

Abstract
The onset of the headache phase during attacks of migraine with aura, which occur in ∼30% of migraineurs, is believed to involve cortical spreading depression (CSD) and the ensuing activation and sensitization of primary afferent neurons that innervate the intracranial meninges, and their related large vessels. The mechanism by which CSD enhances the activity and mechanosensitivity of meningeal afferents remains poorly understood, but may involve cortical metabolic perturbations. We used extracellular single-unit recording of meningeal afferent activity and monitored changes in cortical blood flow and tissue partial pressure of oxygen (tpO2) in anesthetized male rats to test whether the prolonged cortical hypoperfusion and reduction in tissue oxygenation that occur in the wake of CSD contribute to meningeal nociception. Suppression of CSD-evoked cortical hypoperfusion with the cyclooxygenase inhibitor naproxen blocked the reduction in cortical tpO2, but had no effect on the activation of meningeal afferents. Naproxen, however, distinctly prevented CSD-induced afferent mechanical sensitization. Counteracting the CSD-evoked persistent hypoperfusion and reduced tpO2 by preemptively increasing cortical blood flow using the ATP-sensitive potassium [K(ATP)] channel opener levcromakalim did not inhibit the sensitization of meningeal afferents, but prevented their activation. Our data show that the cortical hypoperfusion and reduction in tpO2 that occur in the wake of CSD can be dissociated from the activation and mechanical sensitization of meningeal afferent responses, suggesting that the metabolic changes do not contribute directly to these neuronal nociceptive responses.
SIGNIFICANCE STATEMENT Cortical spreading depression (CSD)-evoked activation and mechanical sensitization of meningeal afferents is thought to mediate the headache phase in migraine with aura. We report that blocking the CSD-evoked cortical hypoperfusion and reduced tissue partial pressure of oxygen by cyclooxygenase inhibition is associated with the inhibition of the afferent sensitization, but not their activation. Normalization of these CSD-evoked metabolic perturbations by activating K(ATP) channels is, however, associated with the inhibition of afferent activation but not sensitization. These results question the contribution of cortical metabolic perturbations to the triggering mechanism underlying meningeal nociception and the ensuing headache in migraine with aura, further point to distinct mechanisms underlying the activation and sensitization of meningeal afferents in migraine, and highlight the need to target both processes for an effective migraine therapy.
Introduction
Migraine is a multifactorial, episodic neurological disorder and the second leading cause of disability worldwide (Vos et al., 2017). It is now well accepted that the onset of migraine headache requires the activation and increased mechanosensitivity of trigeminal afferent neurons that innervate the intracranial meninges and their related large blood vessels (Strassman et al., 1996; Messlinger, 2009; Olesen et al., 2009; Levy, 2012). However, the endogenous processes underlying these afferent responses during a migraine attack are incompletely understood.
Migraine with aura, the second-most prevalent migraine subtype, is thought to involve cortical spreading depression (CSD), an abnormal wave of neuronal and glial activity (Ayata and Lauritzen, 2015). CSD has been suggested to mediate the aura phase (Pietrobon and Moskowitz, 2013), and also contributes to the onset of the headache phase, by promoting prolonged activation and increased mechanosensitivity of meningeal afferents (Zhang et al., 2010; Zhao and Levy, 2015, 2016) and the ensuing activation and sensitization of central trigeminal dorsal horn neurons that receive their input (Zhang et al., 2011b; Melo-Carrillo et al., 2017). The precise mechanisms by which CSD promotes the activation and sensitization of meningeal afferents are unclear, although dural neurogenic inflammation (Bolay et al., 2002; but see Zhao and Levy, 2017) and Pannexin-1-related signaling (Karatas et al., 2013) have been hypothesized.
Migraine with aura is associated with cortical hemodynamic changes, in particular a sustained reduction in cortical blood flow (CBF) contemporaneous with the onset of the headache phase (Olesen et al., 1981, 1982; Lauritzen, 1984; Hadjikhani et al., 2001; Hansen and Schankin, 2017). In animal studies, CSD has been shown to promote a similar prolonged cortical hypoperfusion (Fabricius and Lauritzen, 1993; Fabricius et al., 1995; Piilgaard and Lauritzen, 2009). CSD also leads to transient cortical hypoxia followed by prolonged and milder reduction in cortical tissue partial pressure of oxygen (tpO2) concomitant with the cortical hypoperfusion phase (Takano et al., 2007; Piilgaard and Lauritzen, 2009).
Reduced tissue oxygenation during acute exposure to high altitude can promote headache and trigger a migraine attack (Appenzeller, 1994; Broessner et al., 2016). Normobaric hypoxia has also been shown to trigger migraine in susceptible individuals (Schoonman et al., 2006; Arngrim et al., 2016). These clinical observations together with the notion that reduced blood flow and tissue oxygenation can lead to enhanced activity and responsiveness of primary afferent nociceptors that innervate the cornea, viscera, muscle, and skin (Haupt et al., 1983; Mense and Stahnke, 1983; Longhurst et al., 1991; MacIver and Tanelian, 1992; Hillery et al., 2011) prompted us to examine whether the cortical hypoperfusion and associated reduction in tpO2 that occur following CSD might contribute to the prolonged activation and sensitization of meningeal afferents. Recent work has shown that the inhibition of cortical cyclooxygenase abrogates CSD-evoked cortical hypoperfusion (Gariepy et al., 2017). Here, we investigated whether cyclooxygenase inhibition might also ameliorate the CSD-evoked decreases in tpO2 and interfere with the associated meningeal afferent responses. Having identified such effects, we further tested whether a preemptive increase in CBF to counteract the CSD-evoked hypoperfusion and reduced tpO2, using the ATP-sensitive potassium [K(ATP)] channel opener levcromakalim (Kleppisch and Nelson, 1995; Shin et al., 2003) might also inhibit the enhanced responses of meningeal afferents. Our data suggest that while cyclooxygenase inhibition and K(ATP) channel opening can counteract the CSD-related cortical hypoperfusion and reduced tpO2, they exert distinct inhibitory effects on the associated activation and sensitization of meningeal afferents, thus questioning the direct contribution of these cortical metabolic perturbations to meningeal nociception and the onset of headache in migraine with aura.
Materials and Methods
Animals, anesthesia, and surgical preparation.
