

Abstract
The neurodegenerative disease Machado–Joseph disease (MJD), also known as spinocerebellar ataxin-3, affects neurons of the brain and spinal cord, disrupting control of the movement of muscles. We have successfully established the first transgenic zebrafish (Danio rerio) model of MJD by expressing human ataxin-3 protein containing either 23 glutamines (23Q, wild-type) or 84Q (MJD-causing) within neurons. Phenotypic characterization of the zebrafish (male and female) revealed that the ataxin-3-84Q zebrafish have decreased survival compared with ataxin-3-23Q and develop ataxin-3 neuropathology, ataxin-3 cleavage fragments and motor impairment. Ataxin-3-84Q zebrafish swim shorter distances than ataxin-3-23Q zebrafish as early as 6 days old, even if expression of the human ataxin-3 protein is limited to motor neurons. This swimming phenotype provides a valuable readout for drug treatment studies. Treating the EGFP-ataxin-3-84Q zebrafish with the calpain inhibitor compound calpeptin decreased levels of ataxin-3 cleavage fragments, but also removed all human ataxin-3 protein (confirmed by ELISA) and prevented the early MJD zebrafish motor phenotype. We identified that this clearance of ataxin-3 protein by calpeptin treatment resulted from an increase in autophagic flux (indicated by decreased p62 levels and increased LC3II). Cotreatment with the autophagy inhibitor chloroquine blocked the decrease in human ataxin-3 levels and the improved movement produced by calpeptin treatment. This study demonstrates that this first transgenic zebrafish model of MJD is a valuable tool for testing potential treatments for MJD. Calpeptin treatment is protective in this model of MJD and removal of human ataxin-3 through macro-autophagy plays an important role in this beneficial effect.
SIGNIFICANCE STATEMENT We have established the first transgenic zebrafish model of the neurodegenerative disease MJD, and identified relevant disease phenotypes, including impaired movement from an early age, which can be used in rapid drug testing studies. We have found that treating the MJD zebrafish with the calpain inhibitor compound calpeptin produces complete removal of human ataxin-3 protein, due to induction of the autophagy quality control pathway. This improves the movement of the MJD zebrafish. Artificially blocking the autophagy pathway prevents the removal of human ataxin-3 and improved movement produced by calpeptin treatment. These findings indicate that induction of autophagy, and removal of ataxin-3 protein, plays an important role in the protective effects of calpain inhibition for the treatment of MJD.
Introduction
Machado–Joseph disease (MJD), also known as spinocerebellar ataxia-3, is a neurodegenerative disease characterized by a progressive loss of muscle control and movement, leading to wheelchair dependence and decreased lifespan (Costa and Paulson, 2012). Clinical symptoms include ataxia, dystonia, rigidity, muscle atrophy, and vision and speech disorder (Costa and Paulson, 2012; Rüb et al., 2013). MJD is the most common of the hereditary ataxias found throughout the world (21–28% of autosomal-dominant ataxia; Ranum et al., 1995; Schols et al., 1995; Dürr et al., 1996), with a high prevalence within the Azores of Portugal (Bettencourt et al., 2008) and Indigenous communities of northeast Arnhem Land in Australia (Burt et al., 1996; Martins et al., 2012).
MJD is caused by inheritance of an expanded CAG trinucleotide repeat region within the MJD1/ATXN3 gene on chromosome 14 resulting in expression of the ataxin-3 protein containing a long polyglutamine (polyQ) repeat region (Takiyama et al., 1993; Stevanin et al., 1994). Whilst the ATXN3 gene of healthy subjects contains a short CAG repeat region (12–40 CAG repeats), this region contains >40 CAG repeats in MJD patients (Takiyama et al., 1993; Maciel et al., 1995; Matsumura et al., 1996b). The length of the CAG repeat region is correlated with the severity of the disease (Maciel et al., 1995; Matsumura et al., 1996a).
Neuropathological staining of patient brain samples often reveals the presence of neuronal intranuclear inclusions containing ataxin-3 protein (Schmidt et al., 1998; Yamada et al., 2000; Rüb et al., 2013). Extraction of the proteins within these inclusions has revealed the presence of full-length ataxin-3 as well as smaller ataxin-3 fragments (Goti et al., 2004). The presence of these ataxin-3 protein fragments in patient samples forms the basis of a hypothesis of MJD pathogenesis called the “toxic fragment hypothesis” (Ross, 2002; for review, see Matos et al., 2017) based on previous reports that protein fragments containing long polyQ regions are highly aggregation prone and neurotoxic (Marsh et al., 2000; Morley et al., 2002; Schiffer et al., 2007).
Previous studies have demonstrated that ataxin-3 proteolysis is predominately performed by the calcium-dependent protease calpain (Weber et al., 2017), and that calpain inhibition prevents formation of ataxin-3 fragments in vitro (Haacke et al., 2007; Koch et al., 2011; Hubener et al., 2013). Various groups have investigated whether inhibiting ataxin-3 cleavage through calpain inhibition may be beneficial as a treatment for MJD (Haacke et al., 2007; Koch et al., 2011; Simões et al., 2014). Simões et al. (2014) demonstrated that treating a rodent model of MJD with the calpain inhibitor compound BDA-410 prevents formation of ataxin-3 fragments, neuronal degeneration, and motor deficits (Simões et al., 2014). Despite these positive findings, additional studies are required to confirm whether the beneficial effect of calpain inhibition seen is indeed inhibition of ataxin-3 cleavage, or perhaps another effect of calpain inhibition, such as induction of the macroautophagy protein quality control pathway (Williams et al., 2008).
Small animal models, such as transgenic zebrafish, can be useful for testing potential disease treatments (Zon and Peterson, 2005). One major advantage of using zebrafish embryos and larvae is that they can absorb drugs and small compounds added to the water they live in, decreasing the need to dose each animal individually (Zon and Peterson, 2005; Schiffer et al., 2007; van Bebber et al., 2010; Vaccaro et al., 2012). The small size of the zebrafish, and large offspring numbers (∼300 eggs per week), make obtaining large sample sizes relatively straight-forward (Laird and Robberecht, 2011) and their transparency during development aids observation and imaging of their neuroanatomy and expressed fluorescently tagged proteins (Paquet et al., 2010).
In this study we have established, and characterized, the first transgenic zebrafish model of MJD, identifying relevant disease phenotypes as early as 6 days old through to adult stages. Using these transgenic MJD zebrafish we have gained insight into the potential of calpain inhibition for the treatment of MJD.
Materials and Methods
Transgenesis construct preparation.
The transgenesis expression constructs for driver and responder lines are based on those described by Paquet et al. (2009). The driver line construct contained a HuC (elav) neuronal promoter (Park et al., 2000) driving Gal4 VP16 sequence flanked by tol2 sites, and the responder construct contained a bidirectional UAS driving dsRED expression and a attR1/2 site. We performed recombination using BP clonase (Life Technologies) to insert EGFP-ATXN3 sequence (containing 23 or 84 CAG repeats), into the attR1/2 site. pEGFP-C1-ATXN3-84CAG was a kind gift from Henry Paulson (Addgene, plasmid no. 22123; Chai et al., 2002).
Generation of transgenic zebrafish lines.
