

Abstract
During vertebrate eye morphogenesis, a transient fissure forms at its inferior part, known as the optic fissure. This will gradually close, giving rise to a healthy, spherical optic cup. Failure of the optic fissure to close gives rise to an ocular disorder known as coloboma. During this developmental process, Foxg1 is expressed in the optic neuroepithelium, with highest levels of expression in the nasal optic stalk. Foxg1−/− mutant mice have microphthalmic eyes with a large ventral coloboma. We found Wnt8b expression upregulated in the Foxg1−/− optic stalk and hypothesized that, similar to what is observed in telencephalic development, Foxg1 directs development of the optic neuroepithelium through transcriptional suppression of Wnt8b. To test this, we generated Foxg1−/−;Wnt8b−/− double mutants of either sex and found that the morphology of the optic cup and stalk and the closure of the optic fissure were substantially rescued in these embryos. This rescue correlates with restored Pax2 expression in the anterior tip of the optic fissure. In addition, although we do not find evidence implicating altered proliferation in the rescue, we observe a significant increase in apoptotic cell density in Foxg1−/−;Wnt8b−/− double mutants compared with the Foxg1−/− single mutant. Upregulation of Wnt/β-catenin target molecules in the optic cup and stalk may underlie the molecular and morphological defects in the Foxg1−/− mutant. Our results show that proper optic fissure closure relies on Wnt8b suppression by Foxg1 in the nasal optic stalk to maintain balanced apoptosis and Pax2 expression in the nasal and temporal edges of the fissure.
SIGNIFICANCE STATEMENT Coloboma is an ocular disorder that may result in a loss of visual acuity and accounts for ∼10% of childhood blindness. It results from errors in the sealing of the optic fissure (OF), a transient structure at the bottom of the eye. Here, we investigate the colobomatous phenotype of the Foxg1−/− mutant mouse. We identify upregulated expression of Wnt8b in the optic stalk of Foxg1−/− mutants before OF closure initiates. Foxg1−/−;Wnt8b−/− double mutants show a substantial rescue of the Foxg1−/− coloboma phenotype, which correlates with a rescue in molecular and cellular defects of Foxg1−/− mutants. Our results unravel a new role of Foxg1 in promoting OF closure providing additional knowledge about the molecules and cellular mechanisms underlying coloboma formation.
Introduction
Vertebrate eye development is a multistep process that involves early specification of the eye field followed by bilateral evagination of the diencephalon, giving rise to the optic vesicle. The optic vesicle will then invaginate and form the optic stalk (OS) ventrally and the two-layered optic cup (OC), with an outer layer known as retinal pigment epithelium (RPE) and the inner (close to the lens) neural retina. As the OC grows, the apposed edges of its inferior part, known as the optic fissure (OF), come in close proximity and fuse together to give rise to a fully formed spherical eye structure (Chow and Lang, 2001; Lamb et al., 2007; Fuhrmann, 2010).
The cellular and molecular mechanisms that control OF closure are not fully understood. Errors underlying this process lead to an ocular disorder known as coloboma. Although environmental factors have been implicated in defective OF closure, it is well established that mutations in genes that are normally expressed in the optic vesicle give rise to coloboma (Gregory-Evans et al., 2004; Chang et al., 2006; Williamson and FitzPatrick, 2014).
The study of animal models with colobomatous phenotypes has allowed a better understanding regarding the cellular and molecular basis of the disorder. Among the cellular processes underlying OC formation and OF closure are cell proliferation and apoptotic cell death.
Among the molecules that are involved in coloboma formation is the transcription factor Pax2. Mutations in the PAX2 gene in humans lead to the renal-coloboma syndrome, characterized by renal and ocular malformations, including optic nerve coloboma (Schimmenti et al., 1995; Schimmenti, 2011). Loss of Pax2 in mice leads to a coloboma phenotype due to inability of the edges of the OF to fuse (Torres et al., 1996). New molecular players are continuously added to the list of genes leading to coloboma in mice and humans (Gregory-Evans et al., 2004; Chang et al., 2006; Williamson and FitzPatrick, 2014), including proteins implicated in the HH (Wen et al., 2015), Fgf (Cai et al., 2013), Bmp (Huang et al., 2015), RA (Lupo et al., 2011), and Wnt (Liu and Nathans, 2008; Zhou et al., 2008; Lieven and Rüther, 2011; Liu et al., 2012, 2016; Alldredge and Fuhrmann, 2016) signaling pathways.
Foxg1 is forkhead box transcription factor expressed from early stages of mouse embryonic development in the developing nervous system and is specifically found in the telencephalon, optic chiasm, and retina (Xuan et al., 1995; Huh et al., 1999; Pratt et al., 2004; Fotaki et al., 2006, 2013; Tian et al., 2008). Mice with no functional Foxg1 (Foxg1−/− mutants) die at birth and show severe reduction in the size of telencephalic lobes and eyes (Xuan et al., 1995). In addition, Foxg1−/− eyes display a large ventral coloboma (Huh et al., 1999). Foxg1's role in the developing eye has not been studied in detail. We have recently shown that, in the mouse, Foxg1 is essential for controlling the size of the ciliary margin in the nasal peripheral retina and for suppressing Wnt/β-catenin signaling in this region (Fotaki et al., 2013).
Here, we examine the molecular and cellular causes of the coloboma phenotype of the Foxg1−/− mutant. We found that Wnt8b expression in the wild-type OS is upregulated in this mutant. We hypothesized that, similar to the telencephalon (Danesin et al., 2009), Foxg1 may normally suppress Wnt8b in the nasal OS for proper OC and/or OS formation to take place. We tested this by suppressing Wnt8b expression genetically in Foxg1−/−;Wnt8b−/− double mutants. In accordance with our hypothesis, in the double mutant, we found substantial rescue of the OF closure defect we observed in the Foxg1−/− single mutant. Our results reveal a novel mechanism of OF closure, which relies on Foxg1-mediated suppression of Wnt8b in the nasal OS to maintain balanced apoptosis and normal Pax2 expression in the nasal edges of the fissure.
Materials and Methods
Mice.
All experiments were done according to Home Office regulations (Edinburgh, United Kingdom).
Foxg1+/− heterozygote males were bred to F1 (CBAxC57/B6) females to produce Foxg1+/− heterozygous males and females as previously described (Fotaki et al., 2013). The Wnt8b+/− mice have been previously described (Fotaki et al., 2010). Foxg1+/−;Wnt8b+/− double heterozygotes were generated by intercrossing Foxg1+/− and Wnt8b+/− heterozygous mice.
Embryos.
To generate Foxg1−/− single or Foxg1−/−;Wnt8b−/− double-mutant embryos, timed matings were set up among Foxg1+/− heterozygote or Foxg1+/−;Wnt8b+/− double heterozygote male and female mice, respectively. The day the vaginal plug was detected in females was designated as E0.5.
No gross differences were detected in morphology or marker expression between wild-types (Foxg1+/+) and Foxg1+/− heterozygotes (data not shown). However, unless otherwise stated, in most cases when comparing Foxg1−/− homozygotes with controls, we used wild-type embryos.
Foxg1−/−;Wnt8b−/− double-mutant embryos were compared with two experimental groups: (1) a control group, consisting of wild-type (Foxg1+/+;Wnt8b+/+), single heterozygote (Foxg1+/−;Wnt8b+/+ or Foxg1+/+;Wnt8b+/−), or double heterozygote (Foxg1+/−;Wnt8b+/−) embryos; or (2) a group where the Foxg1 mutation was found in homozygosis (Foxg1−/−) and the Wnt8b allele was either wild-type (Wnt8b+/+) or heterozygous (Wnt8b+/−). This group was collectively named as the Foxg1−/−;Wnt8b+/± mutant group.
Genotyping of mice and embryos.
Foxg1+/− heterozygote mice express one copy of functional β-galactosidase under the control of the Foxg1 promoter and were distinguished from wild-types (Foxg1+/+) by PCR analysis based on detection of the lacZ allele (Xuan et al., 1995). For all embryos younger than E11.0, the Foxg1 mutation was detected by PCR using primers specific for the Foxg1-wild-type allele (Foxg1-wt-F: AGG CTG ACG CAC TTG GAG; Foxg1-wt-R: CAG GGG TTG AGG GAG TAG GT), resulting in an 856 bp PCR product and for the Foxg1-null allele (Foxg1-mt-F: GCT GGA CAT GGG AGA TAG GA; Foxg1-mt-R: GAC AGT ATC GGC CTC AGG AA), resulting in a 650 bp PCR product. For embryos older than E11.0, Foxg1−/− mutants were clearly distinguished by their phenotype as previously described (Fotaki et al., 2013).
The Wnt8b mutation was detected by PCR in both mouse and embryonic tissue as previously described (Fotaki et al., 2010).
Histology and cresyl violet staining.
Mouse embryos were collected on ice-cold PBS buffer and fixed in 4% PFA in 0.1 m phosphate buffer as previously described (Fotaki et al., 2006). Embryos were either embedded in a 1:1 mixture of OCT/sucrose (30%) for cryostat sectioning or in paraffin for microtome sectioning (Fotaki et al., 2013). Embryos used for cell counts were embedded in paraffin and cut at 7 μm horizontal sections. Some sections were stained with 0.2% of cresyl violet acetate (Sigma-Aldrich).
Riboprobe synthesis, in situ hybridization, immunohistochemistry, immunofluorescence, and X-gal staining.
Probes were labeled with digoxigenin according to the manufacturer's instructions (Roche). Riboprobes used for this study were for Axin2 (Fotaki et al., 2011), Bmp7 (Morcillo et al., 2006), Celsr3 (Tissir et al., 2005), Foxg1 (Fotaki et al., 2013), Foxd1 (Herrera et al., 2004), Fzd3 (Montcouquiol et al., 2006), Vangl (Montcouquiol et al., 2006), Vax1 (Bertuzzi et al., 1999), Wnt2b (Fotaki et al., 2013), Wnt3a, Wnt5a (Liu et al., 2003; Ang et al., 2004), and Wnt7b, Wnt8b, as described by Fotaki et al. (2011).
