2 mice, suggesting that this phenomenon inhibits lesion-induced DG hyperactivity. Accordingly, local optogenetic activation of cholinergic terminals in DG completely reversed the CSD sink and DG hyperactivities in both 7 and 30 dpl ChAT-ChR2 mice. This is consistent with evidence suggesting that acetylcholine inhibits synaptic activity of EC inputs in DG (Foster and Deadwyler, 1992) via either presynaptic muscarinic receptor or retrograde endocannabinoid modulation (for review, see Teles-Grilo Ruivo and Mellor, 2013). In our study, DG was particularly sensitive to EC-lesion-induced hyperactivity compared with CA1. As in MCI patients, DG emerged as a preferential place for hyperactivity. Therefore, the fact that cholinergic sprouting is more consistent in DG is not surprising because DG is known for its sparse activity and maintenance of its function presumably requires such abnormal hyperactivity to be controlled.
Reduction of DG hyperactivity may contribute to cognitive recovery through at least two complementary ways. On a behavioral timescale, control of DG hyperactivity may restore appropriate conditions for sparse coding and related pattern separation/memory encoding (Bakker et al., 2012, 2015; Rolls, 2013; Neunuebel and Knierim, 2014). In agreement with this view, DG experimental hyperactivity induces pattern separation and memory-encoding impairments (Jinde et al., 2012). Both functions are known to be associated with septohippocampal cholinergic activation (Toumane et al., 1988; Giovannini et al., 2001). On longer timescales, lowering DG hyperactivity may also lessen the excitotoxic burden generated by excessive glutamate release, which could slow down synaptic loss. In support of this hypothesis, lesioned APOE4 males lacking cholinergic sprouting displayed broader glutamatergic synaptic loss throughout the hippocampus at 30 dpl. Therefore, beyond a direct “online” functional compensation through DG activity normalization, cholinergic sprouting may also slow down disease progression by reducing excitotoxicity generated by glutamatergic hyperactivity.
In conclusion, we have demonstrated that, after bilateral partial EC lesion, cholinergic sprouting in the deafferented DG is necessary and sufficient to mediate recovery of spatial memory, at least until a complete glutamatergic reinnervation occurs. Glutamatergic reinnervation is probably unlikely in AD patients because EC neuronal loss worsens as the disease progresses. Moreover, the cholinergic system also strongly degenerates in advanced stages. Therefore, we hypothesize that the resulting weakening of reactive cholinergic sprouting may represent the signature of the conversion from MCI to AD. The relevance of a cholinergic compensation needs to be demonstrated for advanced stages, but it should certainly be taken into account for earlier stages known to be associated with cholinergic sprouting as MCI (DeKosky et al., 2002). Unfortunately, to our knowledge, no human study has investigated APOE4's effect on cholinergic sprouting. Impaired cholinergic sprouting may contribute to several APOE4-negative effects, including increased hippocampal hyperactivity (Filippini et al., 2009), reduced responsiveness to anticholinesterase (Farlow et al., 1996; Poirier, 1999; Wang et al., 2014), more aggressive MCI (Barabash et al., 2009), and reduced likelihood of reversion (Koepsell and Monsell, 2012), as well as accelerated transition to AD (Xu et al., 2013). If this is confirmed in patients, it implies that APOE4's effects could be partially amyloid independent and linked to a failure of the brain to cope with disease-associated structural changes. Interestingly, we found a complete reversion of APOE4-negative effects in female mice. Better functional recovery was reported after traumatic brain injuries in women (Stein, 2001), suggesting that similar hormonal facilitation occurs in humans. Because MCI mainly appears from the fifth decade onward, such facilitation is probably compromised in female APOE4 patients, as suggested by their higher risk of developing MCI/AD (Altmann et al., 2014). Interestingly, earlier reports suggested that hormone substitution therapies may ameliorate cognitive performance (Henderson et al., 1996) and increase responsiveness to anticholinesterase treatment in patients (Schneider et al., 1996). Therefore, combining cholinergic and hormonal therapies (Newhouse and Dumas, 2015) may be promising for APOE4 female carriers. Potentiation of the cholinergic control on EC–DG hyperactivity through muscarinic-specific or endocannabinoid potentiation (Teles-Grilo Ruivo and Mellor, 2013) may further facilitate the endogenous control of dentate hyperactivity. In conclusion, our results support the use of therapeutic strategies using septohippocampal cholinergic sprouting and related control of EC input activity.
Footnotes
This work was supported by France Alzheimer 68, the University of Strasbourg, the Centre National de la Recherche Scientifique, and the French Ministry of Education and Research. R.G. was supported by a Neurex “Welcome of Researcher” Grant and a Marie Curie Career Integration Grant from the European Research Council. We thank J.-C. Dodart, Celina Zerbinatti, and John Renger (Merck Sharp and Dohme Corporation, Department of Neurosymptomatic Disorders) for providing the initial hAPP-YAC/APOE3-tr and hAPP-YaC/APOE4-tr parental mice; Prof. Feng Guoping from the Massachusetts Institute of Technology for providing ChAT-ChR2 mice; Carole Strittmatter for mice breeding; and Prof. André Dufour for advice on statistical analyses.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Chantal Mathis, Ph.D., Laboratoire de Neurosciences Cognitives et Adaptatives, CNRS-UMR7364 CNRS, Neuropôle de Strasbourg GDR/CNRS2905, 12 rue Goethe, F-67000 Strasbourg, France. chantal.mathis{at}unistra.fr





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}