All animal experiments were performed in accordance with the Animal Ethics Committee of the University of Sydney (K00/2014/575 and 576) and the Animal Ethics Committee of Macquarie University, NSW, Australia (ARA: 2012/050 and 2016/04, respectively). Zebrafish were housed in a standard recirculating aquarium system (Pentair) at 28.5°C. Adult fish were mated and resulting eggs collected for microinjection at the one-cell stage. Each embryo was injected with either the driver or responder transgenesis construct DNA, together with transposase mRNA.
For the experiments described here, driver and responder transgenic zebrafish lines were mated and resulting fish (F1) were studied until adulthood. These F1 fish were then in-crossed and their offspring were used for tracking motor function at the larval stage and for the compound treatment studies.
A mir218-enhancer-Kal4 driver line was used to drive motor neuron-specific expression of the EGFP-ataxin-3 transgenes in some experiments. This driver line involves use of a slit3 regulatory element, previously identified to be a miRNA-218 enhancer (Punnamoottil et al., 2015), driving expression of Kal4 (a version of Gal4) and kaeda fluorescent protein. This mir218-enhancer driver line produces specific motor neuron expression (Laird et al., 2016).
Imaging of whole-mount zebrafish larvae.
For confocal imaging of whole-mount larvae [following 24 h fixation with 4% paraformaldehyde (PFA)] were mounted in 1% agarose and imaged using a Zeiss LSM 710 confocal microscope. Images were taken with a 10× Leica HCX APO U-V-I water-immersion objective. EGFP was excited at 488 nm with an argon laser and dsRED was excited at 557 nm with a Leica white-light laser.
To inspect the motor neurons of the zebrafish larvae for any morphological abnormalities we imaged the lateral view of anesthetized 48 h postfertilization (hpf) zebrafish at 10× on a Leica DMi8 inverted microscope equipped with Leica DFC 365 FX camera. The length of three motor axons (those exiting the spinal cord at the caudal end of the yolksac) was measured using the NeuronJ plugin within ImageJ (NIH). The average motor axon length per animal was then calculated and compared using one-way ANOVA.
Tissue processing and immunohistochemistry.
Adult zebrafish were euthanized through overdose with tricaine anesthetic, transcardially injected with 4% PFA and incubated in PFA for 10 min. The spinal cord and brains of the zebrafish were then removed, fixed overnight in 4% PFA, and cryoprotected in 20% sucrose. The spinal cords and brains were sectioned (30 μm) on a cryostat and mounted in three series onto Superfrost slides. The different series underwent the following staining procedures: cresyl violet staining (standard protocol), ataxin-3 immunostaining, or polyglutamine immunostaining. Ataxin-3 staining involved antigen retrieval (sodium citrate treatment) before standard immunohistochemical protocol using rabbit anti-MJD (a kind gift from H. Paulson, University of Michigan, Ann Arbor, MI) and goat anti-rabbit secondary antibody incubation. Polyglutamine immunostaining was performed in the same manner, but using anti-polyglutamine antibody (1C2, Millipore; 1:800) and biotinylated horse anti-mouse secondary antibody (Vector Laboratories, BA-2000; 1:1000). The stained slides were imaged using a Leica upright DMI4000 microscope. An experimenter, blind to the experimental group, manually counted the number of neurites with polyQ or ataxin-3-positive beading morphology per section.
Motor function testing.
All zebrafish behavioral tracking was performed using a ZebraLab tracking system composed of a ZebraBox for tracking zebrafish embryos and larvae and a ZebraTower for tracking adult fish (Viewpoint). Tracking of 6 d post fertilization (dpf) larva was conducted in 24 multiwell plates within a ZebraBox housed with an enhanced light source under conditions of 6 min light, 4 min dark, and 4 min light. The total distance traveled by each larva within the dark phase was calculated (first light phase was for acclimatization). The swimming behavior of each fish was also recorded monthly from 4 weeks old until 12 months old using the ZebraTower device. The test period involved a 5 min exposure to ambient light within an open tank (770 cm3 containing 400 ml system water).
Survival analysis.
The lifespan of each animal was calculated. A Kaplan–Meier graph of lifespan was then generated and the average survival (days) calculated.
Western blotting.
Protein lysates were prepared from zebrafish larva (3 or 6 dpf) following removal of the protein-rich yolksac. The samples were then lysed in RIPA buffer containing protease inhibitors (Complete ULTRA Tablets, Roche) using a manual dounce. Equal amounts of protein were separated via SDS-PAGE and transferred to PVDF membrane for immunoblot probing. Antibodies used included rabbit anti-MJD (kind gift from H. Paulson), rabbit anti-beclin-1 (11306-1-AP, Proteintech), rabbit anti-p62 (SQSTM1, MBL PM045, MBL Life Science), rabbit anti-LC3B (ab51520, Abcam), mouse anti-spectrin (sc-48382, Santa Cruz Biotechnology), mouse anti-β-actin (A5441, Sigma-Aldrich) and mouse anti-GAPDH (60004-1, Proteintech) for loading controls. The immunoblots were probed with appropriate secondary antibodies (anti-mouse, W4021, and anti-rabbit, W4011, Promega) and visualized by chemiluminescence (Supersignal detection kit, Pierce) using a Bio-Rad GelDoc System. The intensity of the bands within the immunoblot was quantified by Image Studio Lite and the target protein expression level was determined by normalizing against the loading control protein.
Inhibitor compound treatment studies.
Zebrafish embryos (24 hpf) were screened for fluorescence (EGFP and dsRED) indicating that they were positive for the transgene and positive embryos were treated with calpain/caspase inhibitor compounds through addition of the compounds to the E3 solution (Nusslein-Volhard and Dahm, 2002). The four protease inhibitor compounds tested included two calpain inhibitors (ALLN, Calpeptin, Cayman Chemicals), one caspase inhibitor (Z-VAD-FMK, Cayman Chemicals), and one cathepsin inhibitor (CAT-1, Santa Cruz Biotechnology). ALLN inhibits calpain-I, whereas calpeptin inhibits both calpain-I and -II. Each drug was dissolved in dimethyl sulfoxide (DMSO) to make stock solutions that were aliquoted and stored at −20°C. The zebrafish embryos were incubated at 28°C for 2 d within Petri dishes. At 3 dpf, protein lysates were prepared from the zebrafish embryos to allow immunoblot analysis of the lysates.
For the second half of the study, the compound treatment was extended to 6 dpf to allow examination of the effect of the treatment on the 6 dpf motor phenotype. In that case, the treatment was performed in the same way except the tracking was performed before preparation of the protein lysates at 6 dpf.
To test the effect of autophagy inhibition on the motor function and immunoblot results, cotreatment with calpeptin and the autophagy inhibitor compound chloroquine diphosphate (3 mm, Sigma-Aldrich) was also examined. Chloroquine treatment was performed in the same manner as the protease inhibitor compound treatments described above.
Ataxin-3 ELISA.
Ataxin-3 levels were quantified by sandwich ELISA. A 96-well MaxiSorp microplate was coated with rabbit anti-MJD antibody (1:5000, kind gift from H. Paulson, University of Michigan, Ann Arbor, MI) overnight at 4°C, followed by blocking with 3% Bovine Serum Albumin in Phosphate Buffered Saline. Protein samples (20 μg each) and a standard curve of ataxin-3 recombinant protein (Cat No. MBS203345, MYBioSource) were loaded into wells and incubated for 2 h at room temperature. The plate was then incubated with mouse anti-ataxin-3 monoclonal antibody (1:1000; Millipore), followed by anti-mouse HRP antibody (1:2500, Promega). A color reaction was produced by incubating the plate in 1-Step TMB (Cat No. 34028, Thermo) for 15 min, followed by addition of a 2M sulfuric acid stop solution. The plate was then read by a PHERAstar FS plate reader (450 nm, BMG Labtech) and ataxin-3 protein concentration within wells was interpolated from the ataxin-3 standard curve.