Previously described protocols were used for in situ hybridization, immunohistochemistry, immunofluorescence, and X-gal staining (Fotaki et al., 2008, 2011, 2013). Pax2 DAB immunohistochemistry was performed on already X-gal-stained sections of Foxg1+/− embryos (see Fig. 11A,B). DAB immunohistochemistry for β-gal was performed on sections that had already been reacted for Wnt8b in situ hybridization (see Fig. 3). Following incubation with the appropriate secondary biotinylated antibody, DAB immunohistochemistry was performed using the Vectastain ABC kit (Vector Laboratories), as previously described (Fotaki et al., 2006). In situ hybridization sections were in many cases counterstained with Nuclear Fast Red (Vector Laboratories). Immunofluorescence-reacted sections were counterstained with DAPI dilactate (2 μg/ml) (Sigma-Aldrich). All experiments were performed on at least 6 eyes from 3 different embryos for each experimental group (unless otherwise indicated).
All antibodies used for experiments are listed in Table 1. All Alexa-fluorescent antibodies were from Invitrogen/ThermoFisher Scientific.
Antibodies used in this study
Imaging.
DAB and in situ images were taken with a Leica DFC480 camera connected to a Leica DMNB epifluorescence microscope. Fluorescence images were taken with a Leica DM5500B automated upright microscope connected to a DFC360FX camera. Whole embryonic eyes were photographed with a Leica DFC420 camera connected to a Leica M165C stereomicroscope, all from Leica Microsystems. Confocal images were taken with an LSM 510 Axioskop (Leica Microsystems).
Labeling index (LI), mitotic index, apoptotic cell density, and Pax2 cell density counts.
To calculate the LI, we used Foxg1+/+ wild-type controls (n = 4 eyes from 3 different embryos) and Foxg1−/− mutants (n = 5 eyes from 3 different embryos). Using confocal images, we counted the total number of cells counterstained with DAPI and the total number of cells immunostained with BrdU from 3 or 4 dorsal to ventral sections from the nasal and temporal components of the retina. Cell counts were performed manually on merged stacks of confocal images using the software IMARIS 8.0.0 (Bitplane, RRID:SCR_007370), which allows image rotation, facilitating counts in the x-y-z-axis. LI was calculated as the ratio of BrdU-positive to the total number of cells in the nasal and temporal retinae.
To calculate the pHH3 cell surface density (mitotic index), we used Foxg1+/+ wild-type controls (n = 4 eyes from 3 different embryos) and Foxg1−/− mutants (n = 5 eyes from 3 different embryos). The total number of pHH3-positive cells was counted manually in the nasal and temporal retinae of each section from 3 or 4 dorsal to ventral sections. ImageJ (RRID:SCR_003070) was used to outline and calculate the perimeter of the apical surface of the nasal and temporal retinae, where counts were taken from and the volume of each section was calculated by multiplying the thickness of the section (7 μm) by the area.
To calculate the apoptotic cell density in our first experimental setup (Table 2), we used Foxg1+/+ wild-type controls (n = 5 eyes from 3 different embryos) and Foxg1−/− mutants (n = 5 eyes from 3 different embryos); and in our second experimental setup (Table 3), we used control (n = 6 eyes from 4 different embryos), Foxg1−/−;Wnt8b+/± (n = 4 eyes from 2 different embryos), and Foxg1−/−;Wnt8b−/− double mutant (n = 4 eyes from 3 different embryos) embryos. We counted cleaved caspase-3-positive cells from 3 to 5 middle to ventral sections from each eye where OF was detectable and omitted dorsal sections, as cell death detection in these was absent from all our experimental groups. The area of the nasal and temporal retinae, where counts were taken from, was traced using ImageJ, and the volume of each section was calculated by multiplying the thickness of the section (7 μm) by the area.
Apoptotic cell densities in Foxg1+/+ wild-type and Foxg1−/− mutant nasal and temporal retinaea
Apoptotic cell densities in control, Foxg1−/−;Wnt8b+/±, and Foxg1−/−;Wnt8b−/− nasal and temporal retinaea
To calculate the Pax2 cell density at the edges of the OF, we used control (n = 3 eyes from 3 different embryos); Foxg1−/−;Wnt8b+/± mutant (n = 4 eyes from 3 different embryos) and Foxg1−/−;Wnt8b−/− double mutant (n = 3 eyes from 3 different embryos) embryos. We counted Pax2-positive cells from 2 representative mid-sagittal sections within a square of 0.1 mm × 0.1 mm encompassing the edges of the fissure and the average values for the nasal and temporal retinae of each specimen were used for comparisons.
Corrected total cell fluorescence (CTCF) counts.
To quantitate Pax6 and Pax2 expression, we used controls (for E10.5: n = 5 eyes and for E11.5: n = 4 eyes from 3 different embryos); Foxg1−/−;Wnt8b+/± mutants (for E10.5: n = 6 eyes and E11.5: n = 4 eyes from 3 different embryos) and Foxg1−/−;Wnt8b−/− double mutants (for E10.5 and E11.5: n = 4 eyes from 3 different embryos) at E10.5 and E11.5. Using ImageJ, we outlined the nasal and temporal retinae; and for each section, we measured the area, the mean fluorescence, and the integrated fluorescent density, along with several adjacent background readings in 3–6 representative sections per specimen along the ventrodorsal axis. The most dorsal sections were not included in the study as their morphological differences in the mutants are more subtle than those observed in middle and ventral sections. For each section, we calculated the CTCF according to the following formula: CTCF = Integrated density − (Selected area × Mean fluorescence of background readings) (McCloy et al., 2014). The obtained values were divided by 1000.
Statistics.
To compare the LI, mitotic index, and apoptotic cell density between our different experimental groups, we assumed that this follows a normal distribution within the mouse population of the same genotype and that two values were different when the CIs did not intersect. The CIs were calculated using the mean values (LI, mitotic index, or apoptotic cell density), the SE, and the value of the normal distribution that corresponds to at least 95% confidence level (p ≤ 0.05), after applying the Bonferroni correction where appropriate. For our calculations, we used a sampling strategy. We used a two-stage sampling strategy (Shiver and Borders, 1996), in which the primary units are the mouse eyes and the secondary units are the eye sections. This sampling strategy, a widely used methodology in experimental fields such as Forestry, is applied, for example, to calculate total number of trees and/or ratios among specific species (Shiver and Borders, 1996).
Data analysis was performed using R software (version 3.1.2, RRID:SCR_001905) and the package survey for analysis of complex surveys (http://r-survey.r-forge.r-project.org/survey/). The package survey produces an estimate of the mean ratio (LI = Brdu-positive cells/total number of cells; cell density = cell count/volume) and its SE.
To compare the Pax6 and Pax2 CTCF values, we performed ANOVA followed by Bonferroni correction, using the averaged CTCF values along the ventrodorsal axis for the nasal and temporal retina of each specimen analyzed. A similar procedure was followed to calculate the Pax2-positive cell density in the nasal and temporal retinae. The degrees of freedom (df), the F value, and the p value were calculated using the software SPSS (IBM, SPSS Statistics 21, RRID:SCR_002865).
RNA extraction, cDNA synthesis, and PCR arrays.
The OCs of E11.0 wild-type and Foxg1−/− age-matched embryos were dissected out using fine-tip (0.125 mm) tungsten-dissecting probes (WPI) and snap frozen on dry ice. RNA was generated pooling together 6 eyes, dissected out from 3 to 5 different wild-type or Foxg1−/− mutant embryos, and was extracted using the RNeasy micro kit (QIAGEN) following the manufacturer's instructions; 150 ng of RNA from three independent samples of Foxg1+/+ wild-type and Foxg1−/− mutant OCs was used to synthesize cDNA, using the RT2 First Strand Kit (QIAGEN) according to the manufacturer's instructions. The cDNA was mixed with RT2 SYBR Green ROX qPCR mastermix (QIAGEN) according to the manufacturer's instructions, and the mixture was loaded on RT2 Profile PCR array plates for the mouse Wnt signaling pathway [PAMM-043Z] (QIAGEN). The plates were run on a StepOnePlus Real-Time PCR machine (Applied Biosystems/ThermoFisher Scientific). Results obtained from three plates for each group were further processed through the QIAGEN GeneGlobe Data Analysis Centre (http://www.qiagen.com/gb/shop/genes-and-pathways/data-analysis-center-overview-page/).
Results
Expression of Foxg1 in the developing OC and OS at E10.5
Foxg1 expression in the developing mouse retina has been described in detail at E12.5 and E14.5, after completion of OF closure (Huh et al., 1999; Pratt et al., 2004; Tian et al., 2008; Fotaki et al., 2013). To gain understanding about Foxg1 expression in the developing OC before the OF seals, we performed in situ hybridization at E10.5 on horizontal sections throughout the ventral-to-dorsal axis (Fig. 1). Foxg1 expression was detected in the nasal retina and RPE (OC) and in the OS in agreement with previous observations (Hatini et al., 1994). Foxg1 expression was higher in ventral and middle sections compared with dorsal sections (compare Fig. 1a–c with Fig. 1d). In ventral sections, Foxg1 expression appeared more extended toward the midline of the nasotemporal axis than in middle and dorsal sections and reached the nasal edge of the OF (Fig. 1a, arrow). Foxg1 expression in the OC was lower than that in the OS and telencephalon throughout the ventral-to-dorsal axis (i.e., compare intensity of telencephalic and OS to OC signal in Fig. 1A–C). Our results highlight high expression of Foxg1 in the nasal OS and ventral OC, both of which are involved in OF formation.

