

Abstract
Excessive mitochondrial fission is a prominent early event and contributes to mitochondrial dysfunction, synaptic failure, and neuronal cell death in the progression of Alzheimer's disease (AD). However, it remains to be determined whether inhibition of excessive mitochondrial fission is beneficial in mammal models of AD. To determine whether dynamin-related protein 1 (Drp1), a key regulator of mitochondrial fragmentation, can be a disease-modifying therapeutic target for AD, we examined the effects of Drp1 inhibitor on mitochondrial and synaptic dysfunctions induced by oligomeric amyloid-β (Aβ) in neurons and neuropathology and cognitive functions in Aβ precursor protein/presenilin 1 double-transgenic AD mice. Inhibition of Drp1 alleviates mitochondrial fragmentation, loss of mitochondrial membrane potential, reactive oxygen species production, ATP reduction, and synaptic depression in Aβ-treated neurons. Furthermore, Drp1 inhibition significantly improves learning and memory and prevents mitochondrial fragmentation, lipid peroxidation, BACE1 expression, and Aβ deposition in the brain in the AD model. These results provide evidence that Drp1 plays an important role in Aβ-mediated and AD-related neuropathology and in cognitive decline in an AD animal model. Therefore, inhibiting excessive Drp1-mediated mitochondrial fission may be an efficient therapeutic avenue for AD.
SIGNIFICANCE STATEMENT Mitochondrial fission relies on the evolutionary conserved dynamin-related protein 1 (Drp1). Drp1 activity and mitochondria fragmentation are significantly elevated in the brains of sporadic Alzheimer's disease (AD) cases. In the present study, we first demonstrated that the inhibition of Drp1 restored amyloid-β (Aβ)-mediated mitochondrial dysfunctions and synaptic depression in neurons and significantly reduced lipid peroxidation, BACE1 expression, and Aβ deposition in the brain of AD mice. As a result, memory deficits in AD mice were rescued by Drp1 inhibition. These results suggest that neuropathology and combined cognitive decline can be attributed to hyperactivation of Drp1 in the pathogenesis of AD. Therefore, inhibitors of excessive mitochondrial fission, such as Drp1 inhibitors, may be a new strategy for AD.
Introduction
Mitochondria, essential organelles for both life and death, are highly dynamic. They continuously undergo balanced fission and fusion processes, which are termed mitochondrial dynamics (Cho et al., 2010). Mitochondrial dynamics greatly affect mitochondrial functions such as biogenesis, as well as their morphology. Several GTPase proteins, including dynamin-related protein 1 (Drp1), optic dominant atrophy 1 (Opa1), and mitofusin 1/-2 (Mfn1/2), have been identified as regulators of mitochondrial dynamics (Mishra and Chan, 2014). Mitochondrial fission relies on the evolutionary conserved Drp1 protein. Upon stimulation of mitochondrial fission, Drp1 is translocated from the cytosol to the mitochondrial outer membrane and interacts with its receptors, such as mitochondrial fission 1 (Fis1) and mitochondrial fission factor (MFF) to promote the mitochondrial fission process (Lee et al., 2004; Otera et al., 2010). As a regulatory mechanism, various posttranslational modifications such as phosphorylation, nitrosylation, sumoylation, ubiquitination, or GlcNAcylation of Drp1; proteolytic cleavage of Opa1; and ubiquitination or phosphorylation of MFN1/2 have explained the intricate mechanisms of mitochondrial dynamics with different signals (Cho et al., 2010; Cho et al., 2013). Mitochondrial fusion functions as a cell protection mechanism by content mixing, whereas massive mitochondrial fission potentiates cell death (Itoh et al., 2013; Park et al., 2014). Imbalanced mitochondrial dynamics are directly linked to many human diseases including cancer, diabetes, and neurodegenerative diseases (DuBoff et al., 2013; Itoh et al., 2013; Mishra and Chan, 2014). Neurons are particularly dependent on mitochondrial function because of their higher metabolic activity and complex morphology. Because mitochondria are not only the primary producers of reactive oxygen species (ROS), which contribute to mitochondrial dysfunction, but are also pivotal for synaptic development and plasticity, abnormalities in mitochondrial dynamics have been suggested to occur as prominent early events in neurodegenerative diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS) (Itoh et al., 2013; Reddy, 2014).
Membrane-associated oxidative stress, perturbed Ca2+ homeostasis, impaired energy metabolism, apoptosis, and mitochondrial alterations are involved in amyloid-β (Aβ)-mediated neuronal degeneration during the AD process. Mitochondrial dysfunction is an early and prominent feature of AD. In the brains of human AD cases and AD-like cases, Aβ plaques are accumulated in mitochondria and may cause structural and functional abnormalities of the organelles (Lustbader et al., 2004; Cha et al., 2012). In addition, mitochondrial abnormalities are found to arise before Aβ plaque deposition (Xie et al., 2013a, 2013b). Mitochondria from AD brains show fractured cristae, reduced respiratory capacity, and increased fragmentation (Baloyannis, 2006; Cho et al., 2009; Wang et al., 2009a, 2009b). Both the treatment of Aβ and the overexpression of Aβ precursor protein (APP) strongly induce synaptic injury in neuronal cells, as well as massive mitochondrial abnormalities (Cho et al., 2009; Tu et al., 2014). Exposure of oligomeric Aβ or excessive NO can lead to S-nitrosylation of Drp1 (SNO-Drp1) at Cys644, which activates Drp1 GTPase activity and results in the accumulation of excessively fragmented mitochondria (Cho et al., 2009). Moreover, Drp1 activity and the level of SNO-Drp1 were significantly elevated in the brains of sporadic AD cases (Cho et al., 2009; Wang et al., 2009a, 2009b; Manczak et al., 2012). Therefore, adjustment of the imbalance in mitochondrial dynamics may have beneficial effects on mitochondrial structure and function and neuronal survival in AD. However, it remains to be determined whether inhibition of excessive mitochondrial fission is beneficial in mammal models of AD. In the present study, we evaluate the effect of the Drp1 inhibitor on mitochondrial dysfunction in Aβ-challenged neurons and AD-like neuropathology in APP/presenilin 1 (PS1) double-transgenic AD mice.