Real-time PCR.
RNA isolation from 6 dpf zebrafish larvae expressing EGFP-ataxin-3 was performed using TRIzol Reagent (Invitrogen) and RNeasy Micro Kit (Qiagen) according to the manufacturer's instructions. The RNA was subjected to reverse transcription using SuperScript VILO cDNA Synthesis Kit (Invitrogen). Equal amounts of cDNA (100 ng per 50 μl reaction) were amplified using TaqMan Gene Expression Master Kit (ThermoFisher) in an Applied Biosystems StepOnePlus Real-Time PCR System (ThermoFisher). Each sample was run in triplicate. Predesigned TaqMan Gene Expression Assays were used containing primers specific for transgenic ATXN3 (Assay ID Hs00245261_m1), and primers for atp5b (Assay ID Dr0342455_m1), and eef1a (Assay IDDr03432748_m1) as reference genes. Relative transgenic ATXN3 expression was determined using the 2−ΔΔCT calculation method to calculate fold-change (RQ) and normalized using geometric averaging of the reference genes.
Experimental design and statistical analysis.
Experiments using zebrafish embryos and larvae were conducted on both male and female zebrafish (sex not determined at that age) with siblings used for controls within the compound treatment studies. Studies completed on the adult zebrafish included an equal proportion of male and female zebrafish per group. All animals were housed under identical conditions within the study. Data analysis was performed using SPSS Statistical software (v21). A Mantel–Cox test was used to compare the lifespan of the transgenic animals. Comparison between just the EGFP-ataxin-3-23Q and EGFP-ataxin-3-84Q genotypes (including the amount of ataxin-3 cleavage fragments) were made using an independent t test. All other group comparisons were made using one-way ANOVA followed by Tukey post hoc analysis to identify differences. Statistically significant results (p < 0.05) identified by these post hoc tests were indicated by symbols within the figures (*∧#).
Results
A transgenic zebrafish model of MJD has impaired motor function and decreased survival
Two transgenic MJD zebrafish lines were produced that expressed EGFP-ataxin-3-23Q and EGFP-ataxin-3-84Q in a pan-neuronal pattern (Fig. 1A). Confocal images of whole-mount transgenic zebrafish at 2 dpf confirmed that the transgenic zebrafish expressed EGFP (indicative of the EGFP-ataxin-3 transgene), as well as a dsRED reporter, both in a neuronal pattern (Fig. 1B). As expected, the fluorophores were expressed throughout neurons of the spinal cord (including motor neurons, interneurons, and sensory neurons), brain, and cranial nerves (Park et al., 2000).

Transgenic zebrafish model of MJD. A, Transgenic zebrafish were produced by mating fish expressing GAL4 under the control of the HuC promoter with fish carrying bidirectional UAS driving a dsRED expression marker and human ataxin-3 (23Q or 84Q) with a N-terminal EGFP fusion protein. B, Resultant transgenic zebrafish (2 dpf shown here) expressed EGFP-fused human ataxin-3 (containing either 23Q or 84Q) and dsRED in all neurons. Scale bar, 200 μm.
Immunostaining for polyglutamine sequence in sagittal sections from the medulla region of 12-month-old nontransgenic, EGFP-ataxin-3-23Q and -84Q zebrafish revealed the presence of polyglutamine-positive neuritic beading pattern (Fig. 2A). This neuritic beading phenotype was not present within the medulla of nontransgenic and EGFP-ataxin-3-23Q zebrafish (n = 6 each group; F(2,15) = 24.415, p < 0.0001; ANOVA and Tukey post hoc analysis, p < 0.0001; Fig. 2B). Similarly, staining medulla sections with the polyclonal ataxin-3 antibody revealed significantly more of the ataxin-3–positive neuritic beading pattern in EGFP-ataxin-3-84Q than nontransgenic or EGFP-ataxin-3-23Q samples (n = 6 each group; F(2,15) = 15.463, p < 0.0001; ANOVA and Tukey post hoc analysis, p < 0.001; Fig. 2A,C).
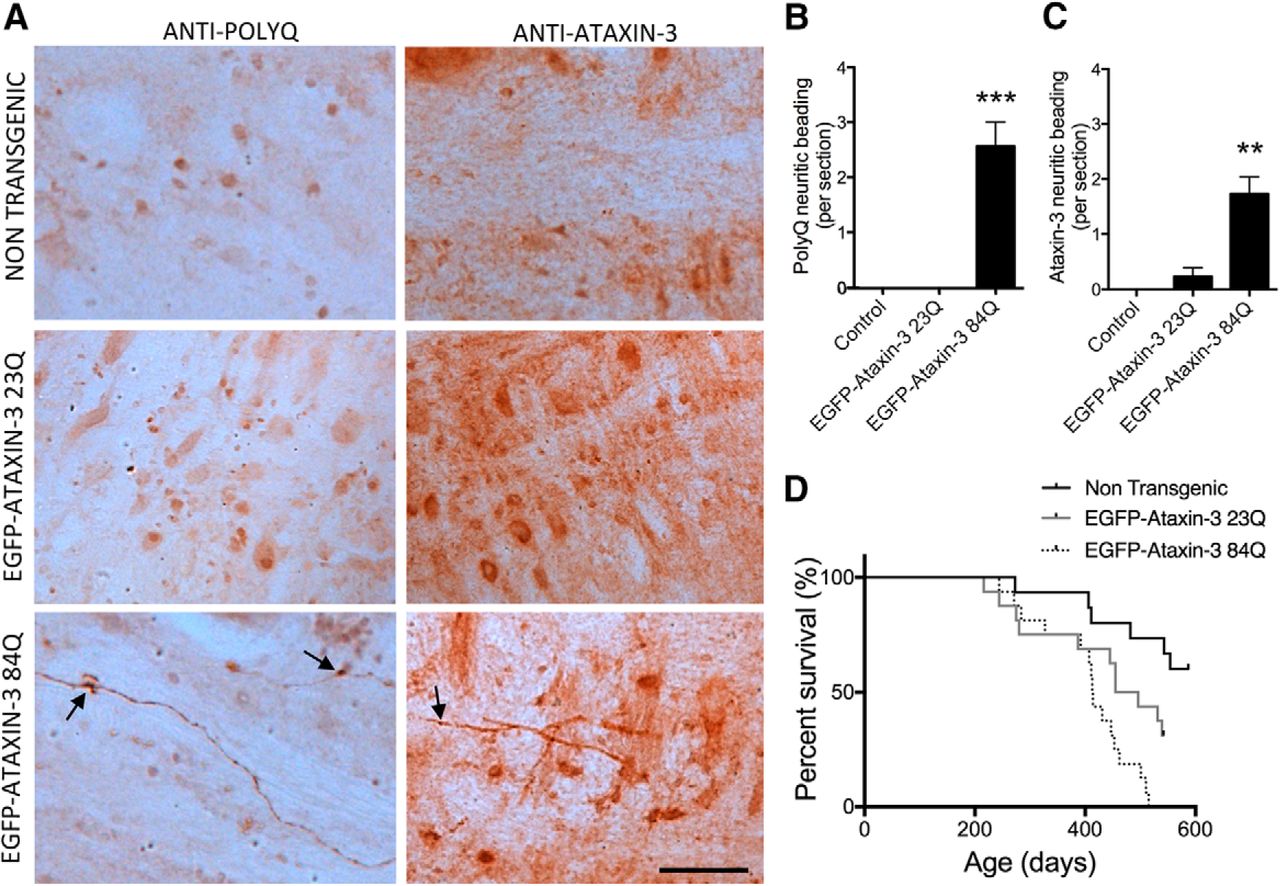
MJD pathology and decreased survival of transgenic MJD zebrafish. A, Immunostaining for polyglutamine and ataxin-3 (antibody recognized both endogenous zebrafish ataxin-3 and transgenic human ataxin-3) in sections throughout the medulla of 12-month-old zebrafish revealed polyglutamine-positive neuritic beading pattern (arrows) in EGFP-ataxin-3-84Q zebrafish only. This neuritic beading pattern was also positive for ataxin-3. Scale bar, 35 μm. B, Quantification of the number of neurites showing the polyglutamine-positive neuritic beading pattern within the medulla region revealed that the phenotype was only present in EGFP-ataxin-3-84Q zebrafish sections (n = 6 each, ***p < 0.0001). C, Quantification of the number of neurites showing ataxin-3-positive beading pattern revealed that it was far more common in EGFP-ataxin-3-84Q zebrafish than nontransgenic and EGFP-ataxin-3-23Q (n = 6 each, **p < 0.001). D, The lifespan of nontransgenic (n = 15), EGFP-ataxin-3-23Q (n = 16) and EGFP-ataxin-3-84Q (n = 16) zebrafish is plotted within a Kaplan–Meier survival graph that reveals significantly different survivals for the three groups, p < 0.0279. Error bars represent mean ± SEM.