The current study used Sprague Dawley rats (males; 250–350 g; Taconic). Rats were housed 3 per cage under a 12 h light/dark cycle, in a temperature- and humidity-controlled room in the animal care facility of the Beth Israel Deaconess Medical Center (Boston, MA). Food and water were available ad libitum. All experimental procedures followed the Guide for Care and Use of Laboratory Animal Resources (NIH publication No. 85–23, revised 1996), were approved by the Animal Care and Use Committee of the Beth Israel Deaconess Medical Center, and were in compliance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. Animals were deeply anesthetized with urethane (1.5 g/kg, i.p.) and mounted on a stereotaxic frame (Kopf Instruments). Core temperature was kept at 37.5–38°C using a homoeothermic control system. Animals were intubated and breathed spontaneously room air enriched with O2. Physiological parameters were collected throughout the experiments using PhysioSuite (Kent Scientific) and CapStar-100 (CWE). Data used in this report were obtained from animals exhibiting physiological levels of oxygen saturation (>95%), heart rate (350–450 beats/min), and end-tidal CO2 (3.5–4.5%). A saline-cooled dental drill was used to perform three separate craniotomies (Fig. 1A). One craniotomy was used to expose the left transverse sinus and the posterior part of the superior sagittal sinus, as well as the adjacent cranial dura, extending ∼2 mm rostral to the transverse sinus. Another small craniotomy (1 × 1 mm) was made in the calvaria over the right hemisphere, and was centered 2 mm caudal and 2 mm lateral to bregma to allow insertion of the recording electrode. An additional small burr hole (diameter, 0.5 mm) was drilled to expose a small area of the frontal cortex to induce CSD (Zhao and Levy, 2015, 2016). The exposed dura was bathed with a modified synthetic interstitial fluid containing 135 mm NaCl, 5 mm KCl, 1 mm MgCl2, 5 mm CaCl2, 10 mm glucose, and 10 mm HEPES, pH 7.2. The femoral vein was cannulated to allow for intravenous administration.
Recording of meningeal afferent activity.
Single-unit activity of meningeal afferents (1 afferent/rat) was recorded from their cell body in the ipsilateral (left) trigeminal ganglion using a contralateral approach, as described previously (Zhao and Levy, 2014, 2015, 2016; Fig. 1A) in which a platinum-coated tungsten microelectrode (impedance, 50–100 kΩ; FHC) is advanced through the right hemisphere into the left trigeminal ganglion using an angled (22.5°) trajectory. The insertion of the recording electrode using this approach does not produce CSD in the left cortical hemisphere (Zhao and Levy, 2015, 2016). Meningeal afferents were identified by their constant latency response to electrical stimuli applied to the dura above the ipsilateral transverse sinus (0.5 ms pulse, 5 mA, 0.5 Hz). The response latency was used to calculate conduction velocity (CV), based on a conduction distance to the trigeminal ganglion of 12.5 mm (Strassman et al., 1996). Neurons were classified as A-δ (1.5 ≤ CV ≤ 5 m/s) or C afferents (CV < 1.5 m/s). Neural activity was digitized and sampled at 10 KHz using power 1401/Spike 2 interface (CED). A real-time waveform discriminator (Spike 2 software, CED) was used to create and store a template for the action potential evoked by electrical stimulation, which was used to acquire and analyze afferent activity.
Assessment of changes in meningeal afferent mechanosensitivity.
Mechanical receptive fields of meningeal afferents were first identified by probing the exposed dura with a 2.0 g von Frey filament (Stoelting). The site of lowest mechanical threshold was further determined using monofilaments that exert lower forces (≥0.02 g). For quantitative determination of mechanical responsiveness, we applied mechanical stimuli to the site with the lowest threshold, using a servo force-controlled mechanical stimulator (Series 300B Dual Mode Servo System, Aurora Scientific), controlled via the power 1401 interface. The stimulus was delivered using a flat-ended cylindrical plastic probe that was attached to the tip of the stimulator arm. One of three probe diameters (0.5, 0.8, or 1.1 mm) was selected for each neuron, depending on the sensitivity of the neuron (Levy and Strassman, 2002). Stimulus trials for testing the mechanosensitivity of the afferent were made using a custom-written script for Spike 2 and consisted of “ramp and hold” stimuli (rise time, 100 ms; stimulus width, 2 s; interstimulus interval, 120 s). Each trial included a threshold stimulus (which normally evoked at baseline 1–3 Hz responses) followed by a suprathreshold stimulus (usually X2 of the threshold pressure). To minimize response desensitization, stimulus trials were delivered every 15 min throughout the experiment (Zhang et al., 2013; Zhao and Levy, 2016). Ongoing afferent discharge was recorded continuously between the stimulation trials. Responses to mechanical stimuli were determined during at least four consecutive trials before the elicitation of CSD. Data were analyzed only from afferents that exhibited consistent responses (variation of <0.5 Hz for threshold responses and <1.5 Hz for suprathreshold responses) during baseline recordings (Zhao and Levy, 2016).
Induction of CSD.
A single CSD episode was induced by quickly inserting a glass micropipette (diameter, 50 μm) ∼2 mm deep in the cortex for 2 s (Zhang et al., 2010). CSD was triggered in the left frontal cortex to avoid potential damage to the meningeal tissue near the tested receptive field of the afferent under study, which could have led to their activation and/or sensitization (Zhao and Levy, 2015).
Measurement of CSD-evoked changes in CBF using laser Doppler flowmetry.
Changes in CBF (Gariepy et al., 2017) were documented using a needle probe (diameter, 0.8 mm) connected to a laser Doppler flowmeter (Vasamedic). The probe was positioned just above the intact dura, ∼1 mm from the mechanical receptive field of the recorded afferent in an area devoid of large pial blood vessels (>100 μm). Such a system (using a near-infrared 785 nm probe and fiber separation of 0.25 mm) is expected to record blood flow changes at a cortical depth of ∼1 mm (Fabricius et al., 1997). The laser Doppler signal was digitized (100 Hz) and recorded using the power 1401/Spike 2. Laser Doppler recordings were obtained with the ambient lights turned off. Baseline data were based on recordings conducted for at least 30 min before CSD.
Measurement of cortical partial pressure of oxygen.