Materials and Methods
Animals and treatment
Hemizygous double-transgenic mice APP/PS1 (APPswe/PSEN1dE9 RRID:MGI:3837130) and age-matched wild-type (WT) littermates were maintained on a C57BL/6J background. All mice were kept on a 12 h light (lights on at 8:00 A.M.)/12 h dark schedule with ad libitum access to food and water. Six-month-old male APP/PS1 mice were divided into three groups (n = 7 in each group): the vehicle administered, 10 mg/kg mitochondrial division inhibitor (mdivi-1)-treated, and 40 mg/kg of mdivi-1-treated groups. The age-matched littermates were divided into two groups (n = 5 in each group: vehicle treated and 40 mg/kg of mdivi-1-treated group). mdivi-1 was administered by oral injection once a day for a month. The Morris water maze (MWM) test began on day 31 of treatment with daily training sessions and lasted until day 34. We conducted the probe test day 35 to assess long-term memory retention. We trained mice for passive avoidance test on day 37 and the test was done on day 38. Animals were anesthetized with zoletil and perfused with PBS (0.9% NaCl) and then killed on day 40. Brain tissue was carefully dissected and some of brains were collected in liquid nitrogen and stored at −80°C. The other brains were fixed in 4% paraformaldehyde for 24 h and then incubated in 30% sucrose at 4°C for immunohistochemistry. This study was reviewed and approved by the Institutional Animal Care and Use Committee of Sungkyunkwan University. All efforts were made to minimize animal suffering and a minimal number of animals was used. We adhered to ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines at all steps of the experiments.
MWM test
The MWM is a white circular pool (120 cm in diameter and 35 cm in height) with a featureless inner surface. The circular pool was filled with water and nontoxic, water-soluble white dye (22 ± 1°C). The pool was divided into four quadrants of equal area. A white platform (8 cm in diameter and 10 cm in height) was centered in 1 of the 4 quadrants of the pool and submerged 1 cm below the water surface so that it was invisible at water level. The pool was located in a test room, which contained various prominent visual cues. The location of each swimming mouse, from the start position to the platform, was monitored by a video tracking system (Ethovision system; Noldus). The experimental group had no water maze pretraining. In the water maze experiments, the day before the experiment was dedicated to swimming training for 120 s in the absence of the platform. The mice were then given 2 trial sessions each day for 4 consecutive days, with an intertrial interval of 15 min, the escape latencies were recorded, and the average of 2 trials was determined. If the mouse did not locate the platform within 120 s, it was permitted to remain on it for 10 s and then removed from the pool by the experimenter. On the fifth day of MWM test, the probe test was conducted for 60 s without the platform in the pool. The latency to reach the former location of the platform, the time spent traveled in the target quadrant, and the frequency of original platform crossings were measured. The point of entry of the mouse into the pool and the location of the platform for escape remained unchanged between trials 1 and 2, but was changed each day thereafter. Behavioral data were analyzed statistically using repeated-measures and multivariate analysis with LSD post hoc test.
Passive avoidance test
Experiments were conducted in a two-compartment apparatus (a bright compartment and a dark compartment divided by a guillotine door). On training day, the mouse was placed in the bright compartment facing away from the dark compartment, allowed to explore for 30 s, and then the guillotine door was lifted. When the mouse entered the dark compartment with all four paws, the guillotine door was closed and the latency to enter was recorded (from the time that the door was lifted). Three seconds after the door was closed, a foot shock (0.5 mA, 2 s) was delivered. Thirty seconds after the foot shock, the mouse was removed. On test day (24 h after training), the mouse was placed in the bright compartment facing away from the dark compartment. After 5 s, the guillotine door was lifted. When the mouse entered the dark compartment with all four paws, the guillotine door was closed and the latency to enter the dark compartment was recorded (test time was a total of 5 min until the mouse entered the dark room). After the test, the mouse was removed and returned to the home cage.
Cell culture, experimental treatments, and measurement of mitochondrial length
SK-N-MC (catalog #HTB-10, RRID:CVCL_0530) and SH-SY5Y (catalog #CRL-2266, RRID:CVCL_0019) neuroblastoma cells were obtained from the American Type Culture Collection. Cells were cultured at 37°C in a 5% CO2 incubator and maintained in DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen). To generate stable cell line, SK-N-MC cells were transfected with pmito-YFP (SK/mito-YFP) using Lipofectamine 2000 according to the manufacturer's protocol (Invitrogen). Stable transfectants were selected by growth in selection medium containing G418 (1 mg/ml) for 10 d. After single-cell dropping, the stable clones were selected under a fluorescence microscope and confirmed by Western blot analysis. Mean mitochondrial length in SK/mito-YFP cells was determined by measuring at least 20 individual mitochondria from images of multiple cells obtained by confocal microscopy. A YFP-fused MitoTracker plasmid (pmito-YFP) was described previously (Park et al., 2014). Aβ (1–42) peptide was purchased from the American Peptide Company. A MitoTracker probe and DAPI dye were purchased from Invitrogen.
Preparation of Aβ oligomers
Aβ (1–42) peptides lyophilized from 1,1,1,3,3,3,-hexafluoroisopropanol were solubilized in DMSO (2 mm) and then in F-12 medium to a final concentration of 125 μm. Aβ (1–42) peptide freshly diluted was used as an Aβ monomer. Aβ (1–42) peptides incubated at 4°C for 24 h was centrifuged at 12,000 × g for 10 min and the resulting supernatant was used as an Aβ oligomer. Prepared oligomeric Aβ was characterized by SDS-PAGE.
Intracellular and mitochondrial ROS measurements
Intracellular ROS levels were measured using a fluorescent dye, 2′,7′-dichlorofluorescein diacetate (H2DCF-DA) (Invitrogen), which was converted to the highly fluorescent 2′,7′-dichlorofluorescein (DCF) in the presence of oxidant. Briefly, cells were plated in 96-well plates. With or without further treatment of Aβ for 24 h, the cells were incubated with H2DCF-DA (5 μm) in serum-free medium for 30 min (excitation/emission wavelength 358/485; Victor X3; PerkinElmer). Relative intracellular ROS ratio was presented as the change in fluorescence of drug-treated samples compared with that of control samples. The level of mitochondrial superoxide anion was detected by using MitoSox-red (Invitrogen, M36008) as instructed by the manufacturer. To detect ROS in mitochondria, we pretreated mdivi-1 (25 μm) for 4 h and continuously treated Aβ (5 μm) for 4 h. After that time, we exchanged media and rat primary cortical neuron cells were incubated with 2.5 μm MitoSox-red for 20 min and then exchanged media. Fluorescence-stained cells were analyzed by confocal microscopy.
Measurement of mitochondria membrane potential and ATP level
Mitochondrial membrane potential was examined with a unique fluorescent cationic dye, JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; BD Biosciences), which detects loss of signal of mitochondrial membrane potential. The depolarized mitochondria were measured using a NucleoCounter NC-3000 apparatus (Chemometec). Cellular total ATP level was detected with an ATP bioluminescence detection kit (Promega) according to the manufacturer's protocol.