We monitored the lifespan of the transgenic animals to determine whether EGFP-ataxin-3-84Q zebrafish have decreased lifespans, reminiscent of human MJD. A Kaplan–Meier survival graph plotting survival curves of EGFP-ataxin-3-23Q (n = 16) and EGFP-ataxin-3-84Q (n = 16) zebrafish is shown in Figure 2D. A log-rank statistical test (Mantel–Cox) comparing the individual group survival curves revealed that the EGFP-ataxin-3-23Q and EGFP-ataxin-3-84Q survival curves were significantly different (p = 0.016). However the survival curves of EGFP-ataxin-3-23Q and EGFP-ataxin-3-84Q were also both significantly different to that of nontransgenic animals (p < 0.0279). The mean survival time of EGFP-ataxin-3-84Q fish is 45 d less than EGFP-ataxin-3-23Q.
To examine whether EGFP-ataxin-3-84Q zebrafish develop motor impairment, we used a behavioral tracking system (ZebraLab, Viewpoint). Six-day-old zebrafish were tracked within 24-well plates during a 4 min darkness response test (Fig. 3A). Images showing the swimming trajectories showed a trend of less movement in the EGFP-ataxin-3-84Q than the nontransgenic and EGFP-ataxin-3-23Q larvae during this testing, with less movements made at fast speed (red) and more time spent inactive (black; Fig. 3A). Quantification of the distance traveled and one-way ANOVA statistical comparison confirmed a significant effect of genotype (F(2,272) = 4.972, p = 0.002). Tukey post hoc analysis revealed that EGFP-ataxin-3-84Q zebrafish swam shorter distances than EGFP-ataxin-3-23Q and nontransgenic zebrafish (p < 0.014; Fig. 3B). Interestingly, expression of the EGFP-ataxin-3 proteins specifically in motor neurons using a mir218 enhancer-Kal4 driver line also affected the distance swum by the zebrafish (F(2,87) = 5.008, p = 0.009, ANOVA; Fig. 3C), producing similar motor impairment to pan-neuronal expression of ataxin-3. Post hoc analysis revealed that the zebrafish expressing EGFP-ataxin-3-84Q within motor neurons (n = 32) swam significantly shorter distances than nontransgenic (n = 36) and those expressing EGFP-ataxin-3-23Q in motor neurons (n = 22; p < 0.049, Tukey post hoc). Inspection for any morphological effects of the expression of EGFP-ataxin-3 in motor neurons revealed a significant difference between the groups at 48 hpf (F(2,61) = 10.838, p < 0.001, ANOVA). Motor neurons in zebrafish expressing EGFP-ataxin-3-84Q zebrafish had a reduced axonal length compared with those in nontransgenic or EGFP-ataxin-3-23Q zebrafish (p < 0.027; Fig. 3D,E).

Motor dysfunction in transgenic MJD zebrafish larvae. A, Six days post fertilization nontransgenic (NT), EGFP-ataxin-3-23Q (23Q), and EGFP-ataxin-3-84Q (84Q) were tracked within 24-well plates during a 4 min darkness response test. Swimming trajectory images reveal movements made at fast speed (red), slow speed (green), and while inactive (black). B, Quantification of total distance swum during the 4 min in darkness revealed that EGFP-ataxin-3-84Q (n = 83) traveled shorter distances than both EGFP-ataxin-3-23Q (n = 73) and control fish (n = 76); *p < 0.014. C, Zebrafish larva (6 dpf) with motor neuron specific (driven by a miRNA-218 enhancer-driven) expression of EGFP-ataxin-3-84Q (n = 32) swim significantly shorter distances than controls (n = 36) and those expressing EGFP-ataxin-3-23Q (n = 22); ∧p < 0.049. D, Motor neurons positive for dsRED were imaged in zebrafish expressing EGFP-ataxin-3-dsRED in motor neurons, driven by the mir218-kal4-kaeda driver line. E, Measurement of the length of the motor axons within the zebrafish revealed that those expressing EGFP-ataxin-3-84Q had significantly shorter axon lengths than nontransgenic and EGFP-ataxin-3-23Q expressing zebrafish (#p < 0.027). Scale bar, 100 μm. Error bars represent mean ± SEM.
Next we evaluated whether the motor dysfunction was sustained and worsened with age. We monitored the swimming behavior throughout adulthood until 12 months old. On each test occasion, we quantified the total distance traveled by each zebrafish during a 5 min period of baseline swimming in light. EGFP-ataxin-3-84Q fish (n = 32) swam consistently shorter distances than nontransgenic (n = 27) and EGFP-ataxin-3-23Q (n = 28) fish (Fig. 4A). Images of the swimming trajectories reveals that the 12-month-old nontransgenic or EGFP-ataxin-3-23Q fish swim quickly (red), whereas the EGFP-ataxin-3-84Q fish spent more time swimming at slow speeds (green; Fig. 4B). Quantification of the total distance swum during the 5 min baseline swimming recording at 12 months old revealed an effect of genotype (F(2,66) = 8.909, p < 0.0001, ANOVA), with the EGFP-ataxin-3-84Q fish (n = 28) swimming shorter distances than both nontransgenic (n = 13) and EGFP-ataxin-3-23Q (n = 28) fish (Fig. 4C; p < 0.004 from Tukey post hoc tests). Further, there was a significant effect of genotype on the swimming of adult (13 week old) zebrafish expressing human ataxin-3 only in motor neurons (F(2,73) = 7.864, p = 0.001, ANOVA), with zebrafish expressing EGFP-ataxin-3-84Q (n = 42) swimming significantly shorter distances than nontransgenic (n = 12) and those expressing EGFP-ataxin-3-23Q in motor neurons (n = 22; Fig. 4D; p < 0.03 from Tukey post hoc tests).