Cortical tpO2 was recorded using a modified Clark-type polarographic oxygen microelectrode (OX-10, Unisense) with a guard cathode. The oxygen electrode was connected to a high-impedance pico-amperometer (PA2000; Unisense) that sensed the currents of the oxygen electrodes. The tpO2 signal was digitized (100 Hz) and recorded using the Power 1401/Spike 2 interface. The microelectrode responded linearly to changes in oxygen concentration and was calibrated before and after each experiment in both air-saturated saline and in an oxygen-free solution consisting of 0.1 m of sodium-ascorbate and 0.1 m NaOH dissolved in saline. This approach has been used previously to record changes in cortical tpO2 in response to CSD (Takano et al., 2007; Piilgaard and Lauritzen, 2009; Thrane et al., 2013). Changes in tpO2 values were recorded at a cortical depth of 100–200 μm, with the electrode placed <0.5 mm away from the laser Doppler flowmetry (LDF) probe. Baseline tpO2 values were recorded for at least 30 min before CSD.
Electrophysiological recording of CSD.
A tungsten microelectrode (impedance, 1–2Ω; FHC) was used to record multiunit activity at a depth of 200–400 μm in the left visual cortex (V1). A silver ground electrode was placed in the neck muscle. Signal was amplified and filtered (20K x, 300 Hz low frequency; 10 KHz high frequency; DAM-80/BMA400, WPI/CWE), and then digitized and sampled at 10 KHz. Cortical activity was recorded simultaneously with CBF changes, with the recording electrode placed <0.5 mm away from the LDF probe.
Drugs.
Naproxen sodium and levcromakalim were purchased from Tocris Bioscience. Naproxen sodium was made in 0.9% immediately before administration (10 mg/kg, i.v). A levcromakalim stock solution (10 mm) was made in DMSO and further diluted with synthetic interstitial fluid (final concentration, 500 μm) immediately before its application. The concentration of levcromakalim expected to be attained at the superficial cortex was estimated to be 100–1000 times lower (Moore et al., 1982; Roth et al., 2014; Zhao et al., 2017).
Data analyses and statistics.
All data were analyzed and plotted using Prism 7 software and are presented as median (IQR). Group data in graphs show all data points and are plotted as box-and-whisker plots with minimum and maximum values. For CSD-related changes in CBF and tpO2, raw data were processed with a custom-written script made for Spike 2. Baseline values were based on the average of the values collected during the 600 s before the induction of CSD. Off-line analyses for afferent responses were conducted using template matching in Spike 2. Criteria used to consider meningeal afferent activation and sensitization responses were based on our previous studies (Zhao and Levy, 2015, 2016). In brief, prolonged increase in the ongoing activity of afferents was considered if the firing rate increased above the upper end point of the 95% CI calculated for the baseline mean for >10 min. Acute afferent activation during the CSD wave was considered as an increased ongoing discharge rate that occurred ≥30 s following the pinprick and lasted ≤120 s. Mechanical sensitization was considered only if the afferent responses changed according to the following criteria: threshold and/or suprathreshold responses increased to a level greater than the upper end point of the 95% CI calculated for the baseline mean; increased responsiveness began during the first 60 min post-CSD; and sensitization lasted for at least 30 min. Changes in CSD-evoked responses of meningeal afferents in the presence of naproxen or levcromakalim were compared with data obtained in control animals using the same surgical approach and CSD induction method. Control data were obtained from experiments in animals treated intravenously with the naproxen vehicle (0.9% saline, 60 min before CSD induction, n = 24), or locally (i.e., continuous dural bathing) with synthetic interstitial fluid (n = 36). The two datasets were comparable and therefore combined to increase power. Differences in CSD-evoked afferent activation and sensitization propensities were analyzed using two-tailed χ2 tests. Within-group and between-group comparisons were made using the Wilcoxon test and Mann–Whitney U test, respectively. Results were considered to be significant at p < 0.05.
Results
CSD-evoked prolonged activation of meningeal afferents coincides with cortical hypoperfusion and reduced tpO2
Protracted cortical hypoperfusion and reduced cortical tpO2 were observed previously (Piilgaard and Lauritzen, 2009; Fordsmann et al., 2013) at time points corresponding to the prolonged activation of meningeal afferents (Zhang et al., 2010; Zhao and Levy, 2015). To examine whether the CSD-evoked cortical metabolic perturbations indeed coincide with prolonged meningeal afferent activation, we used simultaneous recording of meningeal afferent activity with CBF and tpO2 (n = 6). As the example in Figure 1B demonstrates, persistent meningeal afferent activation could be observed during the prolonged post-CSD cortical hypoperfusion and reduced tpO2 stage.

A, Experimental setup: three skull openings (red ovals) were made. A small burr hole was made over the left frontal cortex to elicit a single CSD event using a pinprick (PP) stimulation. Meningeal afferent activity was recorded in the left trigeminal ganglion (TG) using a tungsten microelectrode inserted through a craniotomy made over the contralateral hemisphere. An ipsilateral craniotomy was made to expose a part of the left transverse sinus (TS) and superior sagittal sinus (SSS) and their vicinity to search for mechanosensitive meningeal afferents. Quantitative mechanical stimuli were delivered to the receptive field of afferents using a feedback-controlled mechanical stimulator. A Laser Doppler Flowmeter (LDF) probe was placed over the cortex near the receptive field of afferents to record changes in CBF and validate the induction of CSD noninvasively. In some animals, we also recorded the induction of CSD by monitoring multiunit activity (MUA) of cortical neurons. An oxygen microelectrode was placed in the superficial cortex to record CSD-evoked changes in cortical tpO2. B, Example of raw data recording depicting the activation of a C afferent meningeal afferent (with an overdrawn spike waveform used for data analysis) following the elicitation of CSD with a PP during the prolonged reduction in CBF and tpO2. The afferent recording trace represents a peristimulus time histogram (1 s bin-size). Average ongoing activity rate (spikes/s) are denoted in parentheses. The activation phase and associated cortical hypoperfusion and reduced tpO2 are colored red. LDF values are in arbitrary units (AU). stim, Stimulus.