Primary neuronal cell culture
Primary dissociated cell cultures of hippocampal and cortical neurons were prepared from 18-d-old rat embryos as described previously (Gwon et al., 2012). Briefly, hippocampus and cortex were removed from rat embryos, stripped of blood vessels and meninges, minced, and treated with trypsin (HyClone) for 15 min at 37°C, followed by gentle trituration with a pipette. The dissociated cells were plated at a density of cells on poly-L-lysine-coated cover slides. Cells were maintained in DMEM (Hyclone) supplemented with 10% heat-inactivated (v/v) FBS and 1% (v/v) penicillin/streptomycin (Invitrogen). Twenty-four hours after plating, the culture medium was replaced with the same volume of Neurobasal-A medium (Invitrogen) containing 2% (v/v) B27 supplement (Invitrogen). Half of the medium was replaced every other day. After 14 d in culture, the primary hippocampal neurons were identified using immunofluorescence staining with antibodies and dyes. The cultured neurons were randomly divided into experimental groups and used for in vitro experiments.
Western blot and RT-PCR
Antibodies.
Antibodies used were as follows: anti-BACE1 1:2000 (Cell Signaling Technology catalog #5606S RRID:AB_1903900), anti-full-length APP 1:5000 and C-terminal form 1:2000 (CTF-β) (Sigma-Aldrich catalog #A8717 RRID:AB_258409), and anti-β-Actin 1:5000 (Sigma-Aldrich catalog #A5441 RRID:AB_476744).
Primers.
Primers used were as follows: mouse Bace1 forward 5-TCCTTCCGCATCACCATC-3, reverse 5-ACAGTCGTCTTGGGACGTG-3; mouse Bace1 forward 5-TGCATCGCTACTA CCAGAGG-3, reverse 5-CATGAGGGATGCTCACCAG-3; and mouse β-actin forward 5-GCTGTGCTATGTTGCTCTA-3, reverse 5-CTCGTTGCCAATAGTGATGA-3.
Immunocytochemistry and immunohistochemistry
Immunocytochemical and immunohistochemical analyses were performed as described previously (Baik et al., 2015). Primary neurons were seeded on coverslips coated with poly-D-lysine for 24 h. Cells were then incubated at 37°C with 5 μm Aβ (1–42) oligomer for 6 h with or without pretreatment of mdivi-1 for 4 h. After cells were then incubated at 37°C with 100 nm MitoTracker Red and 5 μm H2DCF-DA for 30 min, washed with PBS, and fixed with 4% paraformaldehyde for 10 min. Cells were washed in PBS and mounted on glass slide in mounting medium (Vector Laboratories) with DAPI (AbD Setotec). For immunohistochemistry, brains of mice were fixed by perfusion with 4% paraformaldehyde (PFA) in PBS and then in PFA + 30% sucrose for 48 h at 4°C. Fixed brains were cut on a microtome (CM3050S; Leica Microsystems) in 45-μm-thick slices and tissue sections were collected in a cold cryoprotectant solution (80 mm K2HPO4, 20 mm KH2PO4, 154 mm NaCl, 0.3 g/ml sucrose, 0.01 g/ml polyvinylpyrrolidone, and 30% v/v ethylene glycol). We prepared brain frozen section slices that were washed 5 times for 3 min in 1 PBS and blocked in 5% FBS with 0.1% Triton X-100 for 1 h, followed by overnight incubation with primary antibody in blocking solution at 4°C. The brain sections were immunostained with primary antibodies against HNE-Michael adducts (Millipore catalog #393207-100UL RRID:AB_566310) and Aβ (6E10; Covance Research Products catalog #SIG-39300-1000; RRID:AB_10175637). MitoTracker red (100 nm dye diluted in PBS; Invitrogen) staining was performed after antibody staining. After the primary antibody-binding step, brain slices were washed 5 times in 1 × PBS for 3 min and incubated with anti-mouse Alexa Fluor 488 or anti-rabbit Alexa Fluor 594 for 2 h, followed by washing 3 times with 1× PBS for 3 min, and then brain slices were mounted with DAPI plus mounting medium on glass slides. All photographs were taken with a confocal microscope (Carl Zeiss, LSM700 RRID:SCR_013672) with a 40× objective.
Presynaptic functional assay (vGlut-pHluorin assay)
pHluorin-tagged synaptic vesicle proteins have proven to be a useful probe for examining the kinetics of endocytosis at nerve terminals (Sankaranarayanan and Ryan, 2000; Sankaranarayanan et al., 2000; Granseth et al., 2006; Kim et al., 2009). Hippocampal CA3-CA1 regions were dissected from postnatal 0- to 1-d-old Sprague-Dawley rats, dissociated, and plated onto poly-ornithine-coated glass for 14–20 d. Coverslips were mounted in a stimulation chamber on a custom-built laser illuminated fluorescence microscope. Cells were perfused with Tyrode's solution at low flow rate (100–200 μl/min). Cells were illuminated using a 488 nm OBIS laser (Coherent Technologies) shuttered by synchronizing the TTL ON/OFF signal from the camera. Fluorescence excitation and collection was through a 40×/1.3 numerical aperture Fluar Zeiss objective using 515–560 nm emission and 510 nm dichroic filters (Chroma). Images were acquired with an iXon ultra 897 (Andor) back-illuminated electron-multiplying charge coupled device camera. Action potentials were evoked by passing 1 ms current pulses via platinum–iridium electrodes from an isolated current stimulator (World Precision Instruments).
Statistics
Data were obtained from at least three independent experiments and are presented as means ± SEM. Statistical analyses were performed using GraphPad Prism 5 software. Statistical significance was determined by Student's t-test for single comparisons or ANOVA for multiple comparisons with appropriate post hoc testing. Sample sizes for all experiments were determined by power analyses based on previously published data.
Results
Inhibition of Drp1 attenuates mitochondrial fragmentation and dysfunction and ROS production in Aβ-treated neurons
Recent studies have shown that excessive mitochondrial fragmentation contributes to AD pathogenesis via mitochondrial dysfunction (Cho et al., 2009; Wang et al., 2012a, 2012b; Park et al., 2014). To investigate the effect of mdivi-1, a Drp1 selective inhibitor, on Aβ-treated cells, SK/mito-YFP cells were incubated with Aβ (1–42) oligomer in the presence or absence of mdivi-1. According to the previous notions, treatment of Aβ induced massive mitochondrial fragmentation. However, the Aβ-mediated mitochondrial fission was highly blocked by treatment of mdivi-1 (Fig. 1A–C). In addition, treatment of mdivi-1 significantly recovered Aβ-mediated mitochondrial dysfunctions, including increased mitochondrial depolarization, ROS production, and decreased ATP levels, in SK-N-MC cells (Fig. 1D–F).