Motor dysfunction in adult transgenic MJD zebrafish. A, Monthly motor tracking revealed that EGFP-ataxin-3-84Q fish consistently traveled shorter distances during behavioral test than control and EGFP-ataxin-3-23Q fish. B, 12-month-old nontransgenic (NT), EGFP-ataxin-3-23Q (23Q), and EGFP-ataxin-3-84Q (84Q) zebrafish were tracked during 5 min of ambient light and their trajectory recorded, colors represent movements made at fast speed (red), slow speed (green), and while inactive (black). C, Quantification of total distance swum during the darkness response test revealed that EGFP-ataxin-3-84Q fish (n = 28) had impaired motor function (decreased swimming distances) than control (n = 13) and EGFP-ataxin-3-23Q fish (n = 28); **p < 0.004, one-way ANOVA and Tukey post hoc tests. D, Adult (3-month-old) transgenic zebrafish with motor neuron specific (driven by a miRNA 218-enhancer) expression of EGFP-ataxin-3-84Q swim significantly shorter distances than control and EGFP-ataxin-3-23Q (n = 12, 42 and 22 respectively; ∧p < 0.03, one-way ANOVA and Tukey post hoc test). Error bars represent mean ± SEM.
Human ataxin-3 is cleaved by calpain within transgenic MJD zebrafish
To confirm that human ataxin-3 protein was being expressed in our transgenic MJD zebrafish, we extracted protein lysates from whole larval transgenic zebrafish (and nontransgenic controls) and performed immunoblot analysis, probing with antibodies against ataxin-3. Immunoblot analysis confirmed that transgenic zebrafish express full-length human ataxin-3 protein of appropriate sizes (72 and 84 kDa for EGFP-ataxin-3-23Q and -84Q, respectively), as well as endogenous zebrafish ataxin-3 (34 kDa; Fig. 5A). Interestingly, smaller ataxin-3-positive cleavage fragments were also present, suggesting that human ataxin-3 undergoes proteolytic cleavage within our transgenic zebrafish, like that identified in MJD patient samples (Goti et al., 2004; Weber et al., 2017).

Human ataxin-3 undergoes proteolytic cleavage within transgenic MJD zebrafish. A, Immunoblot analysis transgenic zebrafish aged 6 dpf confirmed expression of full-length human ataxin-3 protein of appropriate sizes (72 and 84 kDa for EGFP-ataxin-3-23Q and EGFP-ataxin-3-84Q respectively) and endogenously expressed zebrafish ataxin-3 (34 kDa). Interestingly smaller ataxin-3-positive cleavage fragments were also present, suggestive of proteolytic cleavage. B, Quantification of the intensity of ataxin-3-positive cleavage fragments at 6 dpf revealed significantly more of these cleavage fragments (expressed as a proportion of total human ataxin-3) in the EGFP-ataxin-3-84Q samples compared with EGFP-ataxin-3-23Q (n = 11–13); *p = 0.012. FL, Full-length; CF, cleavage fragment; ZF, zebrafish. Error bars represent mean ± SEM.
Quantification of the intensity of the ataxin-3-positive cleavage fragment within multiple immunoblots revealed a similar amount of cleavage fragment in 3 dpf EGFP-ataxin-3-23Q and -84Q zebrafish (data not shown). However quantification of 13 blots from 6 dpf zebrafish revealed that more cleaved ataxin-3 was present in EGFP-ataxin-3-84Q than EGFP-ataxin-3-23Q samples at that age (t(22) = −2.87, p = 0.012, t test; Fig. 5B).
To identify the cause of ataxin-3 proteolysis in our transgenic zebrafish, we tested the effect of treating these zebrafish with a range of protease inhibitor compounds. Control (nontransgenic) or EGFP-ataxin-3-84Q larvae were exposed from 1 to 3 dpf with DMSO (vehicle control) or a chosen protease inhibitor. The protease inhibitor compounds tested included calpain inhibitor compounds (ALLN or calpeptin), a pan-caspase inhibitor (zVAD-fmk), and a cathepsin inhibitor (CAT-1). Proteins lysates were prepared from the zebrafish and immunoblotted for ataxin-3 and cleaved spectrin (decreased levels of cleaved spectrin confirm inhibition of the protease calpain; Fig. 6A). Treatment with calpeptin (calpain I/II inhibitor, 25–100 μm) until 3 dpf produced a dose-dependent decrease in the amount of ataxin-3 cleavage fragments and an increase in the amount of full-length ataxin-3. Exposure to 25–100 μm ALLN (calpain I inhibitor) produced a similar result to calpeptin treatment. Treatment with either a pan-caspase inhibitor compound, zVAD-fmk (10–40 μm) or cathepsin inhibitor (CAT-1) failed to decrease the amount of cleavage fragments or preserve levels of full-length ataxin-3. Quantification of ataxin-3 band intensities from separate immunoblots (n = 3–6) revealed a significant effect of calpeptin treatment on levels of full-length ataxin-3 (F(4,19) = 8.019, p = 0.001, ANOVA) and ataxin-3 cleavage fragments (F(4,19) = 14.357, p < 0.0001, ANOVA). Tukey post hoc analysis revealed that 25 and 50 μm calpeptin significantly increased levels of full-length ataxin-3 (p < 0.04; Fig. 6B) and 100 μm significantly decreased the amount of ataxin-3 cleavage products (p = 0.031; Fig. 6C), both compared with DMSO treatment. Treatment with ALLN produced a similar trend of decreased ataxin-3 cleavage fragments, but the differences in band intensity did not reach significance. Treatment with zVAD-fmk or CAT-1 had no effect on levels of cleavage fragments or the full-length protein. This indicates that ataxin-3 proteolysis is calpain-dependent and not dependent on other proteases such as caspases or cathepsins.

Human ataxin-3 proteolysis is calpain dependent. A, The effect of calpain or caspase inhibition on the presence of ataxin-3 cleavage fragments at 3 dpf was examined by immunoblotting protein lysates from control or EGFP-ataxin-3-84Q larva following 2 d of treatment with DMSO, calpain inhibitor compounds (ALLN or calpeptin), a pan-caspase inhibitor (zVAD-fmk), or a cathepsin inhibitor (CAT-1). The immunoblot was probed with an antibody against cleaved spectrin, ataxin-3, and a loading control (β-actin or GAPDH). The first lane of each immunoblot contained control samples (nontransgenic) and the subsequent lanes contained EGFP-ataxin-3-84Q samples treated with DMSO and increasing concentrations of the protease inhibitor. Calpeptin (50, 100, 200 μm) treatment produced a dose-dependent increase in the amount of full-length ataxin-3 and decrease in ataxin-3 cleavage product. Exposure to ALLN produced a similar result to calpeptin treatment. Treatment with zVAD-fmk or CAT-1 failed to preserve full-length ataxin-3 or decrease the amount of cleavage fragments. B, Quantification of levels of full-length ataxin-3 within separate immunoblots (n = 3–6) for treatment with each drug revealed that 25–50 μm calpeptin significantly increased the amount of full-length ataxin-3 compared with DMSO treatment, *p < 0.04. C, Quantification of the level of ataxin-3 cleavage fragments within samples of zebrafish treated with each protease inhibitor compound revealed a trend of decreased level of cleavage fragments following treatment with calpain inhibitor compounds, with 100 μm calpeptin significantly decreasing levels of the cleavage fragment (p = 0.031). FL, Full-length; CF, cleavage fragment; ZF, zebrafish. Error bars represent mean ± SEM.