Suppression of CSD-evoked cortical hypoperfusion and decreased tpO2 by naproxen is not associated with reduced activation of meningeal afferents
We initially verified that systemic administration of naproxen, which was used to ensure cortical COX (cyclooxygenase) inhibition also under the nerve endings of meningeal afferents branches localized to areas not exposed by the craniotomy, could also block the CSD-evoked cortical hypoperfusion, as we previously have shown using topical administration (Gariepy et al., 2017). Because cortical oxygenation level depends in large part on cortical perfusion (Offenhauser et al., 2005), we further examined whether blocking the CSD-evoked cortical hypoperfusion with naproxen might inhibit the reduction in cortical tpO2. When compared with control animals (n = 22), naproxen treatment (n = 22) augmented the CSD-related acute cortical hyperemic response and blocked, as expected, the protracted cortical hypoperfusion (Fig. 2). Using simultaneous recording of CBF and tpO2, we observed that the inhibitory effect of naproxen on the CSD-evoked cortical hypoperfusion was associated with altered tpO2 responses to CSD (Fig. 3). In the presence of naproxen (n = 6), CSD evoked an initial increase in tpO2 [Δ10.03 mmHg (5.22); W = 6.0; p = 0.03; Fig. 3B, phase 1 and arrowhead in the insert] that was smaller than in vehicle-control animals [n = 11; Δ14.18 mmHg (3.87); U = 7.0; p = 0.007; Fig. 3C]. While in control animals the hyperoxia phase was followed by acute hypoxia [bottom values: 17.66 mmHg (18.64); W = −66.0; p = 0.001], which lasted 110.0 s (70.30; Fig. 3A; phase 2), naproxen blunted this hypoxic response, and instead tpO2 values increased [113.4 mmHg (90.24); W = 21.0; p = 0.015; Fig. 3B, phase 2]. This hyperoxia phase returned to baseline levels at 15–25 min [59.02 mmHg (39.63); W = 7.0; p = 0.29; phase 3, Fig. 3B]. Overall, when compared with control animals, naproxen inhibited both the acute hypoxia (phase 2; U = 0; p < 0.001; Fig. 3D) and prolonged decrease in tpO2 (phase 3; U = 3; p = 0.005; Fig. 3E).

The effect of systemic naproxen on CSD-induced changes in CBF. A, Changes in CBF following induction of CSD in vehicle- and naproxen-treated animals. B, CBF changes during the hyperemia stage, showing the enhancing effect of naproxen. C, CBF changes during the hypoperfusion phase, 10–60 min following CSD, depicting the inhibitory effect of naproxen. p Values are based on a Mann–Whitney U test. PP, Pinprick.

Naproxen inhibits CSD-induced decreases in cortical tpO2. A, B, Time course of changes in tpO2 following CSD induction in control (A) and naproxen-treated animals (B). C–E, Comparisons between CSD-evoked changes in control and naproxen-treated animals during the hyperoxia phase (phase 1), the acute hypoxia phase (phase 2, in control animals), and at 15–25 min post-CSD (phase 3). p Values are based on a Mann–Whitney U test. PP, Pinprick.
We next examined whether the suppressing effects of naproxen on CSD-evoked cortical hypoperfusion and decreased tpO2 might be associated with reduced activation of meningeal afferents. A-δ afferents recorded from naproxen-treated animals (n = 15) had baseline ongoing activity similar to that in control vehicle-treated animals [n = 26; 0.04 spikes/s (0.20) vs 0.05 spikes/s (0.25); U = 189.0; p = 0.87]. Baseline ongoing activity of C afferents was also not different between the naproxen group (n = 16) and control group [n = 34; 1.08 spikes/s (1.58) vs 0.57 spikes/s (1.35); U = 215.0; p = 0.24]. In naproxen-treated animals, CSD evoked acute activation in 5 of 15 A-δ afferents and 6 of 16 C afferents, ratios that were not significantly different from those observed in control animals [9 of 26 A-δ afferents, 14 of 34 C afferents; χ2 = 0.007; p = 0.99 for the A-δ afferent population; χ2 = 0.06; p = 0.80 for the C afferent populations; χ2 = 0.07; p = 0.79 for the combined population]. When compared with the control group, naproxen also did not affect the magnitude of the immediate activation in either A-δ afferents [4.2-fold (3.50) vs 3.6-fold (2.10); U = 22.0; p = 0.99] or in C afferents [1.6-fold (0.50) vs 2.1-fold (1.40); U = 30.0; p = 0.35, Fig. 4C].

Naproxen treatment does not inhibit CSD-evoked acute activation of meningeal afferents. A, B, Examples of experimental trials depicting the acute activation of an A-δ afferent (A) and a C afferent (B) following the induction of CSD using a pinprick (PP) stimulus in animals treated with naproxen. Traces represent a post-stimulus time histogram (1 s bin size). The average ongoing activity rate (spikes/s) is denoted in parentheses. Red areas represent the acute phase of afferent activation during CSD. Raw LDF traces are provided below each recording showing the CSD-evoked increase in CBF. C, D, Comparisons of the magnitude of change in ongoing activity between afferents recorded in control and naproxen-treated animals depicting activated (C) and nonactivated afferents (D). p Values are based on a Mann–Whitney U test.
The likelihood of developing a prolonged increase in ongoing afferent activity following CSD in naproxen-treated animals (Fig. 5A,B, examples) was also not different when compared with that observed in control animals (A-δ afferents: 7 of 15 vs 13 of 26, χ2 = 0.04; p = 0.84; C afferents: 7 of 16 vs 22 of 34, χ2 = 1.96; p = 0.16; combined population: χ2 = 1.427; p = 0.23). Naproxen treatment also did not affect any of the CSD-evoked afferent activation parameters: persistent activation developed in A-δ afferents with a delay of 25 min (40), which was no different than in control animals [10 min (5.0); U = 33.0; p = 0.34, Fig. 5C]. The activation delay observed in C afferents in naproxen-treated animals [15 min (31.25)] was also not different than that in controls [5 min (17.5); U = 67.5; p = 0.58, Fig. 5C]. The duration of CSD-evoked prolonged activation was also unaffected by naproxen: A-δ afferents were activated for 15 min (15) vs 30 min (22.5) in the control group (U = 22.0; p = 0.07, Fig. 5D); C-afferents were activated for 30 min (31.25) vs 35 min (30.0) in the control group (U = 58.0; p = 0.34; Fig. 5D). Finally, when compared with controls, naproxen treatment did not reduce the magnitude of the afferent activation observed in either population [A-δ afferents: 2.3-fold (3.0)] vs [3-fold (3.8); U = 39.0, p = 0.64; C afferents: 1.8-fold (3.0) vs 1.5-fold (0.9); U = 51.0; p = 0.18, Fig. 5E]. In afferents deemed not activated by CSD, the changes in ongoing activity following CSD were also not different between the naproxen and control groups, in both the A-δ afferent (U = 41.0; p = 0.8) and C afferent (U = 35.0; p = 0.2) populations (Fig. 5F).