Foxg1 mRNA expression in the developing optic neuroepithelium. In E10.5 horizontal sections along the ventral-to-dorsal axis, Foxg1 is expressed in the nasal retina (r), RPE, and OS (A–D, a–d). Strong Foxg1 expression is detected in the telencephalon (Tel). a, Arrow indicates the forming OF. N-T, Nasal-temporal axis (b–d, dashed lines). pRPE, Presumptive RPE. Scale bars: A–D, 100 μm; a–d, 50 μm; A, Inset, 200 μm.
Increased Wnt8b expression in the Foxg1−/− OS at E10.5
In the mouse telencephalon, Foxg1 suppresses expression of Wnt molecules (Wnt2b, Wnt3a, Wnt5a, Wnt7b, and Wnt8b), normally confined to the dorsomedial telencephalon and/or cortical hem (Muzio and Mallamaci, 2005; Hanashima et al., 2007; and data not shown). We hypothesized that the same Wnt molecules are also upregulated in the developing optic vesicle leading to defects in OF closure. Using in situ hybridization, we examined expression of these Wnts in wild-type E12.0-E12.5 horizontal sections to determine expression in the OC and OS.
All five Wnt molecules examined were expressed in the dorsomedial telencephalon and/or cortical hem (Fig. 2A,C,E,G,I), as previously described (Richardson et al., 1999; Muzio and Mallamaci, 2005; Fotaki et al., 2010; and data not shown). In the optic neuroepithelium, Wnt8b was detected in a small domain in the OS (Fig. 2B,b, arrowhead) (Roy et al., 2013). Wnt7b-positive cells were observed in the lens (Fig. 2D,d) (Liu et al., 2003; Ang et al., 2004), whereas Wnt2b expression was confined in the peripheral RPE and ciliary margin (Fig. 2F,f) (Liu et al., 2003; Fotaki et al., 2013). Wnt5a was detected in the eyelid epithelium (Fig. 2H,h), as previously reported (Liu et al., 2003; Kumar and Duester, 2010), whereas Wnt3a was not detected in any part of the developing OC or stalk or surrounding periocular mesenchyme (Fig. 2J,j) (Liu et al., 2003).

Expression of Wnt molecules in the developing telencephalon and optic neuroepithelium. E12.5 wild-type horizontal sections depicting expression of Wnt8b (A), Wnt7b (C), Wnt2b (E), Wnt5a (G), and Wnt3a (I) in the dorsomedial telencephalon (dmTel) and cortical hem (ch). Wnt8b is detected in the OS (B, b, arrowhead) and the hypothalamic optic recess (or) (b, arrow). Wnt7b is expressed in the lens (D, d). Wnt7b is also expressed strongly in the diencephalic preoptic area (POA) and anterior hypothalamus (AH) (D). Wnt2b is expressed in the ciliary margin (CM) and peripheral RPE (F, f). Wnt5a is detected in the eyelid epithelium (H, arrow) but, similar to Wnt3a, it is not detected within the optic neuroepithelium (H, h, J, j). Scale bars: A, C, E, G, I, 400 μm; B, D, F, H, J, 200 μm; b, d, f, h, j, 100 μm.
We then studied expression of Wnt2b, Wnt7b, and Wnt8b, which were found expressed in the optic neuroepithelium, in E10.5 horizontal sections of control and Foxg1−/− mutants by means of in situ hybridization. No differences were observed in Wnt2b and Wnt7b expression in the E10.5 OC between wild-types and Foxg1−/− mutants (data not shown). Similar to E12.5, at E10.5 Wnt8b expression in the wild-type developing optic vesicle was restricted to a small domain in the OS (Fig. 3A,a). However in the Foxg1−/− mutant, Wnt8b expression at and near the OS region was found expanded (Fig. 3A′,a′, arrowheads). In controls, Wnt8b expression was found in both the nasal and temporal OS; and nasally it was expressed in Foxg1-positive cells (reflected by positive β-gal staining, expressed under the control of the Foxg1 promoter; Fig. 3B,b). In Foxg1−/− mutants, Wnt8b expansion was observed in the nasal OS, where Foxg1 is normally expressed (Fig. 3B′,b′).

Upregulation of Wnt8b expression in the nasal OS in the Foxg1−/− mutant at E10.5. Wnt8b is expressed in the dorsomedial telencephalon (Tel) (A) and in a small domain in the OS of controls (a, B, b, bracketed area) and is upregulated in the Foxg1−/− mutant telencephalon (A′) and in the nasal OS (a′, B′, b′, arrows) where Foxg1 would normally be expressed (B, b, B′, b′, β-gal staining). Scale bars: A, A′, 200 μm; a, a′, B, B′, 100 μm; b, b′, 50 μm.
Our results indicate that loss of Foxg1 leads to an upregulation of Wnt8b in the developing nasal OS, in a domain where Foxg1 is normally expressed, suggesting that Foxg1 may normally suppress Wnt8b function in this region.
Significant rescue of the coloboma phenotype of the Foxg1−/− mutant in a Wnt8b-null genetic background
Based on expansion of Wnt8b expression in the Foxg1−/− OS at E10.5 (Fig. 3) and the previously described repressor activity of foxg1 on wnt8b in the zebrafish telencephalon (Danesin et al., 2009), we hypothesized that upregulated Wnt8b expression in the OS causes coloboma in the Foxg1−/− mutant. To test our hypothesis, we crossed male and female mice, double heterozygous for the Foxg1 (Foxg1+/−) (Xuan et al., 1995) and Wnt8b (Wnt8b+/−) (Fotaki et al., 2010) alleles, to generate double homozygous embryos Foxg1−/−;Wnt8b−/−.
We first assessed the phenotype of Wnt8b−/− mutant OCs at E15.5. Cresyl violet staining showed similar OC morphology between wild-type and Wnt8b−/− mutants and complete closure of the OF (Fig. 4A,A′). In addition, double immunofluorescence for markers of the proliferating (BrDU and Vsx2) and differentiating (Tuj1 and Islet1) retinal layers did not reveal any gross differences between wild-types and Wnt8b−/− mutants (Fig. 4B,B′,C,C′).

The Wnt8b−/− null mutant shows normal OC morphology and marker expression. Horizontal sections of E15.5 wild-types (Wnt8b+/+) (A–C) and Wnt8b−/− mutants (A′–C′) do not reveal any gross differences among genotypes. Cresyl violet wild-type (A) and Wnt8b−/− mutant (A′) sections reveal normal OC morphology. Double immunofluorescence for BrdU and Tuj1 (B, B′) and Vsx2 and Islet1 (C, C′) shows that the proliferating, outer neuroblastic layer (onbl) (BrdU- and Vsx2-positive cells) and differentiating, inner neuroblastic layer (inbl) (Tuj1- and Islet1-positive cells) retinal layers are similar in wild-types (B, C) and Wnt8b−/− mutants (B′, C′). Scale bars: A, A′, 200 μm; B, B′, C, C′, 200 μm.
We then examined the eye phenotype of control, Foxg1−/−;Wnt8b+/+ or Foxg1−/−;Wnt8b+/− (collectively designated as Foxg1−/−;Wnt8b+/± mutants) and Foxg1−/−;Wnt8b−/− double mutants at E15.5, when OF closure has normally been completed. Control eyes showed normal OC morphology and complete OF closure (Fig. 5A). Foxg1−/−;Wnt8b+/± mutants displayed microphthalmia and ventral coloboma (Fig. 5A′), as previously described (Huh et al., 1999). As predicted by our hypothesis, Foxg1−/−;Wnt8b−/− double mutants displayed a spherical-shaped OC and rescue of the coloboma phenotype (Fig. 5A″).