Calpeptin treatment improves motor function and induces autophagy in MJD zebrafish larva
To investigate whether the presence of ataxin-3 cleavage fragments is a disease-causing feature we wanted to test whether preventing ataxin-3 cleavage with calpeptin treatment prevented the motor phenotype seen in our transgenic MJD zebrafish. Because the motor phenotype is present in the MJD zebrafish at 6 dpf, we treated the zebrafish with DMSO or calpeptin for a longer period, until 6 dpf, and then assessed their motor function. The total distance traveled during a 4 min darkness response test was measured for DMSO-treated nontransgenic, EGFP-ataxin-3-23Q and -84Q larvae, and EGFP-ataxin-3-84Q larvae treated with increasing concentrations of calpeptin (6.25–100 μm; Fig. 7A). One-way ANOVA identified a significant effect of group (F(7,631) = 7.616, p < 0.0001). The DMSO-treated EGFP-ataxin-3-84Q (n = 151) swam shorter total distances than the DMSO-treated nontransgenic (n = 68) and EGFP-ataxin-3-23Q (n = 69) larvae (Tukey post hoc, p < 0.023). Treatment of EGFP-ataxin-3-84Q larvae with calpeptin (25–100 μm) significantly improved their swimming compared with DMSO-treated EGFP-ataxin-3-84Q (n = 71–122, Tukey post hoc, p < 0.001). Treatment of the EGFP-ataxin-3-84Q zebrafish with the calpain inhibitor compound ALLN produced similar improvements in distances swum (data not shown).

Calpeptin treatment rescues the motor phenotype of MJD zebrafish at 6 dpf, concurrent with a decrease in human ataxin-3 protein levels and induction of autophagy. A, The total distance swum during a 4 min darkness response test is shown for DMSO-treated nontransgenic (n = 69), EGFP-ataxin-3-23Q (n = 68), and EGFP-ataxin-3-84Q larva (n = 151), and EGFP-ataxin-3-84Q larva treated with increasing concentrations of calpeptin (6.25–100 μm: n = 26–105). DMSO-treated EGFP-ataxin-3-84Q swam significantly shorter distances than the DMSO-treated nontransgenic and EGFP-ataxin-3-23Q larva (*significant p < 0.023). EGFP-ataxin-3-84Q larva treated with 25–100 μm Calpeptin swam significantly longer distances compared with their DMSO-treated siblings; ***p < 0.001. B, Immunoblots of calpeptin-treated MJD zebrafish revealed that calpeptin treatment decreased levels of ataxin-3 in a dose-dependent manner. Probing the immunoblot for beclin-1, LC3-I/II, and p62 revealed that calpeptin also decreased levels of p62 and beclin-1 increased the amount of LC3-II. C, Quantification of band intensity of ataxin-3 cleavage fragment levels from separate immunoblots (n = 3–8) revealed a dose-dependent decrease, with a significant decrease following treatment with 50 and 100 μm calpeptin compared with DMSO-treated EGFP-ataxin-3-84Q zebrafish (**p < 0.008). D, Quantification of full-length ataxin-3 levels revealed a significant decrease in full-length ataxin-3 levels following treatment with 25–100 μm calpeptin compared with DMSO EGFP-ataxin-3-84Q (**p < 0.01). E, Quantification of the intensity of the p62 band revealed that a significant decrease in p62 levels following treatment with 25–100 μm calpeptin compared with DMSO-treated EGFP-ataxin-3-84Q, **p < 0.01, n = 3–8. F, ELISA assay of 6 dpf protein lysates confirmed that calpeptin treatment (50 μm) did significantly reduce levels of human ataxin-3 in EGFP-ataxin-3-84Q zebrafish compared with DMSO treatment (n = 4–5, *p = 0.036). FL, Full-length; CF, cleavage fragment; ZF, zebrafish. Error bars represent mean ± SEM.
We checked whether the amount of ataxin-3 cleavage fragments was still decreased after 5 d of calpeptin treatment using immunoblot analysis of protein lysates from 6 dpf DMSO and calpeptin-treated EGFP-ataxin-3-84Q. Treatment of EGFP-ataxin-3-84Q zebrafish with calpeptin resulted in decreased amounts of the ataxin-3 cleavage fragments compared with DMSO-treated, and surprisingly also decreased levels of full-length ataxin-3 (Fig. 7B). Probing the membrane for the autophagy markers beclin-1, LC3-II, and p62 revealed decreased levels of beclin-1 and p62 and increased levels of LC3-II. We quantified the ataxin-3 band intensities from separate ataxin-3 immunoblots (n = 3–8) and one-way ANOVA revealed a significant effect of calpeptin treatment on the amount of ataxin-3 cleavage fragments (F(7,31) = 6.703, p < 0.0001; Fig. 7C) and full-length ataxin-3 (F(7,31) = 7.279, p < 0.0001; Fig. 7D). Tukey post hoc analysis revealed that 50–100 μm calpeptin significantly decreased levels of ataxin-3 cleavage fragments (p < 0.008), and 25–100 μm calpeptin significantly decreased levels of full-length ataxin-3 (p < 0.01), both compared with DMSO treatment. Quantification of the intensity of the p62 bands from separate immunoblots (n = 3) also revealed an effect of treatment (F(7,31) = 6.884, p < 0.0001, ANOVA; Fig. 7E). Treatment with calpeptin (50 and 100 μm) significantly decreased levels of p62 compared with DMSO treatment (Tukey post hoc, p < 0.008), with treatment with 25 μm approaching significance (p = 0.053; Fig. 7E).
To confirm that calpeptin treatment had decreased the level of human ataxin-3 protein within the transgenic zebrafish we developed an ELISA assay to measure levels of human ataxin-3 protein in our samples. This ELISA quantification confirmed a significant effect of calpeptin treatment on levels of human ataxin-3 within our transgenic zebrafish (F(3,15) = 9.296, p = 0.001, ANOVA; Fig. 7F). Tukey post hoc analysis revealed that EGFP-ataxin-3-84Q zebrafish treated with calpeptin (50 μm) had significantly lower levels of human ataxin-3 protein than those treated with DMSO (n = 4–5; p = 0.036; Fig. 7F). In fact, calpeptin treatment had decreased levels of human ataxin-3 protein to that of nontransgenic animals (reduced from 0.876 to 0.028 ng/ml).