Naproxen treatment does not inhibit CSD-evoked prolonged activation of meningeal afferents. A, B, Experimental trials depicting the CSD-evoked prolonged activation of an A-δ afferent (A) and a C afferent (B) in animals treated with naproxen. C–E, Naproxen treatment did not affect the onset latency of the afferent activation (C), its duration (D), or its magnitude (E). Changes in ongoing activity rate in nonactivated afferents in naproxen-treated animals were not different than those in controls (F). p Values are based on a Mann–Whitney U test. PP, Pinprick.
Naproxen inhibits CSD-evoked meningeal afferent mechanosensitization
We previously proposed that the activation and mechanosensitization of meningeal afferents that occur following CSD are mediated via distinct mechanisms (Zhao and Levy, 2016). We therefore examined next in 8 A-δ and 7 C afferents whether the inhibitory effects of naproxen on the cortical hypoperfusion and decreased tpO2 might be associated with amelioration of the post-CSD mechanosensitization response. Because the characteristics of the CSD-evoked sensitizations of the A-δ and C afferents are similar (Zhao and Levy, 2016), we combined the data from the two afferent populations for comparisons with the data obtained from vehicle-treated animals (10 A-δ afferents and 13 C afferents). At baseline, the number of dural mechanosensitive receptive fields and the mechanical responsiveness of afferents recorded in naproxen-treated animals were similar to those in the control group (Table 1). Unlike its ability to inhibit the CSD-evoked activation of meningeal afferents, naproxen blocked the afferent sensitization responses, at both the threshold level [3 of 15 vs 13 of 23; χ(2) = 4.97; p = 0.03; Fig. 6C] and the suprathreshold level [2 of 15 vs 13 of 23; χ(2) = 7.09; p = 0.008; Fig. 6]. When sensitization was defined as enhanced responsiveness at either stimulus level, there were also fewer sensitized afferents in the naproxen group [4 of 15 vs 17 of 23; χ(2) = 9.084; p < 0.003]. When the responses of all sensitized and nonsensitized units were combined, naproxen also had an overall inhibitory effect on the changes in threshold responses [Δ0 spike/s (0.6) vs Δ0.39 spikes/s (1.43); U = 90.5; p = 0.01, Fig. 6G] and suprathreshold responses [0.86-fold (0.5) vs 1.2-fold (0.5); U = 22.0; p = 0.004, Fig. 6H].
Baseline mechanical response properties of meningeal afferents in animals treated with vehicle, naproxen, or levcromakalim

Naproxen inhibits CSD-induced mechanical sensitization of meningeal afferents. A, B, Examples of experimental trials depicting the responses (post-stimulus time histogram; 0.1 s) of two C afferents to threshold (TH) and suprathreshold (STH) mechanical stimuli (green traces) applied to the dural receptive field during baseline recording and then at 30 min following CSD elicitation in control (A) and naproxen-treated animals (B). C–F, Note the lack of sensitization following naproxen treatment. Individual threshold and suprathreshold responses of sensitized and nonsensitized afferents in control and naproxen-treated animals. G, H, Magnitudes of CSD-related threshold (G) and suprathreshold (H) changes in mechanosensitivity depicting the inhibition of sensitization by naproxen. p Values are based on a Mann–Whitney U test.
The K(ATP) channel opener Levcromakalim counteracts CSD-evoked cortical hypoperfusion and reduced tpO2
Naproxen may inhibit the CSD-evoked afferent mechanosensitization indirectly, by blocking the cortical hypoperfusion and reduced tissue oxygenation. However, it is also possible that the inhibition of sensitization is independent of these cortical metabolic effects, since prostanoids (elaborated by CSD), whose synthesis is blocked by naproxen, exert a direct sensitizing effect on meningeal afferents (independent of metabolic effects; Zhang et al., 2007), and naproxen also inhibits afferent sensitization induced by a mixture of inflammatory mediators (Levy et al., 2008). We therefore asked next whether opposing the CSD-evoked cortical hypoperfusion and the associated decrease in tpO2 using an alternative, cyclooxygenase-independent approach might also inhibit the afferent sensitization response. We postulated that local application of a potent cortical vasodilator could serve this purpose and tested the effects of levcromakalim, a selective K(ATP) channel opener with strong cortical vasorelaxation properties (Reid et al., 1995). In preliminary experiments, we observed that the maximum cortical vasodilatory effect of levcromakalim occurs within 15–25 min. Therefore, to counteract the CSD-evoked persistent hypoperfusion, we used a pretreatment approach and started levcromakalim treatment 30 min before CSD elicitation. Using this protocol, before CSD induction, levcromakalim (n = 28) increased basal CBF [196.10% (72.7); W = 231.0; p < 0.0001, Fig. 7A]. Levcromakalim did not inhibit the elicitation of CSD (n = 3, Fig. 7C), which was associated with a significant cortical hyperemic response, albeit smaller in magnitude when compared with the response in control animals [105.3% (1.9) vs 180.3% (61.6); U = 0.0; p < 0.0001; Fig. 7E]. In the presence of levcromakalim, there was a decrease in CBF following CSD, but values did not attain the hypoperfusion level observed in control animals (Fig. 7F; U = 51.0; p < 0.0001); up to the 45 min post-CSD period, values were not different from those observed during the predrug baseline period [99.56% (39.67); W = −8.0; p > 0.99] and increased afterward [119.1% (54.9)] at 60 min post-CSD (W = 366.0; p < 0.0001).