mdivi-1 inhibits Aβ-mediated mitochondrial fragmentation and dysfunction in neuroblastoma cells. A–C, SK-N-MC cells stably expressing mito-YFP (SK/mito-YFP) were treated with Aβ (1–42) oligomer (10 μm) in the presence or absence of mdivi-1 (25 μm) for 4 h. Then, the cells were imaged using a fluorescence microscope (A), cells with fragmented mitochondria were counted (B), and mitochondrial length was measured (C). D–F, SK-N-MC cells were treated with Aβ (10 μm) in the presence or absence of mdivi-1 (25 μm) for 8 h. D, Then, the alteration of mitochondrial membrane potential was monitored by the Mito-Probe JC-1 assay. E, Intracellular ROS level was measured by a H2DCF-DA fluorescence ROS detection assay. F, Next, the cellular total ATP level was examined by an ATP bioluminescence detection assay. Scale bar, 10 μm. Data are presented as the means ± SEM (n = 4). One-way ANOVA followed by Turkey post hoc comparisons tests were performed in all statistical analyses (*p < 0.05, **p < 0.01, ***p < 0.001).
To further investigate the effect of mdivi-1 on Aβ-induced mitochondrial fission and dysfunction, primary cultured rat hippocampal neurons were exposed to Aβ (1–42) oligomer with or without mdivi-1 and mitochondrial morphology was examined with a MitoTracker. Treatment of Aβ alone strongly increased mitochondrial fragmentation in hippocampal neurons, whereas cotreatment with mdivi-1 significantly reduced the Aβ-mediated mitochondrial fission (Fig. 2A,B). Nonetheless, total mitochondrial intensity was not altered in Aβ or mdivi-1-treated cells, indicating that Aβ or mdivi-1 did not affect mitochondrial biogenesis (Fig. 2C). Next, we measured cellular ROS levels in response to Aβ (1–42) oligomer and mdivi-1 by using the fluorescent probes H2DCF-DA and MitoSox Red, which preferentially detect hydroxyl radical and mitochondrial superoxide, respectively. Aβ-mediated intracellular ROS generation was suppressed by treatment of mdivi-1 in primary cultured hippocampal neurons (Fig. 2D). Treatment of mdivi-1 also caused a significant reduction of Aβ-mediated mitochondrial superoxide generation (Fig. 2E). Together, these results suggest that inhibition of Drp1 using mdivi-1 attenuates Aβ-induced mitochondrial fragmentation, depolarization, and ROS accumulation in neurons.
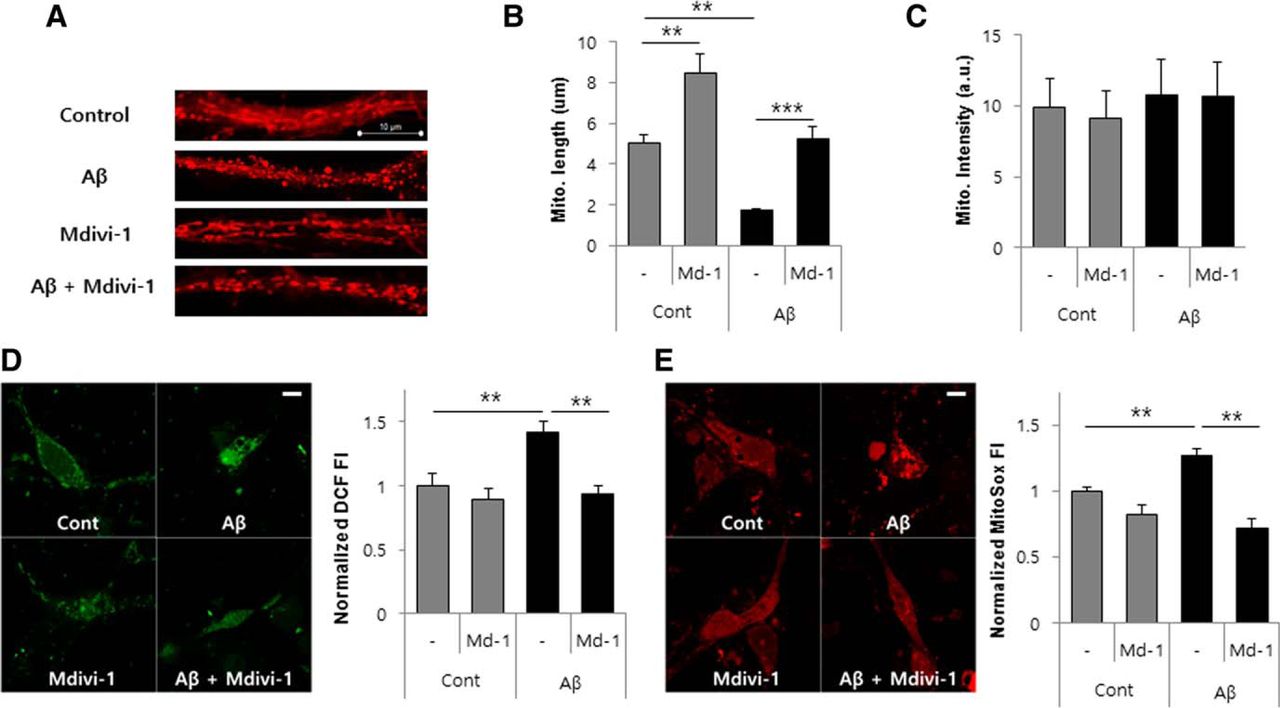
mdivi-1 inhibits mitochondrial fragmentation and ROS production in Aβ-treated primary hippocampal neurons. A–D, Primary hippocampal neurons treated with Aβ (1–42) oligomer (5 μm) in the presence or absence of mdivi-1 (25 μm) for 6 h and then the mitochondria were stained with a selective mitochondrial probe (100 nm) (MitoTracker). Mitochondria were imaged by a confocal microscopy (A) and the length (B) and intensity (C) of the mitochondria were measured in confocal 3D images. Intracellular ROS accumulation of Aβ-treated hippocampal neurons was assayed using H2DCF-DA fluorescent dye (D). E, Mitochondrial ROS induction by Aβ was detected by MitoSox Red staining in hippocampal neurons. Scale bar, 20 μm. Data are presented as the means ± SEM (n = 4). One-way ANOVA followed by Turkey post hoc comparisons tests were performed in all statistical analyses (*p < 0.05, **p < 0.01, ***p < 0.001).