Rescue of the coloboma phenotype of the Foxg1−/− mutant in a Foxg1−/−;Wnt8b−/− null background. OC images of control embryos showing normal OF closure (A), Foxg1−/−;Wnt8b+/+ single mutants with a large ventral coloboma (A′), and Foxg1−/−;Wnt8b−/− double mutants displaying rescue of the coloboma phenotype (A″). Cresyl violet-stained sections of control (B), Foxg1−/−;Wnt8b+/+ single (B′), and Foxg1−/−;Wnt8b−/− double mutants (B″) revealed that the double-mutant OC and optic nerve (B″, arrow) resemble that of the control (B, arrow), rather than that of the single mutant (B′). In the single mutant, the OS does not form normally as indicated by B′ (two arrows), resulting in abnormal formation of the optic nerve. High-power images of coronal sections of control (C–E) and Foxg1−/−;Wnt8b−/− double mutants (C′–E′) in anterior (C, C′), mid-lenticular (D, D′), and posterior (E, E′) levels show failure of the OF to seal completely in the double mutant anterior and mid-lenticular levels (C′ and D′, respectively, arrowheads). Scale bars: A, A′, A″, 20 μm; B, B′, B″, 200 μm; C–E, C′-E′, 100 μm.
Gross analysis of OC morphology did not reveal a large ventral gap in any of the Foxg1−/−;Wnt8b−/− double-mutant eyes examined (n = 12). Cresyl violet staining showed a spherical OC and optic nerve in control embryos (Fig. 5B), an abnormal OC with an elongated retina with foldings and with no clearly identifiable optic nerve in Foxg1−/−;Wnt8b+/+ mutants (100% of all eyes examined; n = 22) (Fig. 5B′) (Pratt et al., 2004) and an OC and optic nerve resembling that of controls in Foxg1−/−;Wnt8b−/− double mutants (Fig. 5B″). Sequential coronal sections of Foxg1−/−;Wnt8b−/− double-mutant eyes (n = 6) revealed that 50% of the OCs had complete OF closure anteriorly and at the mid-lenticular level and 100% at the posterior pole (Fig. 5C′–E′). At anterior and mid-lenticular levels, 50% of the OCs showed unfused tips in the form of small gaps or indentations (Fig. 5C′,D′), which were never observed in controls (n = 18 eyes) (Fig. 5C,D). The indentations anteriorly included both the interior and exterior aspects of the Foxg1−/−;Wnt8b−/− neural retina (Fig. 5C′, arrowheads), whereas at mid-lenticular level it either included both (data not shown) or just the interior aspect (Fig. 5D′, arrowhead). The edges of the OF of the Foxg1−/−;Wnt8b−/− OC at the posterior pole were fused and resembled those of controls (Fig. 5E,E′).
Our results collectively show a spectacular rescue of the OC and OS/nerve morphology and of the OF closure defect observed in the Foxg1−/− mutant in a genetic background lacking Wnt8b expression and strongly suggest that Wnt8b expression normally needs to be suppressed by Foxg1 for normal OC and OS development to take place.
Nasotemporal defects are not rescued in the Foxg1−/−;Wnt8b−/− double-mutant retina
Foxg1 is crucial for specification of the nasal retina, and its loss leads to an abnormal expansion of the temporal expression of Foxd1 nasally (Huh et al., 1999; Tian et al., 2008). We hypothesized that the rescue of the double-mutant Foxg1−/−;Wnt8b−/− OC morphology (Fig. 5B″), may be the result of a rescue in nasotemporal retinal patterning. In situ hybridization for Foxd1 expression at E11.5 and E15.5 revealed restricted expression in the temporal retina in controls (Fig. 6A,B) and expanded expression in the nasal domain of Foxg1−/−;Wnt8b+/± retinae (Fig. 6A′,B′). In the Foxg1−/−;Wnt8b−/− double mutant, we observed a similar expansion of Foxd1 to that observed in the Foxg1−/−;Wnt8b+/± retinae at E11.5 and E15.5 (Fig. 6A″,B″). Results were similar at E10.5 and consistent for all specimens analyzed (8 eyes from 4 different embryos for each experimental group, across all ages), indicating that rescued morphology of the OC in Foxg1−/−;Wnt8b−/− double mutants occurs despite the retention of nasotemporal patterning defects.

Upregulation of Foxd1 expression in the Foxg1−/− and in the Foxg1−/−;Wnt8b−/− double mutant nasal retina. Foxd1 mRNA expression in horizontal E11.5 (A–A″) and E15.5 (B–B″) sections is detected in the temporal retina of controls (A, B) but is found upregulated throughout the temporal and nasal retinal domains in Foxg1−/−;Wnt8b+/± (A′, B′) and Foxg1−/−;Wnt8b−/− double mutants (A″, B″). Scale bars: A, A′, A″, 100 μm; B, B′, B″, 200 μm.
The Foxg1−/−;Wnt8b−/− double-mutant OC morphology at E10.5 resembles that of the Foxg1−/− single mutant
To understand when in development we first start to observe morphological differences between controls, Foxg1−/−;Wnt8b+/± mutants and Foxg1−/−;Wnt8b−/− double mutants, we examined the morphology and marker expression of the optic neuroepithelium in these experimental groups at E10.5. Using as markers Coup-TFI, which labels the retina and the OS (Tang et al., 2010), and Mitf, which labels the RPE (Nguyen and Arnheiter, 2000), we first observed major morphological differences between wild-types and Foxg1−/− single mutants along the nasotemporal axis (Fig. 7). Coup-TFI expression was detected throughout the ventral OC (Fig. 7A,A′), whereas in dorsal sections it showed a hightemporal-lownasal gradient (Fig. 7D,D) (Tang et al., 2010) in wild-types and Foxg1−/− mutants. Coup-TFI expression revealed that in ventral sections the Foxg1−/− OC displayed an abnormal flattened shape (Fig. 7A′) compared with the wild-type U-shaped OC (Fig. 7A). In addition, the forming OF in wild-types (Fig. 7C, asterisk) was not detectable in the Foxg1−/− mutant (Fig. 7C′, ?). Coup-TFI expression in the OS, although similar between wild-types and Foxg1−/− mutants, revealed an abnormally enlarged distance between nasal and temporal OS in the Foxg1−/− mutant (Fig. 7C′, double arrow). Regarding Mitf expression, this was limited to the RPE in wild-types and Foxg1−/− mutants (Fig. 7B,B′,E,E′) but revealed a thickened nasal RPE in the Foxg1−/− mutant in both ventral (Fig. 7B′) and dorsal (Fig. 7E′) sections.

The shape of the nasal OC is compromised in Foxg1−/− mutants. Double immunofluorescence for Coup-TFI and Mitf in wild-type (Foxg1+/+) ventral (A–C) and dorsal (D–F) and Foxg1−/− ventral (A′–C′) and dorsal (D′–F′) E10.5 horizontal sections. Coup-TFI is found in the ventral (A, A′) and dorsal (D, D′) retina (r) and in the OS in both wild-types (A, D) and Foxg1−/− mutants (A′, D′). Mitf is found in the RPE in ventral and dorsal sections in both wild-types (B, E) and Foxg1−/− mutants (B′, E′). B, B′, Brackets indicate the presumptive RPE (pRPE) in wild-types and Foxg1−/− mutants, respectively. C, *OF in wild-types, which is not clearly visible in Foxg1−/− mutants (C′, ?). C′, Double arrow indicates the abnormally enlarged distance between nasal and temporal OS in the Foxg1−/− mutant. Arrowhead indicates the lack of invagination of the nasal OC. F, F′, Dashed lines indicate the nasotemporal axis. pMLC2 expression in wild-types (G) and Foxg1−/− mutants (G′) is detected along the RPE (G, G′, small arrows) and in the hinge region (G, G′, vertical arrow). Scale bars: A–F, A′–F′, 100 μm; G, G′, 50 μm.
We then examined our three experimental groups described above using well-established markers for the retina, RPE, and OS [Pax6: in the retina with highperipheral-to-lowcentral gradient and a lowventral-to-highdorsal gradient and in the RPE (Walther and Gruss, 1991; Bäumer et al., 2003)]; [Coup-TFII: in the RPE and OS (Tang et al., 2010; Eiraku et al., 2011)]; [Pax2: in the OS and central retina with a highventral-to-lowdorsal gradient (Nornes et al., 1990; Püschel et al., 1992; Bäumer et al., 2003)] to determine differences in optic neuroepithelium morphology and marker expression. Results described below were consistent for all specimens from the same experimental group (6 eyes from 3 different embryos for each experimental group).
In terms of morphology, the ventral OC of the Foxg1−/−;Wnt8b−/− double mutant was similar to that of the Foxg1−/−;Wnt8b+/+ mutant, showing a flattened appearance and lack of the U-shape formation in controls due to lack of nasal invagination of the OC (Fig. 8A″,C″, arrows, A′,C′). However, in dorsal sections, Foxg1−/−;Wnt8b−/− double-mutant OCs resembled the control OC shape (Fig. 8B,B″,D,D″). To determine whether the changes in Foxg1−/− OC morphology may be attributed to defects in the formation of the “hinge” region at the nasal RPE-retinal transition, we examined expression of the phosphorylated myosin light chain 2 (pMLC2), which has been implicated in RPE stiffness and in shaping the OC (Eiraku et al., 2011; Carpenter et al., 2015). However, we did not observe any gross differences in pMLC2 expression between E10.5 wild-type and Foxg1−/− mutant OCs at the hinge region that may account for the changes observed in the mutant nasal OC shape (Fig. 7G,G′).