Effects of the K(ATP) channel opener levcromakalim on CSD elicitation and CSD-evoked CBF changes. A, Levcromakalim increased basal CBF. B, C, Examples of the combined recording of intracortical neural multiunit activity (MUA; post-stimulus time histogram; 1 s in middle panels) and CBF (bottom panels) showing a similar induction of CSD in control (B) and levcromakalim-treated animals (C). Note the initial depolarization (increased cortical activity; arrows) followed by the suppression and gradual recovery of neural activity. Also note the CSD-evoked cortical hyperemic response in the control animal and the diminished response in levcromakalim-treated animal (arrowheads). D, Time course changes of CBF in control and levcromakalim-treated animals following PP-induced CSD. Note that despite the decrease in CBF in levcromakalim-treated animals, values remained near predrug baseline values (denoted as 100%) unlike the marked decrease in control animals. E, CBF changes in control and levcromakalim-treated animals during the hyperemia stage. F, CBF changes during the hypoperfusion phase, 10–60 min following CSD. p Values are based on a Mann–Whitney U test. PP, Pinprick.
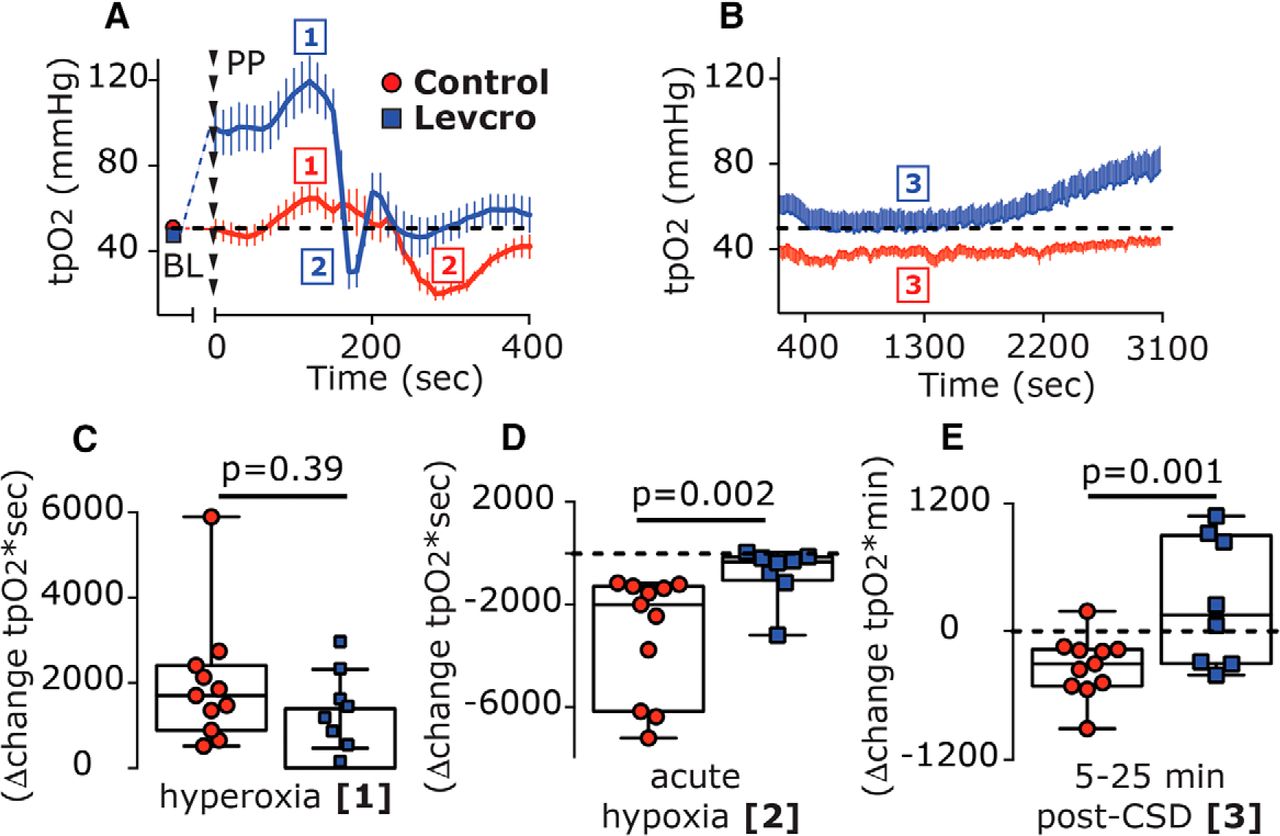
We next tested whether levcromakalim might also affect the CSD-evoked changes in cortical tpO2 (n = 8). Before CSD elicitation, levcromakalim increased the basal tpO2 [Δ43.55 mmHg (35.3); W = 36.0; p = 0.008; Fig. 8A]. In the presence of levcromakalim, CSD gave rise to a further, brief increase in tpO2 [peak, 120.5 mmHg (56.3); W = 36.0; p = 0.008; Fig. 8A, phase 1] with a magnitude similar to that observed in control animals [Δ18.22 mmHg (10.32) vs Δ14.18 mmHg (3.87); U = 24.0; p = 0.11]. While the duration of this hyperoxic phase was shorter than that in control animals [70.0 s (30.0) vs 100 s (50.0); U = 11.0; p = 0.05], overall the hyperoxia phase [area under the curve (AUC)] was not different between the two groups (U = 33.0; p = 0.39; Fig. 8C). Following the brief hyperoxia, tpO2 values decreased transiently, reaching hypoxic values [18.5 mmHg (26.36)] that were not different from those in control animals [17.66 mmHg (18.64); U = 32.0; p = 0.35]. However, when compared with control animals, levcromakalim shortened this hypoxic phase [20 s (27.5) vs 110 s (70.0); U = 3.5; p = 0.0002], yielding a reduced hypoxic response [246.6 mmHg*sec (676.0) vs 2001.0 mmHg*sec (4883.9); U = 7.0; p = 0.002; Fig. 8D]. Levcromakalim also prevented the prolonged reduction in tpO2 when compared with control animals (U = 15.0; p = 0.001; Fig. 8B, Phase 3, E), with values not different than baseline at the 5–10 min time point [49.49 mmHg (43.2); U = 26.0; p = 0.46] and the 10–20 min time point [47.16 mmHg (44.51); U = 31.0; p = 0.64].