Inhibition of Drp1 restores Aβ-mediated synaptic depression in hippocampal neurons
Next, we determined whether these morphological displacements are related to functional synaptic physiology at CNS synapses. To monitor synaptic vesicle exocytosis with high accuracy at nerve terminal, we used vGlut-pHluorin (vG-pH) expressed in primary cultured hippocampal neurons; PHluorin, a mutant form of GFP with fluorescence that can be quenched by protonation (pKa∼7.1) (Sankaranarayanan et al., 2000), is attached to the luminal region of synaptic vesicle transmembrane protein (vGlut1) and vG-pH is quenched in the resting state of synaptic vesicle which is in acidic condition (∼pH 5.5). By action-potential-driven synaptic vesicle exocytosis, it regains fluorescence with the alkaline extracellular environment. Neurons transfected with vG-pH were pretreated with DMSO (control), mdivi-1, Aβ, and mdivi-1 with Aβ for 3 h. Subsequently, we measured vG-pH responses to 100 action potential stimuli at 10 Hz across boutons of neuron. Signal was normalized with entire synaptic vesicle pool by rapid application of NH4Cl. Application of Aβ (1–42) oligomer resulted in ∼50% depression of exocytosis of synaptic vesicle, whereas vehicle (control) and mdivi-1-treated neurons remained unchanged (Fig. 3). Synaptic depression by Aβ oligomers was significantly restored by treatment of mdivi-1, suggesting that Aβ-induced mitochondria dysfunction causes impairment of functional synaptic physiology at CNS synapses and this disruption is restored by inhibition of mitochondrial fission by treatment with the Drp1 inhibitor mdivi-1.

mdivi-1 suppresses synaptic depression in Aβ-treated primary cultured hippocampal neurons. A, Representative images of primary hippocampal neurons transfected with vG-pH in each condition. Top images are the resting condition, middle images are ΔF images of the 100 action potential. Bottom is the visualization of all synaptic boutons by applying NH4Cl. B, Representative traces of synaptic vesicle exocytosis stimulated by 100 action potential from control, mdivi-1-treated, Aβ-treated, and mdivi-1+Aβ-treated neurons. Neurons transfected with vG-pH were stimulated at 10 Hz 10 s with or without mdivi-1 and Aβ. Intensities were normalized to the peak NH4Cl response. C, Mean values of amplitudes of 100 action potential response in control (n = 9 cells), mdivi-1-treated (n = 8), Aβ-treated (n = 15), and mdivi-1+Aβ-treated (n = 12) neurons. Scale bar, 5 μm. Data represent means ± SEM. One-way ANOVA followed by Turkey post hoc comparisons tests were performed in all statistical analyses (**p < 0.001).
Inhibition of Drp1 improves learning and memory in APP/PS1 mice
Because synaptic dysfunction mediated by Aβ causes molecular and cellular basis for cognitive deficits and neuropathological features in AD, we next investigated the effect of Drp1 inhibition in learning and memory deficits using AD model mice, APP/PS1-overexpressing Swedish APP (APP695swe) mutants, PS1 mutants (PS1-dE9), and age-matched WT littermates. We examined target searching strategy and the spatial reference memory using the MWM. This is the most common behavioral test used to determine hippocampal spatial memory deficits. The MWM latency period measured the time required for the animal to swim to a submerged platform after being trained to learn the location of the platform, with shorter times reflecting improved memory. Compared with vehicle-treated APP/PS1 mice, mdivi-1-treated APP/PS1 mice revealed significantly shorter latency to find the hidden platform on day 4 of the training session (Fig. 4A). Moreover, in the probe test on the following day 5, mdivi-1-treated APP/PS1 mice showed an increased staying time in the target quadrant (Fig. 4B) and the number of times crossing the target after removing the platform than the vehicle-treated mice (Fig. 4C,D). However, there was no considerable difference in the swimming speed among these groups (Fig. 4E), suggesting that the observed results in spatial learning and memory represent cognitive functions, but not altered motivation or motility of animals. We further assessed the effect of mdivi-1 on memory impairment with the passive avoidance test, a fear-motivated test to measure hippocampal-dependent associative learning of the animal. Performance in the passive avoidance test was assessed by the latency of entering the shock-paired dark compartment in the vehicle-administered APP/PS1 mice and mdivi-1-administered APP/PS1 mice groups. Compared with the vehicle-treated group, our data showed that the latency to enter the dark compartment in the mdivi-1-treated APP/PS1 group was significantly increased (Fig. 4F). However, mdivi-1 treatment on WT littermates did not improve learning and memory functions compared with the vehicle-treated control WT group.

Treatment of mdivi-1 attenuates hippocampal memory impairment in APP/PS1 mice. APP/PS1 mice and age-matched WT littermates were administered with vehicle or mdivi-1 (10 mg/kg, 40 mg/kg) daily for 4 weeks. The effect of mdivi-1 on learning and memory in APP/PS1 mice versus vehicle-treated control mice by MWM test and passive avoidance test was determined. The average latency to escape to the hidden platform during each day of training sessions (A), the time spent in the target quadrant where the hidden platform was previously placed during probe trial (B), the mean number of mice crossing the target quadrant during probe trial (C), typical swimming traces during probe test (D), and visual swimming speed test (E) of MWM test were performed. F, Latency time in passive avoidance task was measured. Data represent means ± SEM (n = 7 per group). One-way ANOVA followed by Bonferroni's post hoc comparisons tests were performed in statistical analyses (*p < 0.05, **p < 0.01, ***p < 0.001 compared with the vehicle-treated control group).
mdivi-1 restores mitochondrial length and decreases ROS production and gliosis in APP/PS1 mice
Our findings showed that mdivi-1 inhibits ROS generation, mitochondrial fragmentation, and synaptic depression in Aβ-treated cultured neurons (Figs. 1, 2, 3), as well as deficits of learning and memory of APP/PS1 mice (Fig. 4). We confirmed these observations in the brain tissues of midivi-1-administered APP/PS1 mice. To examine the mitochondrial length and the level of lipid peroxidation, the brain tissues were analyzed by immunohistochemistry with a MitoTracker and anti-4-HNE-Michael adduct antibody. Quantitative analysis indicated that mitochondrial length is dramatically shortened in the APP/PS mice compared with age-matched WT littermates and the lipid peroxidation level is also highly increased in the brain tissue of APP/PS mice compared with WT mice (Fig. 5A–C). Treatment of mdivi-1 (40 mg/kg) notably elongates the mitochondrial length in the brain of APP/PS1 and WT mice compared with that of the vehicle-treated mice (Fig. 5A). Figure 5B shows representative confocal microscopy images of 4-HNE-Michael adduct staining in the brain tissues of APP/PS1 mice. Treatment of mdivi-1 significantly reduced 4-HNE fluorescence intensity (Fig. 5C), suggesting that mdivi-1 inhibits mitochondrial fragmentation and lipid peroxidation in the brains of APP/PS1 mice. Next, we determined gliosis of APP/PS1 mice by staining for both activated microglia (Iba-1) and astrocytes (GFAP) (Fig. 5D,E). APP/PS1 mice showed the characteristic robust staining for both activated microglia (Iba-1) and astrocytes (GFAP). However, the mdivi-1-treated APP/PS1 mice had significantly less immunoreactivity for Iba-1 and GFAP compared with vehicle-treated APP/PS1 mice.