Marker analysis of the optic neuroepithelium in control, Foxg1−/−;Wnt8b+/+, and Foxg1−/−;Wnt8b−/− E10.5 horizontal sections. Coup-TFII (green) and Pax2 (magenta) immunofluorescence in control (A, B), Foxg1−/−;Wnt8b+/+ (A′, B′), and Foxg1−/−;Wnt8b−/− (A″, B″) sections. Coup-TFII is found in the OS and the presumptive RPE (pRPE), indicated in A, A′, A″ (brackets) in ventral sections and the RPE in dorsal sections (B, B′, B″) in controls (A, B), single (A′, B′) and double mutants (A″, B″). Pax2 is found in the OS (A, C) and in the region of the retina (r) that surrounds the forming OF in control ventral sections (A, C, magenta bracketed areas). In Foxg1−/−;Wnt8b+/+ single-mutant ventral sections, Pax2 is found in the OS (A′, C′), but expression in the retina is shifted toward the temporal domain, as indicated in A′, C′ (magenta bracket). In the Foxg1−/−;Wnt8b−/− double mutant, Pax2 is expressed in the OS (A″, C″) and throughout the retina in ventral sections (A″, C″, magenta bracket). In dorsal sections, Pax2 expression is found in the region that will give rise to the optic disc (OD) (B, D, B′, D′, B″, D″) in all three genotypes. Dorsal Pax2 expression in the Foxg1−/−;Wnt8b+/+ mutant is shifted to the temporal retina, as with ventral sections (B′, D′). Pax6 (green) and Pax2 (magenta) immunofluorescence in control (C, D), Foxg1−/−;Wnt8b+/+ (C′, D′), and Foxg1−/−;Wnt8b−/− (C″, D″) sections. Pax6 expression is found in the retina and RPE throughout the ventrodorsal axis. In Foxg1−/−;Wnt8b+/+ mutants, the size of the ventronasal Pax6+ domain is clearly enlarged (C′, green bracket) compared with that of controls (C) and Foxg1−/−;Wnt8b−/− double mutants (C″, green bracket). A, A′, A″, Green brackets indicate nasal Pax2-negative expression, which corresponds to Pax6-positive expression in C, C′, C″, whereas the magenta brackets indicate the Pax2 expression domain. A′, C′, A″, C″, Arrows indicate the lack of a clear constriction where the OC invagination forms nasally in single and double mutants, respectively. E, Graph represents the difference in the mean values of the CTCF for Pax6 and Pax2 in the nasal and temporal retinal domains along the ventrodorsal axis. *p = 0.04. An ANOVA was performed to define the statistical significance of the difference of the results (F). ncontrol = 5 eyes from 3 different embryos; nFoxg1−/−;Wnt8b+/+ = 6 eyes from 3 different embryos; nFoxg1−/−;Wnt8b−/− = 4 eyes from 3 different embryos. Scale bars, 100 μm.
In terms of molecular profile, our marker analysis revealed that, in the Foxg1−/−;Wnt8b+/+ single mutant, the Pax2-positive domain seemed reduced in the nasal retina in ventral sections compared with that of controls (compare areas indicated by magenta brackets in Fig. 8A,C and Fig. 8A′,C′), whereas Pax6 expression seemed expanded nasally in both ventral and dorsal sections compared with that of controls (compare areas indicated by green brackets in Fig. 8C,C′ and Fig. 8D–D′). However, in Foxg1−/−;Wnt8b−/− double mutants, Pax2 expression ventrally expanded throughout the nasal and temporal retinal domain (Fig. 8A″, magenta bracketed area), similar to that of controls, whereas Pax6 staining was restricted to the peripheral retina both nasally and temporally and resembled more that of control staining (Fig. 8C″, green bracketed area, D″).
To quantitate the above observations, we measured the CTCF for the Pax6 and Pax2 cells in the nasal and temporal retinae (for details, see Materials and Methods). The Pax6 CTCF was found increased in the Foxg1−/−;Wnt8b+/+ single mutant compared with that of controls and Foxg1−/−;Wnt8b−/− double mutants (ANOVA; df, 2; F = 4.433, p = 0.036), and the difference reached statistical significance between single and double mutants (Fig. 8E,F). In the case of Pax2, although the CTCF was found reduced in the Foxg1−/−;Wnt8b+/+ single mutant compared with that of controls and double mutants, the difference was not significant (ANOVA; df, 2; F = 0.5; p = 0.619) (Fig. 8E).
Our results show that, at E10.5, ventral OC morphology in the Foxg1−/−;Wnt8b−/− double mutant resembles more that of Foxg1−/−;Wnt8b+/+ single mutants than that of controls, despite the fact that Pax6 expression is significantly reduced in these mutants compared with that of the Foxg1−/−;Wnt8b+/+ single mutant.
Morphological and molecular alterations in OF development in Foxg1−/− embryos are rescued in Foxg1−/−;Wnt8b−/− double mutants by E11.5
We then examined the morphology and marker expression of the developing OC at E11.5, using the same markers as for E10.5 embryos (Coup-TFII/Pax2 and Pax6/Pax2) (Fig. 9). As for the E10.5 embryos, results described below were consistent for all specimens from the same experimental group (6 eyes from 3 different embryos for each experimental group).
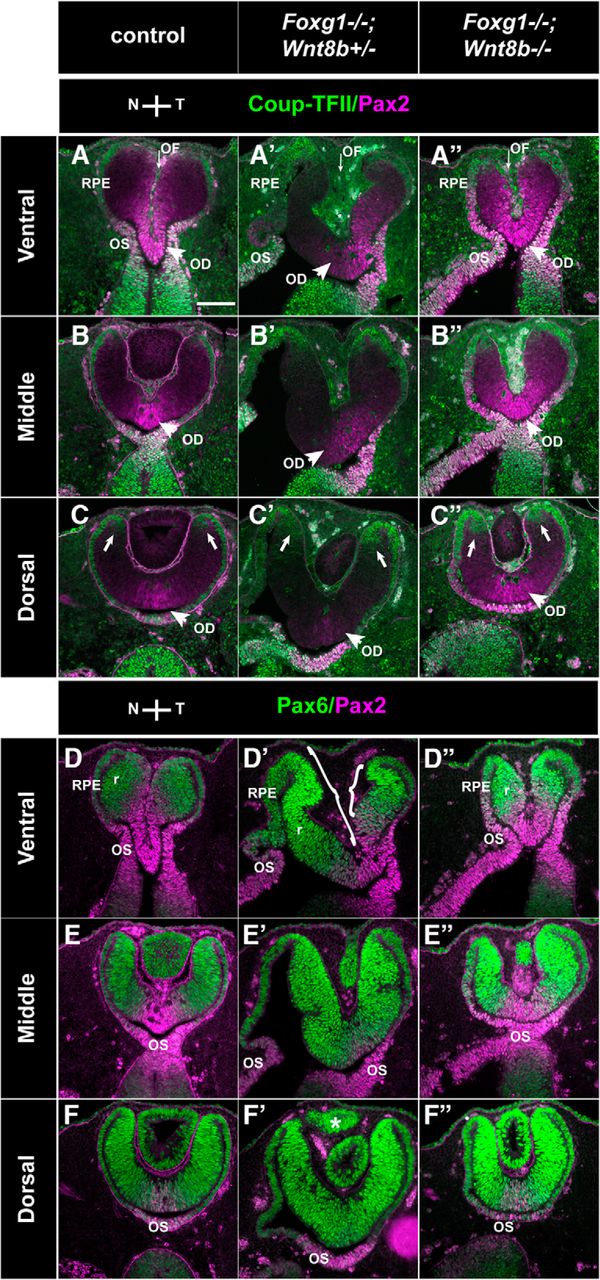
Marker analysis of the optic neuroepithelium in control, Foxg1−/−;Wnt8b+/±, and Foxg1−/−;Wnt8b−/− E11.5 horizontal sections. Coup-TFII (green) and Pax2 (magenta) immunofluorescence in control (A–C), Foxg1−/−;Wnt8b+/± (A′–C′), and Foxg1−/−;Wnt8b−/− (A″–C″) sections. In ventral and middle sections, Coup-TFII is restricted in the RPE (A–A″, B–B″), whereas in dorsal sections, it is expanded at the tips of the peripheral retina (C–C″, arrows). In ventral sections, Pax2 is detected at the apposed edges of the OF in controls (A) and Foxg1−/−;Wnt8b−/− double mutants (A″). In Foxg1−/−;Wnt8b+/± mutants, Pax2 expression is found in the temporal, but not the nasal, edges of the OF (A′). Pax2 is also detected in the optic disc (OD) marked with arrowheads in all three different genotypes (A–A″, B–B″, C–C″) and OS (A–A″, D–D″, E–E″). A–A″, Thin arrows indicate the OF. Pax6 (green) and Pax2 (magenta) immunofluorescence in control (D–F), Foxg1−/−;Wnt8b+/± (D′–F′), and Foxg1−/−;Wnt8b−/− (D″–F″) sections. Pax6 expression is found in the retina (r) and RPE throughout the ventrodorsal axis. Pax6 staining reveals that the size of nasal and temporal retina is similar in controls (D–F) and Foxg1−/−;Wnt8b−/− double mutants (D″–F″). In Foxg1−/−;Wnt8b+/± mutants, the size of the nasal retina is clearly enlarged compared with that of the temporal retina (compare size of bracketed areas in D′). F′, *Presence of abnormal ciliary margin tissue in the Foxg1−/−;Wnt8b+/± mutant, as previously described (Fotaki et al., 2013). Scale bars, 100 μm.
At E11.5, OC morphology in Foxg1−/−;Wnt8b+/± mutants was severely compromised in ventral and middle sections (Fig. 9A′–E′), whereas the appearance of dorsal OC sections resembled more that of controls (Fig. 9C,C′,F,F′). Coup-TFII expression revealed a thickened RPE in ventronasal sections in this mutant (Fig. 9A′). The gap between the nasal and temporal edges of the OF was much greater in Foxg1−/−;Wnt8b+/± mutants compared with controls (compare the OF region indicated by an arrow in Fig. 9A and Fig. 9A′) and Pax2 expression along the adjoining sides was reduced temporally and was absent nasally (Fig. 9A′,D′). Similar to controls (Fig. 9D–F), Pax6 expression in Foxg1−/−;Wnt8b+/± mutants was detected in the retina and RPE (Fig. 9D′–F′). However, in ventral sections, Pax6 expression revealed a greater nasal domain compared with the Pax6-positive temporal domain (compare the bracketed nasal and temporal retinae in Fig. 9D′).
In contrast, in Foxg1−/−;Wnt8b−/− double mutants, the anatomy of the OC resembled that of controls along the ventrodorsal axis (Fig. 9A″–F″). In ventral sections, the OC displayed a clearly centrally placed OF between the juxtaposed nasal and temporal sides (Fig. 9A″,D″). Although the distance between the edges of the OF in the Foxg1−/−;Wnt8b−/− double mutant was not as narrow as that of controls, it was greatly reduced compared with that of the Foxg1−/−;Wnt8b+/± mutant (compare the OF region indicated by an arrow in Fig. 9A″ to that in Fig. 9A,A′). The Coup-TFII staining revealed a normal RPE appearance ventronasally and not the thickening observed in Foxg1−/−;Wnt8b+/± mutants (Fig. 9A″). In ventral sections, Pax2 expression in the OC was increased compared with Foxg1−/−;Wnt8b+/± mutants and surrounded both the nasal and temporal edges of the Foxg1−/−;Wnt8b−/− OF (Fig. 9A″,D″). Similar to controls, Pax6 expression was detected in the OC and RPE (Fig. 9D″–F″).
Similar to E10.5, we quantitated expression of Pax6 and Pax2 using the corrected total cell fluorescence along the ventrodorsal axis. In agreement with our observations, we found a significant increase in Pax6 fluorescence (ANOVA; df, 2; F = 17.29; p = 0.001) (Fig. 10A,C) and a significant decrease in Pax2 fluorescence (ANOVA; df, 2; F = 23.665; p < 0.0001) (Fig. 10B,C) in Foxg1−/−;Wnt8b+/± mutants compared with that of controls and double mutants. In addition, Pax2 fluorescence was significantly increased in double mutants compared with that of controls (Fig. 10B,C).