To ensure that the decreased levels of human ataxin-3 and endogenous zebrafish p62 protein were due to protein clearance, rather than altered gene expression, we performed real-time PCR of mRNA samples extracted from our DMSO and calpeptin-treated EGFP-ataxin-3-84Q larva and controls. First, we checked levels of human ATXN3 mRNA in control (nontransgenic), EGFP-ataxin-3-23Q and EGFP-ataxin-3-84Q (with and without 50 μm calpeptin treatment) 6 dpf samples. We found a significant effect of group (F(3,11) = 8.919, p = 0.002, ANOVA), but post hoc analysis revealed that all transgenic MJD zebrafish (including calpeptin-treated) had similar levels of human ATXN3 mRNA, but no human ATXN3 mRNA was detected in nontransgenic zebrafish (Fig. 8A; n = 3–4, p < 0.041). This demonstrates that calpeptin treatment had not affected ataxin-3 protein levels within EGFP-ataxin-3-84Q zebrafish through changes in human ATXN3 mRNA levels. Real-time PCR for zebrafish p62 (SQSTM1) mRNA levels in samples from nontransgenic controls, EGFP-ataxin-3-23Q and EGFP-ataxin-3-84Q (with and without 50 μm calpeptin treatment) revealed an effect of experimental group (F(3,11) = 9.272, p = 0.002, ANOVA) and post hoc analysis identified that EGFP-ataxin-3-84Q zebrafish had higher p62 mRNA levels than control and EGFP-ataxin-3-23Q larva (n = 3–4, Tukey post hoc p < 0.031; Fig. 8B). Calpeptin treatment did not affect the p62 mRNA levels of EGFP-ataxin-3-84Q zebrafish (p = 0.976; Fig. 8B). This suggests that the decrease in p62 levels seen following calpeptin treatment is due to protein degradation processes rather than gene downregulation.

Real-time PCR revealed that changes in human ataxin-3 and p62 levels following calpeptin treatment were not present at an mRNA level. A, Real-time PCR of human ATXN3 mRNA levels in control (nontransgenic, n = 3), EGFP-ataxin-3-23Q (n = 3) and EGFP-ataxin-3-84Q (with and without 50 μm calpeptin treatment, n = 3 and 4, respectively) revealed similar human ataxin-3 levels in all transgenic MJD zebrafish samples. All transgenic ataxin-3 zebrafish groups had significantly higher human ATXN3 levels than control (∧p < 0.041), with no significant decrease in human ATXN3 mRNA levels in EGFP-ataxin-3-84Q larva receiving calpeptin (50 μm) treatment. B, Real-time PCR of p62 mRNA levels in control, EGFP-ataxin-3-23Q and EGFP-ataxin-3-84Q (with and without calpeptin treatment) samples revealed that EGFP-ataxin-3-84Q zebrafish have higher p62 mRNA levels than control and EGFP-ataxin-3-23Q larva; #p < 0.031. Calpeptin treatment did not effect the p62 mRNA levels of EGFP-ataxin-3-84Q zebrafish. n values were the same as in A. Error bars represent mean ± SEM.
We then cotreated EGFP-ataxin-3-84Q zebrafish larvae with calpeptin and the autophagy flux inhibitor, chloroquine (3 mm) to confirm that the low levels of human ataxin-3 and p62 protein found following calpeptin treatment were due to induced autophagic clearance. Cotreatment of the EGFP-ataxin-3-84Q zebrafish larva with calpeptin and chloroquine prevented the decreases in human ataxin-3 (both full-length and cleavage fragment) and p62 levels produced by calpeptin treatment alone (Fig. 9A), indicating that the removal of human ataxin-3 and p62 is dependent on the autophagy pathway. The chloroquine cotreatment also prevented the improvement in motor function produced by calpeptin treatment in these EGFP-ataxin-3-84Q zebrafish (F(2,325) = 16.414, p < 0.0001, ANOVA, followed by Tukey post hoc analysis of chloroquine/calpeptin versus calpeptin alone, p < 0.0001; Fig. 9B).

Autophagy inhibition prevents the beneficial effect of calpain inhibition. A, Immunoblot analysis of 6 dpf EGFP-ataxin-3-84Q larva treated with vehicle (DMSO, first 3 lanes) compared with calpeptin (50 μm) and cotreatment of calpeptin (50 μm) and chloroquine (3 mm). Whereas calpeptin treatment resulted in removal of human ataxin-3, cotreatment with calpeptin and the autophagy inhibitor chloroquine resulted in no loss of human ataxin-3. The addition of chloroquine also prevented the decrease in p62 levels seen with calpeptin treatment alone. B, Measuring the total distance swum by 6 dpf EGFP-ataxin-3-84Q zebrafish larva during 4 min response to darkness test revealed that although calpeptin treatment (50 μm) increased the distance swum by EGFP-ataxin-3-84Q larva (*p = 0.002), addition of chloroquine to the calpeptin treatment blocked the beneficial effect of calpeptin (**p < 0.0001). FL, full-length; CF, cleavage fragment; ZF, zebrafish. Error bars represent mean ± SEM.
Discussion
In this study we have developed the first transgenic zebrafish model of MJD. We aimed to develop a novel small animal model of MJD to aid testing of possible disease treatments and chose zebrafish because of the advantages that zebrafish offer for drug treatment studies.
Characterizing the disease phenotypes that developed in our transgenic zebrafish revealed a range of differences between the transgenic zebrafish expressing EGFP-ataxin-3-23Q and EGFP-ataxin-3-84Q, including motor impairment at ages as early as 6 dpf, which provided an assay that could be used for testing disease treatments. We also found that EGFP-ataxin-3-84Q zebrafish had impaired movement throughout aging, particularly at middle age, relevant to the human disease, and consistent with rodent models of MJD (Colomer Gould, 2012).
The motor phenotype that we identified in our EGFP-ataxin-3-84Q zebrafish larvae involved a lack of accelerated swimming during periods of darkness. We found that at 6 dpf our EGFP-ataxin-3-23Q and -84Q zebrafish have similar swimming speeds and behavior at baseline (in a well-lit setting), but in darkness the EGFP-ataxin-3-23Q and nontransgenic accelerate due to an escape response (MacPhail et al., 2009), whereas the EGFP-ataxin-3-84Q do not. A similar motor phenotype was present in zebrafish larvae that only expressed EGFP-ataxin-3-84Q in motor neurons. This finding indicates that the motor phenotype was not an aberrant stress response, as higher centers in the brain that control stress were not expressing the human ataxin-3-84Q protein in the motor neuron specific line. Further, examination of the motor neuron morphology revealed decreased axonal lengths for motor neurons expressing EGFP-ataxin-3-84Q compared with EGFP-ataxin-3-23Q and nontransgenic controls. Similar axonopathies have been reported in zebrafish expressing mutant proteins known to cause amyotrophic lateral sclerosis and related diseases (Lemmens et al., 2007; Laird et al., 2010, 2016; Hogan et al., 2017). These finding suggest that motor neurons play an important role in the motor impairment produced by polyQ-expanded ataxin-3 and that cell autonomous toxicity is produced by the polyQ expanded ataxin-3.