Levcromakalim inhibits CSD-evoked cortical hypoxia. A, Time course changes (median and IQR) in cortical tpO2 values at baseline (BL) and during the acute hyperoxia and hypoxia phases in response to CSD induction with pinprick (phases 1 and 2, respectively) in control and levcromakalim-treated animals. Note the shorter duration of the hypoxia phase following levcromakalim treatment. B, The prolonged, post-CSD decrease in tpO2 (phase 3) in control animals and its amelioration by levcromakalim are shown. Dashed line represents predrug baseline values. C–E, Differences between control and levcromakalim-treated animals in the hyperoxic response (AUC, phase 1; C), acute hypoxia (AUC, phase 2; D), and the prolonged decrease in tpO2 at 5–25 min post-CSD (AUC, phase 3; E). p Values are based on a Mann–Whitney U test. Levcro, levcromakalim; PP, pinprick.
Levcromakalim inhibits CSD-evoked activation but not sensitization of meningeal afferents
Given its ability to counteract the cortical metabolic perturbations evoked by CSD, we next determined the effect of levcromakalim treatment on the development of CSD-evoked afferent activation in 9 A-δ and 11 C afferents. In the presence of levcromakalim, baseline ongoing activity in A-δ afferents [0.13 spikes/s (0.31)] and in C afferents [0.28 (0.89)] were not different from that in control animals (U = 91.0, p = 0.32 and U = 156.0, p = 0.41, respectively). Levcromakalim treatment, however, significantly inhibited the acute afferent activation [control group: A-δ afferents, 0 of 9 vs 9 of 26; χ(1) = 4.19, p = 0.04; C afferents, 1 of 11 vs 14 of 34; χ(2) = 3.85, p = 0.049); combined population: 1 of 20 vs 23 of 60; χ(2) = 7.937; p = 0.005; Fig. 9C]. Levcromakalim also inhibited the prolonged afferent activation when compared with control group [A-δ afferents, 1 of 9 vs 13 of 26; χ(2) = 4.21; p = 0.04; C afferents, 3 of 11 vs 22 of 34; χ(2) = 4.717; p = 0.03; combined population: 4 of 20 vs 35 of 60; χ(2) = 8.82; p = 0.003; Fig. 9D].

Levcromakalim inhibits CSD-evoked acute and prolonged meningeal afferent activation. A, B, Examples of single-unit recording of an A-δ afferent (A) and a C afferent (B), combined with laser Doppler flowmetry, depicting the lack of acute and prolonged afferents following the elicitation of CSD with a pinprick in animals treated with levcromakalim. C, In the presence of levcromakalim, only one C afferent became acutely activated (open square outside the minimum/maximum box). D, Prolonged activation was noted only in one A-δ and three C afferents (open circle and squares outside the minimum/maximum box). PP, Pinprick.
Finally, we tested the effect of levcromakalim on the development of CSD-evoked mechanical sensitization in 7 A-δ afferents and 10 C afferents, which had a similar number of identified dural mechanosensitive receptive fields and baseline mechanical responsiveness as the afferents tested in the control group (Table 1). Unlike its effect on the CSD-evoked afferent activation, levcromakalim treatment did not inhibit the sensitization response (Fig. 10A, example). When compared with control experiments, levcromakalim did not reduce the relative number of afferents that were deemed sensitized when using the threshold [10 of 17 vs 13 of 23 affected afferents; χ(2) = 0.04; p = 0.85] or the suprathreshold [6 of 17 vs 14 of 23 affected afferents; χ(2) = 1.1, p = 0.29] stimuli. When sensitization was defined as enhanced responsiveness at either stimulus level, the ratio of sensitized/nonsensitized was also not different when compared with that observed in control animals [11 of 17 vs 17 of 23; χ(2) = 0.2, p = 0.66]. When compared with controls, levcromakalim also did not decrease the magnitude of the sensitization responses to threshold stimuli [Δ1.73 spikes/s (3.36) vs Δ1.3 spikes/s (2.18); U = 38.5; p = 0.10; Fig. 10D] or suprathreshold stimuli [1.39-fold (0.5) vs 1.37-fold (0.54); U = 34.0; p = 0.54; Fig. 10E].

Levcromakalim does not inhibit CSD-evoked mechanical sensitization of meningeal afferents. A, An example of an experimental trial depicting the CSD-evoked increase in mechanical responsiveness of a C afferent recorded from a levcromakalim-treated animal. B, C, Note that CSD-evoked sensitization at both the threshold and suprathreshold levels (poststimulus time histogram; 0.1 s). Individual threshold (B) and suprathreshold (C) responses of sensitized and nonsensitized afferents. D, E, Comparisons between the magnitudes of CSD-related changes in mechanosensitivity at the threshold (D) and suprathreshold levels (E) depicting similar CSD-evoked sensitization responses in control and levcromakalim-treated animals. p Values are based on a Mann–Whitney U test. AU, Arbitrary units; Levcro, levcromakalim; STH, suprathreshold; TH, threshold.
Discussion
Spreading cortical hypoperfusion has been demonstrated during the early stage of migraine with aura and likely reflects a CSD-related vascular event (Ayata and Lauritzen, 2015), and decreased cortical oxygenation has been suggested to trigger migraine headache (Schoonman et al., 2006; Arngrim et al., 2016). In our CSD rodent model, the cortical hypoperfusion and decreased tpO2 coincided with the emergence of the prolonged activation of meningeal afferents, suggesting a potential interaction. However, a key finding of this study was that despite exerting a powerful inhibitory effect on the protracted cortical hypoperfusion and the related decrease in oxygenation, naproxen did not ameliorate the activation of meningeal afferents in response to CSD. Our data thus point to a dissociation between these CSD-related cortical metabolic perturbations, or downstream processes, and the mechanism responsible for driving meningeal afferent activity. The ineffectiveness of naproxen was somewhat surprising in light of its ability to reduce the persistent activation of meningeal afferents following application of a mixture of inflammatory mediators to their meningeal receptive field (Levy et al., 2008)—a model of meningeal nociception (Strassman et al., 1996) and migraine pain (Oshinsky and Gomonchareonsiri, 2007; Edelmayer et al., 2009; De Felice et al., 2013). This discrepancy suggests that the prolonged activation of meningeal afferents in the wake of CSD occurs via a distinct mechanism, one that does not involve local nociceptive actions of these classical inflammatory mediators (Strassman et al., 1996) or vasoconstricting prostanoids that mediate the cortical hypoperfusion following CSD (Shibata et al., 1992; Gariepy et al., 2017).