Quantitation of Pax6 and Pax2 expression in E11.5 retinal sections. An ANOVA was performed to define the statistical significance of the difference in the mean values of the CTCF for Pax6 (A) and Pax2 (B) in the nasal and temporal retinal domains along the ventrodorsal axis. C, Graph represents these differences. Asterisks indicate corresponding p values. n = 4 eyes from 3 different embryos for all three groups analyzed.
The above results show an amelioration of OC and OS morphology in the Foxg1−/−;Wnt8b−/− double mutant by E11.5, with wild-type levels of OC Pax6 expression and a significant increase in nasal OC Pax2 expression compared with that of wild-types and Foxg1−/−;Wnt8b+/± mutants.
To validate rescue of Pax2 expression in the edges of the Foxg1−/−;Wnt8b−/− OF, we further examined sagittal sections immunostained for Pax2. First, we examined Pax2 expression in Foxg1−/− single mutants and wild-type littermates at E12.0, when OF closure is initiated in controls (Fig. 11). In contrast to wild-type expression (Fig. 11A–C), in Foxg1−/− retinae, the anterior tip was Pax2-negative and located at a distance from the Pax2-positive-posterior OF tip (Fig. 11C′). Similar results were obtained with in situ hybridization for Vax1 (Fig. 11D,D′,E,E′), which is also expressed at the edges of the OF (Bäumer et al., 2002) and, when mutated, gives rise to a coloboma phenotype in mice (Hallonet et al., 1999). However, similar expression of Bmp7, a gene essential for OF formation (Morcillo et al., 2006), was observed in both the anterior and posterior tips of wild-types and Foxg1−/− mutants along the proximodistal axis (Fig. 11F,F′,G,G′).

OF marker expression in control (Foxg1+/+; Foxg1+/−) and Foxg1−/− mutant sagittal sections. At E11.5, Foxg1 (A, B, blue staining) is expressed in the anterior (A) edge in controls (Foxg1+/−), whereas Pax2 (A, B, brown staining) is detected in both the anterior and posterior (P) edges of the OF (A, B). At E12.0, Pax2 immunofluorescence reveals normal anterior and posterior expression at the OF edges in the wild-type (Foxg1+/+) (C), whereas in the Foxg1−/− mutant, the anterior domain of expression is lost and the posterior is maintained (C′). Similarly, Vax1 mRNA wild-type anterior expression (D, E) is compromised in the Foxg1−/− mutant (D′, E′), whereas posterior expression is intact (D, D′, E, E′). Bmp7 mRNA expression is intact in both the anterior and posterior edges of the OF in the Foxg1−/− mutant (F′, G′) similar to the wild-type (F, G). A, D, D′, F, F′, More proximal sections to B, E, E′, G, G′, respectively. Scale bars: A, B, 100 μm; C, C′, 100 μm; D–G, D′–G′, 100 μm.
In Foxg1−/−;Wnt8b−/− E11.5 sagittal sections (4 eyes from 3 different embryos in the sagittal plane), Pax2 expression was present in both the anterior and posterior tips of the Foxg1−/−;Wnt8b−/− OF (Fig. 12A″) along the proximodistal axis (3 of 4 eyes examined), similar to controls (Fig. 12A). In one case, anterior Pax2 staining in distal sections was limited to only a few cells (data not shown). However, in the Foxg1−/−;Wnt8b+/± mutant, Pax2 expression was not detected in the anterior OF (Fig. 12A′), in agreement with the results we observed in the Foxg1−/− single mutant (Fig. 11C,C′). This was further confirmed by counting the Pax2-positive cells at the edges of the OF within a square area of 0.01 mm2 (for details, see Materials and Methods). The Pax2-positive cell density in the nasal edge of the OF was significantly reduced in the Foxg1−/−;Wnt8b+/± mutant compared with that of controls and double mutants (ANOVA; df, 2; F = 141,374; p < 0.0001) (Fig. 12B). No differences were observed in temporal Pax2-positive cell density between groups (ANOVA; df, 2; F = 3,669; p = 0.081).

Pax2 expression in the anterior and posterior tips of the OF. Pax2 expression in E11.5 sagittal control (A), Foxg1−/−;Wnt8b+/± mutant (A′), and Foxg1−/−;Wnt8b−/− double mutant (A″) sections showing that the normal Pax2 expression in the anterior (A) and posterior (P) edges of the control OF (A) is only found posteriorly in the Foxg1−/−;Wnt8b+/± mutant (A′) but is rescued in the Foxg1−/−;Wnt8b−/− double mutant (A″). ANOVA revealed that the Pax2-positive cells within a 0.01 mm2 area at the edges of the nasal retina is significantly reduced (p < 0.0001) in the Foxg1−/−;Wnt8b+/± mutants compared with that of controls and double mutants (B). ncontrol = 3 eyes from 3 different embryos; nFoxg1−/−;Wnt8b+/± = 4 eyes from 3 different embryos; nFoxg1−/−;Wnt8b−/− = 3 eyes from 3 different embryos. Scale bars, 100 μm.
These results confirm that loss of Wnt8b function in the Foxg1−/− null background leads to a specific increase in Pax2 expression in the anterior (nasal) tips of the OF, strongly suggesting that this may contribute to the rescue of the coloboma phenotype in the Foxg1−/−;Wnt8b−/− double mutant.
Cell proliferation is not altered in the Foxg1−/− mutant at E10.5
OC formation and OF closure rely on balanced cell proliferation and cell death (Morcillo et al., 2006; See and Clagett-Dame, 2009; Cai et al., 2013; Noh et al., 2016).
To understand whether aberrant OC cell proliferation may be involved in coloboma formation in the Foxg1−/− mutant, we calculated the LI (BrDU-positive cells over total number of cells) in nasal and temporal retinae of E10.5 wild-type and Foxg1−/− mutant OCs (for a detailed description, see Materials and Methods). To define the border between nasal and temporal retina, we used as a guide Foxg1+/− heterozygous and Foxg1−/− homozygous sections immunostained for β-galactosidase (β-gal) (Xuan et al., 1995), normally found in the nasal retina (Fig. 13A,A′, dashed line). Our cell counts did not reveal any significant differences in the LI ± SE among wild-type (0.45170 ± 0.05608) and Foxg1−/− mutant (0.45398 ± 0.03902) nasal retina or wild-type (0.42959 ± 0.03825) and Foxg1−/− mutant (0.43340 ± 0.05842) temporal retina. In addition, immunohistochemistry for phosphorylated histone H3 (pHH3), a mitosis (M)-phase marker (Hans and Dimitrov, 2001), did not result in any differences in the cell distribution or the mitotic index (number of pHH3-positive cells per surface area ± SE) (for details, see Materials and Methods) between E10.5 wild-type (0.03483 ± 0.00454) and Foxg1−/− mutant (0.03418 ± 0.00266) nasal retina or wild-type (0.03133 ± 0.00337) and Foxg1−/− mutant (0.03686 ± 0.00772) temporal retina. Finally, based on the suggestion that precocious differentiation of neural progenitors at the apposed edges of the OF may result in failure of the fissure to seal (Lohnes et al., 1994), we performed immunohistochemistry for β-tubulin III (Tuj1), a marker of early born neurons (Memberg and Hall, 1995). However, we did not observe any Tuj1-positive cells in the E10.5 developing OC or OS of wild-types or Foxg1−/− mutants (data not shown).