Administration of mdivi-1 reduces mitochondrial fragmentation and ROS production in the brain of APP/PS1 mice. A, APP/PS1 mice were administered with mdivi-1 (10 and 40 mg/kg) daily for 38 d and control mice were give vehicle. After brain dissection, the tissues were stained with DAPI and MitoTracker for 20 min. Then, the fluorescence images (63× and zoom in 10×) were captured by a confocal microscopy and length of mitochondrial length was measured. B, C, APP/PS mice were administered with mdivi-1 (10 mg/kg, 40 mg/kg) daily for 38 d. After brain dissection, the tissues were stained with DAPI and anti-4-HNE antibody. Then, the fluorescence images (40×) were captured by confocal microscopy (B) and the fluorescence intensity of 4-HNE was measured (C). D, E, Brain sections of the indicated groups were stained with anti-Iba-1 antibody, anti-NeuN antibody, anti-GFAP antibody, and anti-MAP2 antibody and immunohistochemical staining in hippocampus (CA1) was imaged with a confocal microscope. Scale bar, 20 μm. Data represent means ± SEM (n = 5 per group). One-way ANOVA followed by Turkey post hoc comparisons tests were performed in all statistical analyses (*p < 0.05, **p < 0.01, ***p < 0.001 vs the vehicle-treated control group).
mdivi-1 diminishes expression of BACE1 and decreases accumulation of Aβ plaques in APP/PS1 mice
It has been suggested previously that ROS positively contribute to BACE1 expression and generation of Aβ. Therefore, we further investigated the effect of mdivi-1 on protein expression related to Aβ generation. APP/PS1 mice were administered with mdivi-1 for 4 weeks and the expression of BACE1 and APP CTF-β were examined. BACE1 protein and mRNA and APP CTF-β levels were significantly increased in APP/PS1 mice compared with WT mice (Fig. 6A–D). As shown in Figure 6, mice treated with mdivi-1 (40 mg/kg) showed significantly lower levels of both BACE1 and APP CTF-β compared with the untreated control mice (Fig. 6A–D). However, the full-length APP protein level was not significantly affected by mdivi-1 treatments (Fig. 6A). The protein levels were calculated as a ratio against the loading control (Fig. 6B,C). These results implicate that mdivi-1 downregulates Aβ generation by reducing ROS generation in the brain of APP/PS1 mice. Because a decreased level of BACE1 resulted in reduction of Aβ, we investigated the effect of mdivi-1 on Aβ amyloidosis. Consistently, APP/PS1 mice administered with mdivi-1 showed significant reduction of accumulation of Aβ plaques in both hippocampus and cortex in the brain of APP/PS1 mice compared with that of control-treated mice (Fig. 7A–C).
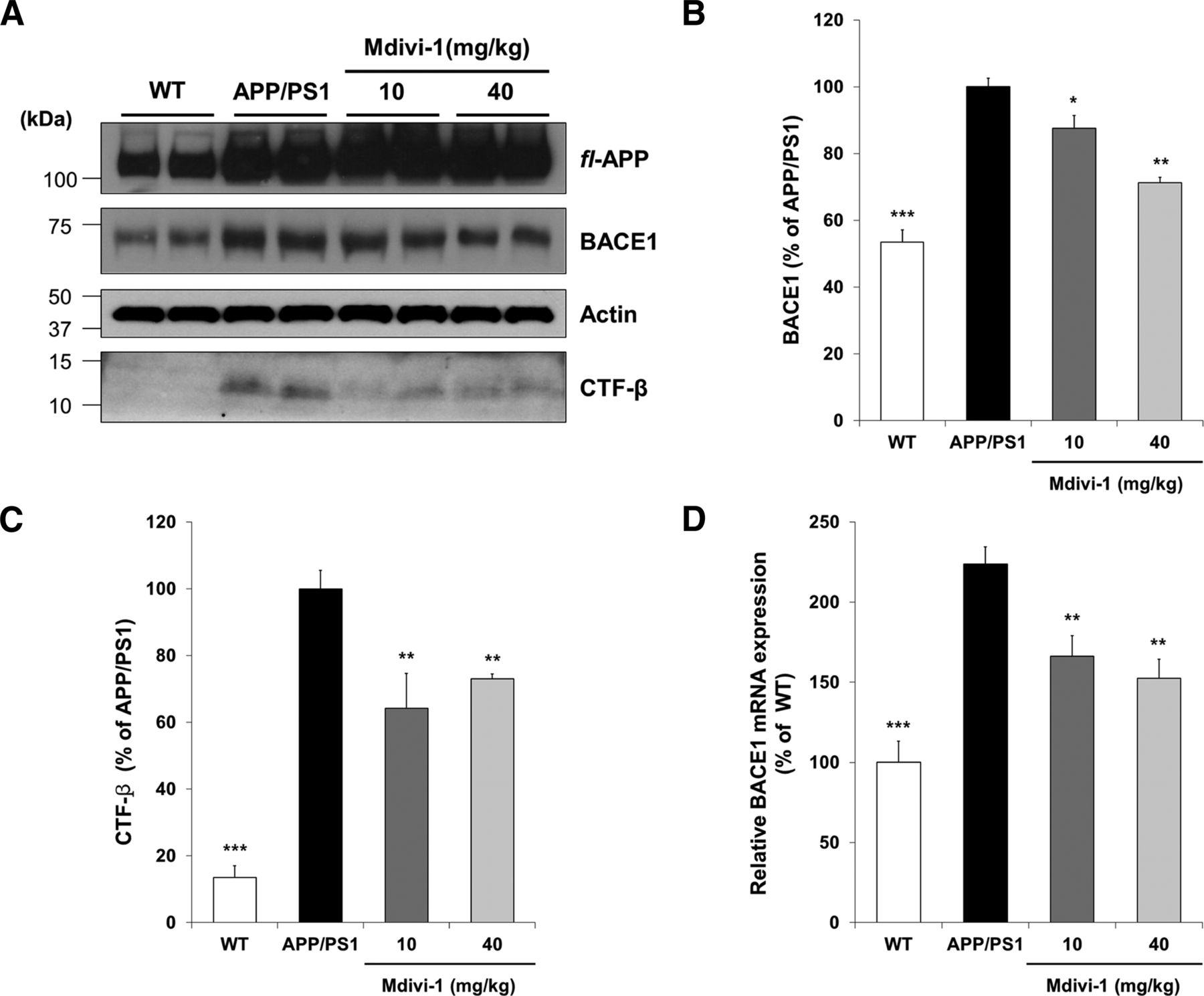
mdivi-1 reduces the expression of BACE1 and APP CTF-β in the brain of APP/PS1 mice. APP/PS1 mice were administered with mdivi-1 (10 mg/kg, 40 mg/kg) daily for 38 d. A, Expression of BACE1 and sAPPβ was examined by Western blot analysis with whole-brain protein extracts. B, C, Optical density of BACE1 (B) and sAPPβ (C) protein by Western blotting was quantified with Image software. D, Expression level of BACE1 mRNA was determined by quantitative RT-PCR analysis with mRNA extracted from whole-brain tissues. Values are means ± SEM (n = 6 per group). One-way ANOVA followed by Turkey post hoc comparisons tests were performed in all statistical analyses (*p < 0.05, **p < 0.01, ***p < 0.001 vs the vehicle-treated control group).