Our transgenic MJD zebrafish also developed signs of ataxin-3 neuropathology. Immunostaining sections from the brains of the transgenic MJD zebrafish at the 12-month-old endpoint revealed a neuritic beading-staining pattern (positive for both ataxin-3 and polyglutamine) within the medulla of EGFP-ataxin-3-84Q zebrafish. This staining pattern was not present in the EGFP-ataxin-3-23Q or control fish. Interestingly, a neuritic beading-staining pattern has been previously reported within the medullary white matter of patients with MJD (Seidel et al., 2010) and Huntington's disease (Rüb et al., 2014). We did not find any signs of neuronal intranuclear inclusions in our ataxin-3 or polyglutamine stained zebrafish brains and spinal cords, which contrasts with previous findings in transgenic MJD mice (Cemal et al., 2002; Goti et al., 2004; Bichelmeier et al., 2007), but this may be because our staining was performed on samples from 12-month-old zebrafish (middle aged), not from natural end-stage disease. Like other animal models of MJD, our MJD zebrafish also have decreased survival compared with EGFP-ataxin-3-23Q and nontransgenic control fish. Interestingly, the EGFP-ataxin-3-23Q also had decreased survival compared with nontransgenic controls, perhaps due to increased total ataxin-3 levels (zebrafish plus human ataxin-3).
Another relevant proteinopathy identified within our transgenic MJD zebrafish was the presence of ataxin-3-positive cleavage fragments within immunoblots of protein lysates extracted from the transgenic MJD zebrafish larva. Although these fragments were present within samples from all transgenic ataxin-3 zebrafish, the EGFP-ataxin-3-84Q samples contained a significantly higher proportion of cleavage fragments than the EGFP-ataxin-3-23Q samples. Similar ataxin-3 cleavage fragments have previously been reported in MJD patient samples (Goti et al., 2004; Weber et al., 2017), experimental animals (Colomer Gould et al., 2007; Hubener et al., 2013), and MJD cell cultures (Koch et al., 2011; Hubener et al., 2013; Weber et al., 2017). Although there was some variability in the amount of full-length or cleavage fragments present within different samples, no correlation was found between the level of cleavage fragments or full-length ataxin-3 and the degree of motor impairment.
To confirm that the ataxin-3 fragments seen in vivo within our MJD zebrafish protein lysates were the product of proteolytic cleavage we tested whether treatment with protease inhibitor compounds could prevent their formation. Treatment with the calpain inhibitor calpeptin from 24 hpf to 3 dpf resulted in preservation of full-length ataxin-3. This result is consistent with previous reports that treatment with calpain inhibitor compounds could prevent human ataxin-3 from being cleaved (Haacke et al., 2007; Koch et al., 2011), including a report that treating a mouse model of MJD with the calpain inhibitor compound BDA-410 reduced levels of cleaved human ataxin-3 and improved fine motor function and grip strength (Simões et al., 2014). A recent study by Weber et al. (2017) also elucidated the sites of calpain cleavage within the ataxin-3 protein and demonstrated that the cleavage fragments are aggregation prone and neurotoxic.
Exposing the MJD zebrafish (EGFP-ataxin-3-84Q) to calpeptin (25–100 μm) until 6 dpf improved the motor function of the MJD zebrafish to the same level as the ataxin-3 (23Q), and immunoblot analysis at 6 dpf revealed that calpeptin treatment had resulted in complete removal of the human ataxin-3. The decrease in full-length ataxin-3 is closely linked with the concentration that improved the motor behavior of the MJD zebrafish. Quantification of the level of human ataxin-3 protein in the zebrafish samples using an ELISA assay confirmed a complete removal of the human ataxin-3 protein in the calpeptin-treated MJD zebrafish. This is the first time that complete clearance of polyQ expanded human ataxin-3 protein has been achieved by treatment with a calpain inhibitor compound in vivo. We confirmed via real-time PCR that the decreased ataxin-3 protein levels were not due to decreased human ATXN3 mRNA levels, suggesting that calpeptin treatment results in removal of human ataxin-3 through a protein clearance pathway.
We then examined levels of markers for the protein quality control pathway macroautophagy (beclin-1, LC3-II, and p62) in the EGFP-ataxin-3-84Q larva, with and without calpeptin treatment. We found that levels of the autophagy adaptor protein p62 and beclin-1 were dramatically decreased by calpeptin treatment, whereas LC3-II levels were elevated. These results indicate that autophagic flux was induced (indicated by depleted p62 levels) and that autolysosomes were being cleared successfully by calpeptin treatment. Treatment with the autophagy inhibitor chloroquine, in addition to calpeptin, blocked the decrease in ataxin-3 levels (both full-length ataxin-3 and the cleavage fragments), as well as p62 protein levels, and prevented the rescue of motor function seen with calpeptin treatment alone.
These results show that the clearance of ataxin-3 protein and improved motor function produced by calpeptin treatment is dependent on activity of the autophagy pathway. This finding is in line with the previous finding of Williams et al. (2008) that autophagy is activated by calpain inhibition in COS-7 cells transfected with mutant huntingtin containing 74Q. Likewise, Simões et al. (2014) have reported that treatment of cultured MJD granule cells with a novel calpain inhibitor BDA-410 resulted in decreased presence of ataxin-3 cleavage fragments as well as a small decrease in full-length ataxin-3 levels. Although Simões et al. (2014) did not explore what had caused the decrease in ataxin-3 protein following BDA-410 treatment, they did suggest that it was likely protein clearance by a quality control pathway.
The ability of calpain inhibition to completely remove the polyQ expanded human ataxin-3, as seen within our study, suggests that calpain inhibition warrants further investigation as a treatment for MJD. This is a promising avenue for disease treatment because the effect of polyQ expansion within ataxin-3 appears to be through a toxic gain-of-function mechanism (Paulson et al., 1997). The beneficial effect of removal of polyQ expanded ataxin-3 from cells through gene silencing (Alves et al., 2008, 2010) and autophagy induction (Nascimento-Ferreira et al., 2011, 2013) were both previously demonstrated to have potential for the treatment of MJD, suggesting that decreased ataxin-3 levels may be tolerated. Therefore drug-mediated autophagy induction is an important option to investigate for MJD treatment. A recent report by Onofre et al. (2016) identified that autophagy function is impaired in MJD patients (decreased autolysosome formation) further suggesting that drug-mediated autophagy induction may be helpful in the treatment of MJD.
In summary, we report the generation and characterization of the first transgenic zebrafish model of MJD. We demonstrate that this model develops relevant signs of the human disease including the development of motor impairment, decreased survival, formation of ataxin-3 cleavage fragments, and ataxin-3 neuropathology. We found that treatment with calpain inhibitor compounds had a protective effect on our in vivo model of MJD and that the protective effect of calpain inhibition was likely autophagy induction and removal of the human ataxin-3 protein, rather than just prevention of ataxin-3 proteolysis. Our results demonstrate the importance of further investigation into the beneficial effect of calpain inhibition for the treatment of MJD. Finally, we propose that by using small animal models of MJD, such as our transgenic MJD zebrafish, we can test whether other drugs may have similar effects and have potential as a much needed treatment for MJD.
Footnotes
This work was supported by the National Health and Medical Research Council of Australia (AP1069235), the MJD Foundation and Anindilyakwa Land Council, Australia, Macquarie University Research Development Grant, the Swedish SCA Network, and The Snow Foundation and BitFury (N.J.C.). We thank Henry Paulson for the MJD antibody and EGFP-ataxin-3 plasmids, Reinhard Köster for the HuC-GAL4 and bidirectional UAS constructs, and staff members (past and present) of the zebrafish facilities at the Brain and Mind Centre and Macquarie University for the care of the zebrafish described within this paper.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Angela Laird, Centre for Motor Neuron Disease Research, Faculty of Medicine & Health Sciences, 2 Technology Place, Macquarie University, Sydney, NSW 2109, Australia. angela.laird{at}mq.edu.au





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}