Apoptotic cell density is significantly reduced in the nasal retina in the Foxg1−/− mutant. β-gal staining defines the border between the nasal and temporal retinae (r) (A, A′, dashed line) in Foxg1+/− heterozygote controls (A) and Foxg1−/− mutants (A′). Cleaved caspase-3 immunohistochemistry was used to detect retinal cells undergoing apoptotic cell death in wild-types (Foxg1+/+) (B) and Foxg1−/− mutants (B′). Mean apoptotic cell density values were normalized as a percentage to the wild-type nasal value (100% apoptotic density) (C). Scale bars, 50 μm.
Apoptotic defects in the nasal retina in the Foxg1−/− mutant at E10.5 are partially rescued in the Foxg1−/−;Wnt8b−/− double mutant
To determine whether abnormal apoptosis is associated with the OC phenotype of Foxg1−/− mutants, we examined the apoptotic density in E10.5 nasal and temporal wild-type and Foxg1−/− horizontal sections, using immunohistochemistry for cleaved caspase-3 (Noh et al., 2016). Our cell counts revealed a significantly lower density of apoptotic cells (p < 0.05) in the Foxg1−/− nasal retina compared with that of wild-types (Fig. 13; Table 2), with a mean value reaching 35 pp (percentage points) of the wild-type nasal values. No differences in apoptotic density were observed among genotypes in the temporal retina (Fig. 13; Table 2).
The correlation between decreased nasal apoptosis and failed OF closure in Foxg1−/− mutants led us to hypothesize that the rescue of OF closure in the Foxg1−/−;Wnt8b−/− double mutant observed at E11.5 (Fig. 9A″,D″) is associated with restoration of normal apoptotic levels. We analyzed apoptotic cell density in three experimental groups: (1) controls, (2) Foxg1−/−;Wnt8b+/± mutants, and (3) Foxg1−/−;Wnt8b−/− double mutants. In nasal retina, and in accordance with our hypothesis, we found an increase of 24pp in mean apoptotic density in Foxg1−/−;Wnt8b−/− double mutants compared with Foxg1−/−;Wnt8b+/± mutants, which was statistically significant (p < 0.05) (Fig. 14; Table 3), although the increase did not quite reach the level observed in our control samples (p < 0.05) (Fig. 14; Table 3). No significant differences were observed in temporal apoptotic cell density between controls, Foxg1−/−;Wnt8b+/± mutants, and Foxg1−/−;Wnt8b−/− double mutants (Table 3).

Partial rescue of the apoptosis phenotype of the Foxg1−/− mutant in the Foxg1−/−;Wnt8b−/− double mutant. Cleaved caspase-3 immunohistochemistry labels retinal cells undergoing apoptotic cell death in control (A), Foxg1−/−;Wnt8b+/± (A′), and Foxg1−/−;Wnt8b−/− (A″) E10.5 horizontal sections. A, A″, Arrows indicate the OF, which starts to form in the control and Foxg1−/−;Wnt8b−/− double mutant. A′, Dashed arrow indicates a less-clear formation of the OF in Foxg1−/−;Wnt8b+/+single mutants. Mean apoptotic cell density values were normalized as a percentage to the control nasal value (100% apoptotic density) (B). Scale bars, 50 μm.
Our results show that, at E10.5, there is significant increase in nasal apoptotic cell density in the Foxg1−/−;Wnt8b−/− double mutant compared with the Foxg1−/−;Wnt8b+/± mutant, consistent with the idea that this may be partly responsible for the amelioration of the Foxg1−/−;Wnt8b−/− OF morphology at E11.5.
Upregulation of Wnt/β-catenin signaling target molecules in the Foxg1−/− OC and OS
Wnt molecules signal mainly through the Wnt/β-catenin or the planar cell polarity pathways (for review, see Loh et al., 2016). To gain insight into which Wnt signaling pathway is affected by the upregulation of Wnt8b observed in the OS of the Foxg1−/− mutant, we examined downstream targets of the Wnt/β-catenin and the planar cell polarity pathways in the OC and OS.
First, we performed a PCR array analysis and profiled the expression of 84 genes related to Wnt-mediated signal transduction, using RNA extracted from OCs of E11.0 wild-type and Foxg1−/− embryos. Of 12 Wnt-signaling target genes included in the array (Table 4, asterisk), the only one that showed a significant change in expression was c-Jun, a downstream target of the Wnt/β-catenin pathway (Mann et al., 1999), with a 2.5-fold upregulation (p = 0.01) in Foxg1−/− OCs (Table 4). This upregulation became apparent at the protein level by E12.0, when we detected a clear increase in c-Jun expression in the Foxg1−/− retina (Fig. 15A,A′) (results were consistent in n = 6 from 3 different control and Foxg1−/− mutant eyes).
PCR array results for 84 genes related to Wnt-mediated signal transduction, showing the average ΔCt values for each experimental group, fold changes, and the p values of these changesa

OC and OS Wnt-signaling target gene analysis in controls and Foxg1−/− mutants at E12.5. c-Jun protein expression is upregulated in the Foxg1−/− OC (A′) compared with expression in controls (A). No differences in Fzd3 (B, B′), Celsr3 (C, C′), or Vangl2 (D, D′) mRNA expression are observed between Foxg1−/− mutants and controls. Axin2 expression is upregulated in the nasal component of the OS in Foxg1−/− mutants (E′, arrowheads) compared with controls (E, arrow) and mirrors upregulated expression of Wnt8b in the same region (compare E with F and E′ with F′). Scale bars: A, A′, 100 μm; B–E, B′-E′, 200 μm; F, F′, 100 μm.
We then analyzed mRNA expression by means of in situ hybridization in the OS of wild-type and Foxg1−/− mutants of molecules implicated in the planar cell polarity pathway (Fzd3, Celsr3, and Vangl3) (Fig. 15B–D,B′–D′) (Tissir et al., 2005; Montcouquiol et al., 2006), as well as Axin2 (Fig. 15E,E′), a read-out of the Wnt/β-catenin signaling pathway (Jho et al., 2002). Axin2 showed a clear upregulation in the OS of the Foxg1−/− mutant (Fig. 15E′), similar to the upregulation observed in Wnt8b expression (Fig. 15F,F′), which became evident as early as E10.5 (data not shown).
These results show that Wn8b upregulation in the Foxg1−/− OS results in upregulation of the Wnt/β-catenin signaling pathway through overexpression of the downstream targets c-Jun and Axin2 in the Foxg1−/− mutant OC and OS, respectively.
Discussion
Our data unravel a novel mechanism of OF closure, which relies on Foxg1-mediated suppression of Wnt8b in the nasal OS, resulting in balanced apoptosis and normal Pax2 expression in the nasal edges of the fissure (Fig. 16A–C). This newly described role of Foxg1 in OC formation is independent of its function as a retinal nasotemporal determinant.