mdivi-1 reduces cerebral Aβ load in the brain of APP/PS1 mice. APP/PS1 mice were administered with mdivi-1 (10 mg/kg, 40 mg/kg) daily for 38 d. Brain sections from vehicle-treated or mdivi-1-treated APP/PS1 mice were incubated with an antibody against Aβ (6E10) and visualized by DAB staining. The amyloid plaques in hippocampus and cortex were identified with microscopy (A) and the neurite plaques in hippocampus (B) and cortex (C) were quantified. Scale bar, 800 μm. Values are means ± SEM (n = 5 per group). One-way ANOVA followed by Turkey post hoc comparisons tests were performed in all statistical analyses (**p < 0.01).
No adverse effects observed in mdivi-1-treated APP/PS1 mice
The body weights measured at the end of the dosing period were indistinguishable from those of vehicle-administered APP/PS1 mice or mdivi-1-treated APP/PS1 mice (Fig. 8A), which verified the toxicity of mdivi-1 in APP/PS1 mice. In addition, there were no differences in major organ weights among the study cohort, including brain weight (Fig. 8B). This observation suggests that overt toxic effects of chronic mdivi-1 administration were not present in these animals.

There was no adverse effect of chronic mdivi-1 treatment in APP/PS1 mice. To assess the toxicity of mdivi-1, APP/PS1 mice were given 0, 10, or 40 mg/kg/d mdivi-1 orally for 40 d. mdivi-1-administered or vehicle-treated male mice from 7 months of age were assessed for final body weights and organs weights. A, Body weights measured at the end of the dosing period were indistinguishable from vehicle-administered APP/PS1 mice or mdivi-1-treated APP/PS1 mice, which verified the toxicity of mdivi-1 in APP/PS1 mice. B, In addition, there were no differences in major organ weights among the study cohort, including brain weight. There was no statistically significant change among any of the group within the measures as determined by one-way ANOVA with Tukey's post hoc analysis.
Discussion
There is substantial evidence supporting that abnormal mitochondrial dynamics has a crucial role in pathogenesis of diverse neurodegenerative diseases, including AD, HD, PD, and ALS (Haun et al., 2013a, 2013b; Mishra and Chan, 2014). In addition to Aβ depositions and neurofibrillary tau tangles, enhanced mitochondrial fission and synaptic loss are the important histopathological changes seen in the AD brain (Cho et al., 2009; Reddy et al., 2012). Moreover, excessive mitochondrial fragmentation can contribute to AD pathogenesis through oxidative stress, synaptic damage, and neuronal death (Cho et al., 2009; Wang et al., 2014). Therefore, restoration of mitochondrial fission/fusion imbalance may have therapeutic value (Brooks et al., 2009; Liu et al., 2015). Nonetheless, it was not known whether inhibition of excessive fragmentation of mitochondria is advantageous in mammal models of AD. In the present study, we determined the effect of Drp1 inhibition using mdivi-1 on Aβ-mediated mitochondrial dysfunction and AD-associated neuropathology in cultured neurons and APP/PS1 double-transgenic AD mice. Our results illustrate that inhibition of Drp1 suppresses mitochondrial fission, mitochondrial depolarization, ROS production, and ATP reduction in Aβ-treated neurons. Moreover, Aβ-induced synaptic depression was significantly restored by inhibiting excessive mitochondria fragmentation with mdivi-1, a Drp1 inhibitor, which suggests that Aβ-mediated mitochondrial disruption causes impairment of functional synaptic physiology at neuronal synapses. Furthermore, mdivi-1 administration significantly reduced lipid peroxidation, BACE1 expression, Aβ deposition, and synaptic loss in the brain tissues of APP/PS1 mice. As a result, memory deficits in AD mice were rescued by Drp1 inhibition.
Increasing evidence has revealed that abnormalities in mitochondrial dynamics are early events in several neuronal diseases including AD (Wang et al., 2008, 2009b; Manczak et al., 2010, 2011; Calkins et al., 2011), HD (Wang et al., 2009a, 2009b; Costa et al., 2010; Song et al., 2011; Haun et al., 2013a, 2013b), PD (Kamp et al., 2010; Wang et al., 2012a, 2012b), ALS (Song et al., 2013), and multiple sclerosis (Kalman et al., 2003). In particular, abnormal mitochondrial dynamics and synaptic dysfunction, including Aβ and hyperphosphorylated tau interaction with Drp1, increased mitochondrial fission, disturbed axonal transport, and synaptic degeneration, have been increasingly observed in neurons of AD patients and mouse models of AD with disease progression (Manczak et al., 2012). Progressively accumulated Aβ in mitochondria is associated with increased oxidative stress, reduced mitochondrial membrane potential and ATP production, disturbed mitochondrial calcium regulation, secretion of mitochondrial apoptotic factors, decreased mitochondrial motility, and increased mitochondrial fission (Lustbader et al., 2004; Yao et al., 2011; Cha et al., 2012). Although the precise molecular mechanism underlying excessive mitochondrial fragmentation in AD brain is not been well understood, several molecular mechanisms have been proposed. Reddy's group investigated the interaction of Drp1 with Aβ and phosphorylated tau and Drp1 GTPase activity in the brain specimens from AD patients and AD mice (Manczak et al., 2011; Manczak et al., 2012). Their study showed a progressively increased interaction among Drp1, Aβ, and phosphorylated tau, which enhanced GTPase enzymatic activity of Drp1 and resulted in excessive mitochondrial fragmentation. Furthermore, it was revealed that S-nitrosylation of Drp1 at Cys644 caused mitochondrial fragmentation in brain neurons from AD and HD patients (Cho et al., 2009; Haun et al., 2013a, 2013b). Cys644 residue resides within the GTPase effector domain of Drp1, which affects both oligomer formation and GTPase activity of Drp1 (Zhu et al., 2004; Cho et al., 2009). S-nitrosylation of Drp1 induces its dimerization and activates its GTPase activity; however, mutation of Cys644 to Ala (C664A) abrogates these effects of NO (Cho et al., 2009). Moreover, blocking of Drp1 S-nitrosylation using C664A mutant prevented Aβ-mediated mitochondrial fission, synaptic damage, and neuronal death. These findings suggest that Aβ enhances Drp1 activity and causes excessive mitochondrial fission, leading to mitochondrial dysfunction and neuronal death in AD progression. Findings from these studies also suggest that inhibitors of mitochondrial fission could be potential therapeutic targets for reducing Drp1 GTPase activity in patients with AD. Consistently, we observed that inhibition of Drp1 GTPase activity prevented Aβ-mediated mitochondrial fission, mitochondrial depolarization, ROS production, and ATP reduction in cultured neurons.