Schematic summary of main findings and proposed Foxg1 regulatory network in the developing eye. A, In wild-types, Foxg1 is normally expressed in the nasal OC and OS, Wnt8b is expressed in the nasal and temporal OS, and Pax2 is expressed in the nasal and temporal OS and in the nasal and temporal edges of the OF. Apoptosis is normally observed in the nasal and temporal edges of the OF (brown dots). B, In Foxg1−/− mutants, loss of Foxg1 results in abnormal upregulation of Wnt8b nasally, increased suppression of Pax2, and decreased apoptotic cell death in the nasal edges of the fissure. C, In Foxg1−/−;Wnt8b−/− double mutants, loss of both Foxg1 and Wnt8b results in rescued Pax2 expression in the nasal edges of the OF, although Pax2 expression is stronger distally than proximally. Apoptosis is increased compared with the Foxg1−/− mutant (B), but it is still significantly below the control values (A). D–F, A model of possible molecular interactions showing how Foxg1 function and loss of function affects OC and OF formation. —|, Repression; ->, activation. Our model cannot predict whether the molecular interactions are direct or indirect.
We hypothesized that, similar to foxg1 function in the zebrafish telencephalon (Danesin et al., 2009), mouse Foxg1 suppresses Wnt8b function in the developing optic neuroepithelium for proper OC formation to take place. Indeed, wild-type Wnt8b expression in the OS was upregulated in the Foxg1−/− mutant as early as E10.5. This was observed in the nasal stalk, which is the region of the developing optic neuroepithelium with highest Foxg1 expression levels at this developmental stage (Hatini et al., 1994; current study). Our genetic experiment allowed us to evaluate the in vivo effects of loss of Wnt8b expression in a Foxg1-null genetic background. The remarkable rescue of the OC and OS morphology and the substantial sealing of the OF by E15.5 in the Foxg1−/−;Wnt8b−/− mutant are in agreement with our hypothesis. Future experiments will show whether mouse retinal Foxg1 suppresses Wnt8b by direct binding, similar to zebrafish telencephalon (Danesin et al., 2009), or whether the suppression is indirect, supported by the lack of Foxg1 binding sites within the Wnt8b promoter region in mouse neural stem cells (Bulstrode et al., 2017).
Foxg1 is expressed in the nasal retina of vertebrates (Hatini et al., 1994; Takahashi et al., 2003; Picker et al., 2009), and work in chick and zebrafish has established Foxg1's role as an early determinant of retinal nasotemporal polarity exerting an antagonistic effect on Foxd1 (Yuasa et al., 1996; Takahashi et al., 2003), a temporal retina determinant that is abnormally expressed in the Foxg1−/− nasal retina (Huh et al., 1999; Tian et al., 2008). The fact that abnormal Foxd1 expression in the nasal retina is still observed in the Foxg1−/−;Wnt8b−/− mutant reveals that the coloboma phenotype is not a secondary defect to the abnormal Foxg1−/− nasotemporal patterning.
Although the shape of the Foxg1−/−;Wnt8b−/− OC resembles that of the Foxg1−/− mutant at E10.5, its Pax6 and Pax2 expression profiles mimic those of controls. By E11.5, the Foxg1−/−;Wnt8b−/− OC morphology resembles that of controls, with normal Pax6 and elevated Pax2 expression levels. The OC normally undergoes a series of morphological changes, from a flattened to a spherical shape (Lamb et al., 2007; Eiraku et al., 2011; Eiraku and Sasai, 2012). Our data support the idea that, in the Foxg1−/−;Wnt8b−/− mutant, OC formation is delayed, resulting in closure of the OF at a later developmental point, at ∼E15.5 rather than at E13.5 (Hero, 1989).
Pax2 is required for OF closure (Torres et al., 1996), and loss of Pax2 expression in the nasal (anterior) edge of the OF in the Foxg1−/− mutant may account for failure of the OF edges to fuse. This is further supported by the fact that, in the Foxg1−/−;Wnt8b−/− mutant with 100% OF fusion in proximal sections, there is 100% rescue of Pax2 expression in the anterior OF proximally, whereas in distal sections, where Pax2 expression is not fully recovered, we observe less efficient rescue (Fig. 16A–C).
Precise regulation of cellular events is crucial for the development of the OC and OS. At early stages of OC development, intense cell proliferation takes place, which is associated with the invagination of the optic vesicle and the appearance of the OF (Calvente et al., 1988). We hypothesized that changes in OC morphology in the Foxg1−/− mutant may result from aberrant proliferation, which may be rescued in the Foxg1−/−;Wnt8b−/− mutant. However, the lack of difference in the LI, mitotic index, and β-tubulin-III expression between wild-types and Foxg1−/− mutants at E10.5 argued against the idea that aberrant proliferation and/or premature differentiation is involved in the early Foxg1−/− OC morphological defects.
Programmed cell death (apoptosis) in the nasal and temporal edges adjoining the OF normally occurs during mouse OC formation and OF closure (Hero, 1989; Ozeki et al., 2000). Apoptosis is first detected in ventronasal retina in the region of the presumptive OF at E9.5 and is then found in the OF edges, becoming undetectable after the edges fuse at E13.5 (Ozeki et al., 2000). In addition, either increased or decreased OF apoptosis has been found in mouse mutants with a coloboma phenotype (Cai et al., 2013; Noh et al., 2016). This evidence strongly supports balanced apoptosis at the edges of the OF as a major determinant of proper OF closure. Our data show significantly reduced levels of caspase-3-mediated apoptosis in the Foxg1−/− ventronasal retina, which strongly suggest a requirement for Foxg1 in promoting apoptotic cell death at the nasal edge of the fissure. Reduction in apoptosis is observed in Bmp7−/− mouse mutants, which fail to form OF (Morcillo et al., 2006). However, normal Bmp7 expression in the edges of the Foxg1−/− fissure indicates that Foxg1 OF function is independent of Bmp7. In the Foxg1−/−;Wnt8b−/− double mutant, apoptosis in the nasal retina was significantly higher to that of Foxg1−/−;Wnt8b+/± mutants, suggesting that Wnt8b overexpression is a contributing factor to the reduction in apoptosis we observe in the Foxg1−/− embryos.
Wnt8b activates Wnt/β-catenin signaling (Lee et al., 2006), and our data suggest that upregulation of this signaling cascade results in coloboma. Although our present data do not provide a direct link between the observed changes in apoptosis and Wnt/β-catenin signaling, it is interesting that the only downstream target with upregulated expression in the Foxg1−/− OC was c-Jun. c-Jun protects cells from excessive apoptotic activity (Wisdom et al., 1999; Shaulian and Karin, 2002), and its upregulation in the Foxg1−/− mutant OC may result in reduction of apoptosis in the nasal edge of the OF compromising fissure closure.
Although our array analysis in the OC did not reveal differences between wild-types and Foxg1−/− mutants in Axin2 expression, a read-out of the Wnt/β-catenin pathway (Jho et al., 2002), Axin2 was found upregulated in the Foxg1−/− OS, at the same sites where Wnt8b upregulation was observed. This is in agreement with the current notion that Wnt proteins are locally acting signaling molecules (Alexandre et al., 2014; Farin et al., 2016; Loh et al., 2016) and suggests that the rescued OC morphology in the Foxg1−/−;Wnt8b−/− mutant may be a secondary effect to a primary rescue in OS formation. This idea is further supported with the following model of molecular interactions in the developing eye at ∼E11.5 (Fig. 16D–F).
Foxg1 in the nasal ciliary margin controls Wnt2b levels, which in turn result in normal transcriptional activation of Pax6. In parallel, Foxg1 in the nasal stalk keeps Wnt8b levels in check, resulting in normal Pax2 expression in the ventral retina, due to low transcriptional repression. Finally, Pax6 and Pax2 levels are balanced through reciprocal inhibition, as previously described (Schwarz et al., 2000) (Fig. 16D). When Foxg1 function is abolished, Pax6 expression is upregulated, resulting in an expanded ciliary margin (Fotaki et al., 2013), whereas Pax2 expression is significantly reduced (this study), resulting in failure of the OF to form properly (Fig. 16E). In the Foxg1−/−;Wnt8b−/− mutant, Pax2 levels are significantly increased compared with those of single mutants and controls (this study), resulting in proper OF closure. Although Pax6 expression should still be elevated in the double mutant, our quantitation analysis reveals similar levels of expression to those of controls. A possible explanation for this is that, in the double mutant, Pax2 expression is more elevated than that of Pax6; and when it suppresses Pax6, it reduces it to normal values (Fig. 16F). Although currently unavailable, we predict that a Pax2-overexpressing mouse strain crossed to the Foxg1-mutant background will phenocopy the Foxg1−/−;Wnt8b−/− phenotype in line with our hypothesis.
Our observations that upregulated Wnt/β-catenin signaling associates with coloboma formation seem to be at odds with the fact that a coloboma phenotype is also observed in cases when the Wnt/β-catenin signaling is reduced, as in the case of humans with mutations in the Wnt receptor gene FZD5 (Liu et al., 2016) and Fzd5−/− null mice, which show increased apoptosis and increased Pax2 expression in the OC (Liu and Nathans, 2008). However, Wnt/β-catenin signaling must also be increased in Dkk1+/− mice with reduced levels of the Wnt antagonist Dkk1 and in mice with a loss of function of Axin2, a negative regulator of the Wnt/β-catenin signaling, both of which also show a coloboma phenotype (Lieven and Rüther, 2011; Alldredge and Fuhrmann, 2016). This all suggests that Wnt/β-catenin signaling needs to be tightly regulated in the OC and OS for proper OF closure, and unbalanced expression (overexpression or underexpression) of its components leads to coloboma.
Our work uncovers a novel action of Foxg1 in limiting Wnt/β-catenin signaling in the OS for proper OC and OS formation and OF closure to take place and provides additional knowledge regarding the molecular players and cellular mechanisms underlying coloboma formation.
Footnotes
This work was supported by Medical Research Council MR/J013137/1, Wellcome Trust 085065, and Medical Research Scotland Vacation Scholarship. We thank the CBS-BRR University of Edinburgh animal house staff for invaluable help with mouse maintenance; John Mason for the Wnt8b+/− mutant mice; the following researchers for sharing plasmids for riboprobe synthesis: André Goffinet (Celsr3, Fzd3, Vangl3), Eloisa Herrera (Foxd1), and Robert Hindges (Vax1), Thomas Theil (Bmp7); the Developmental Studies Hybridoma Bank, University of Iowa (Department of Biological Sciences, Iowa City, IA) for the Islet1 (generated by Thomas Jessell and Susan Brenner-Morton) and Pax6 (generated by Atsushi Kawakami) antibodies; Heinz Arnheiter for the rabbit polyclonal Mitf antibody; Wai-Kit Chan and Viv Allison for technical support during the final phases of the project; Anisha Kubasik-Thayil for invaluable help with the confocal imaging and the use of the IMARIS software; Emily Osterweil's laboratory for kindly providing access to the StepOnePlus Real-Time PCR machine; Mike Molinek for the excellent upkeep of our communal laboratory; Zrinko Kozic for bioinformatics advice; and members of our laboratories for discussions and support.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Vassiliki Fotaki, Edinburgh Medical School, Biomedical Sciences, Centre for Integrative Physiology, Hugh Robson Building, George Square, Edinburgh, EH8 9XD, United Kingdom. vfotaki{at}ed.ac.uk
This is an open-access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}