Mitochondria residing at synapses are usually older and are therefore much more vulnerable to cumulative damage mediated by deleterious factors such as Aβ compared with nonsynaptic mitochondria (Du et al., 2010). Moreover, early deposition of Aβ occurs in mitochondria residing in synapses, which might make synaptic mitochondria much more vulnerable to Aβ toxicity (Dragicevic et al., 2010; Du et al., 2010). Accordingly, synaptic mitochondria are functionally compromised and show greater vulnerability to damage compared with nonsynaptic mitochondria in AD mice (Dragicevic et al., 2010; Du et al., 2010). These observations indicate that dysfunction of synaptic mitochondria occurs early in the progress of AD and disturbance of synaptic mitochondria plays a key role in Aβ-mediated mitochondrial and neurosynaptic toxicity. Therefore, we determined whether inhibition of mitochondrial fission would be beneficial for Aβ-mediated synaptic dysfunction. Treatment of Aβ oligomers induced significant depression of exocytosis of synaptic vesicle; however, mdivi-1-treated neurons showed normal synaptic exocytosis. Aβ oligomers-mediated synaptic depression was significantly protected by application of mdivi-1, indicating that Aβ-induced mitochondria dysfunction causes impairment of functional synaptic physiology. Therefore, we first proved that Aβ oligomer-mediated functional synaptic failure was through excessive mitochondrial fission.
mdivi-1, a derivative of quinazolinone, has been identified as a cell-permeable selective inhibitor of Drp1 (Cassidy-Stone et al., 2008). mdivi-1 inhibits GTPase activity by blocking the self-assembly of Drp1 and mitochondrial fission. The mdivi-1 molecule has been investigated in several experimental animal models for seizures (Xie et al., 2013a, 2013b), epilepsy (Qiu et al., 2013), cerebral ischemia/reperfusion (Zhang et al., 2013; Liu et al., 2015), rhabdomyolysis (Tang et al., 2013), HD (Manczak and Reddy, 2015), and diabetes (Huang et al., 2015). Another Drp1 inhibitor, P110, has been studied in an HD animal model (Guo et al., 2013). The Drp1 inhibitors exhibited protective effects on affected cells and tissues by ameliorating excessive mitochondrial fragmentation and maintaining the normal mitochondrial functions in all of these investigated animal models of diseases. However, it was not clear whether mdivi-1 ameliorates neuropathology and memory decline by improved neuropathology in AD mice models. In this study, we exploited APP/PS1 double-transgenic mice overexpressing both an APP695swe mutant and a PS1 mutant (PS1-dE9), which are highly associated with early-onset AD. APP/PS1 mice showed progressively increased Aβ deposition, modest neuron loss, dendritic spin loss, and cognitive deficits in spatial learning and memory (Radde et al., 2006; Serneels et al., 2009; Rupp et al., 2011; Bittner et al., 2012). We found that the administration of mdivi-1 could improve learning and memory performance in APP/PS1 mice, suggesting that Drp1-mediated mitochondria fragmentation is required for impairment of learning and memory in the APP/PS1-transgenic mice. Expectedly, we also observed reduced lipid peroxidation and mitochondrial fission in the brain tissues of midivi-1-administered APP/PS1 mice. Previous studies reported that oxidative stress is known to be associated with increased BACE1 expression, γ-secretase activity, and Aβ (Tamagno et al., 2002; Jo et al., 2010; Gwon et al., 2012). Therefore, excessive mitochondrial fission-mediated mitochondrial oxidative stress could be an upstream regulator of BACE1 expression, APP processing, and amyloid pathology, resulting in declined learning and memory in AD. Notably, we had determined that inhibition of Drp1 activity by mdivi-1 attenuated both BACE1 expression and accumulation of Aβ, as well as memory decline in APP/PS1 mice, which suggested that inhibition of excessive mitochondrial fragmentation might have important roles in alleviating Aβ production and deposition in AD. Despite these results, the role of Drp1 in mdivi-1-mediated amelioration of pathogenesis of Alzheimer's disease is further elucidated.
Together, our studies in Aβ-treated neurons and in vivo animal model of AD suggest that neuropathology and combined cognitive decline can be attributed, at least in part, to hyperactivation of Drp1 in the pathogenesis of AD. Our data further indicate that inhibitors of excessive mitochondrial fission, such as Drp1 inhibitors, may be effective strategies for preventing and halting the progression of AD.
Footnotes
This work was supported by the Korean Health 21 R&D Project (Grants HI14C2539 and HI16C1176), the Korea–UK Collaborative Alzheimer's Disease Research Project, Ministry of Health and Welfare, Republic of Korea (Grant A120196), and the Basic Science Research Program through the National Research Foundation of Korea (NRF), the Ministry of Education, Science and Technology, Republic of Korea (Grants 2012R1A5A2A28671860 and 2015R1A2A1A01003530).
The authors declare no competing financial interests.
- Correspondence should be addressed to either of the following: Dong-Hyung Cho, Graduate School of East-West Medical Science, Kyung Hee University, Yongin, Korea, dhcho{at}khu.ac.kr; or Dong-Gyu Jo, School of Pharmacy, Sungkyunkwan University, Suwon 16419, Korea, jodg{at}skku.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}