The cortical hyperemia that occurs during the CSD wave is likely driven in part by the acute activation of meningeal afferents, and the ensuing release of vasodilatory neuropeptides such as CGRP (Busija et al., 2008). We found that naproxen also increased the initial cortical hyperemia, which likely mediated the conversion of the acute hypoxia phase to hyperoxia, but these effects did not influence the acute activation of the afferents. These data support the view that cortical vasodilation and its related increases in tissue oxygenation do not drive meningeal afferent activity (Levy and Burstein, 2011), and that during the hyperemic phase of the CSD there is a concurrent release of opposing vasoconstricting prostanoids.
During the prolonged cortical hypoperfusion phase, CSD also gives rise to persistent vasodilatation of the middle meningeal artery (Bolay et al., 2002), a vascular response proposed to be mediated by prolonged activation of dural afferents and downstream signaling via a central parasympathetic reflex (Bolay et al., 2002). Importantly, a recent study (Karatas et al., 2013) demonstrated that this CSD-evoked meningeal vasodilatory response could be blocked by systemic administration of naproxen. That naproxen did not inhibit the CSD-evoked activation of meningeal afferents together with previous data showing its inability to block parasympathetic outflow to the cranium (Akerman et al., 2012) suggests therefore that CSD-evoked dural vasodilatation may not be driven by enhanced ongoing meningeal afferent activity. Our present results thus call into question the use of dural artery vasodilation as a surrogate of increased meningeal afferent activity and the activation of the migraine pain pathway in preclinical studies.
We found that counteracting the protracted cortical hypoperfusion and prolonged decrease in tpO2 by preemptively increasing CBF using levcromakalim was associated with inhibition of the acute and prolonged afferent activation phases. We propose, however, that the inhibitory effect of levcromakalim on these afferent responses did not involve the normalization of CBF or tpO2 given that naproxen, which abrogated the cortical metabolic perturbations following CSD, failed to inhibit the afferent activation. We cannot exclude the possibility that the cortical vasodilation and increased tpO2 induced by levcromakalim, before the onset of CSD, contributed somehow to the blockade of the post-CSD afferent activation. However, the finding that levcromakalim did not affect the basal ongoing activity of afferents suggests that the vascular effects of levcromakalim were unlikely to mediate to the inhibition of the CSD-evoked afferent activation.
Cortical neurons also express K(ATP) channels (Karschin et al., 1997), and their prolonged activation by levcromakalim could have resulted in diminished cortical excitability (Soundarapandian et al., 2007), potentially affecting the elicitation of CSD. However, levcromakalim did not inhibit the induction of CSD, and particularly not the initial strong depolarization before the prolonged suppression of cortical neural activity. K(ATP) activation in cortical neurons could, nonetheless, reduce the release of pronociceptive transmitters, such as glutamate (Zhao et al., 2013), which may contribute to the acute activation, and potentially to the prolonged activation, of meningeal afferents. Cortical astrocytes also express K(ATP) channels (Thomzig et al., 2001), and their activation could potentially decrease the high extracellular K+ level that is attained during the passage of the CSD wave (Kraig and Nicholson, 1978) by enhancing K+ “spatial buffering” (Kofuji and Newman, 2004). In response to CSD, elaborated K+ in the cortical extracellular space has been suggested to diffuse into the leptomeninges and promote the acute activation of meningeal afferents and potentially also their prolonged activation, at least in part (Pietrobon and Moskowitz, 2013; Zhao and Levy, 2017). Activation of astrocytic K(ATP) channels, leading to decreased parenchymal K+, thus could have potentially inhibited the CSD-evoked afferent activation. Our recent finding that K+-evoked acute activation of meningeal afferents was not associated with mechanical sensitization (Zhao and Levy, 2017) is also in agreement with the observation that levcromakalim did not inhibit the CSD-evoked mechanical sensitization response. Finally, activation of K(ATP) channels expressed at the peripheral nerve endings of meningeal afferents could have also contributed to the inhibition of the afferent activation by reducing their overall excitability (Chi et al., 2007). However, the finding that levcromakalim did not reduce basal ongoing discharge or inhibit the mechanical sensitization argues against such a mechanism.
We found that naproxen distinctly inhibited the CSD-evoked mechanical sensitization of meningeal afferents. That levcromakalim treatment greatly reduced the acute hypoxia phase and normalized the prolonged cortical hypoperfusion and decreased tpO2, but did not inhibit the enhanced mechanical responsiveness, suggests, however, that the mechanism underlying the mechanical sensitization following CSD is unrelated to the changes in cortical perfusion and tpO2. It should be noted that while levcromakalim treatment normalized CBF and tpO2 (to values not different than those observed before CSD), it did not inhibit the process that led to their decreases following CSD. We therefore propose that the mediators that contribute to these metabolic changes (likely cyclooxygenase derived; see below) are responsible for the afferent sensitization rather than the cortical hypoperfusion and abnormally reduced tpO2 per se. It is possible that the acute hypoxic phase that occurred in levcromakalim-treated animals (albeit for only a fraction of the time noted in control animals) may have contributed somehow to the afferent sensitization process. However, we also observed brief cortical hypoxia in some naproxen-treated animals, further suggesting a dissociation between the acute hypoxic phase and the afferent changes.
The discrepancy between the effects of naproxen and levcromakalim on the CSD-evoked afferent activation and mechanical sensitization responses supports the view that these two migraine-related nociceptive responses are mediated by distinct mechanisms (Levy and Strassman, 2004; Zhang et al., 2011a, 2013; Zhao and Levy, 2016). The site of action of naproxen and the nature of the cyclooxygenase-derived factors that distinctly promote the sensitization of meningeal afferents following CSD require further studies. The release of cyclooxygenase-derived sensitizing mediators from cortical neurons and glial limitans astrocytes that interface with the meninges (Xu et al., 2004) could contribute to the afferent sensitization. The mode of cortex-to-meninges transport of cyclooxygenase-derived prostanoids could involve diffusion or bulk flow via the glymphatic system (Iliff et al., 2012; Schain et al., 2017). The transport of sensitizing factors from the parenchymal interstitial space into the subarachnoid CSF could influence primarily leptomeningeal afferents and meningeal afferents that terminate within arachnoid granulations (von Düring and Andres, 1991; Fricke et al., 1997). Additional flow of sensitizing mediators from the subarachnoid CSF into the dural lymphatics system (Raper et al., 2016; Louveau et al., 2017) could affect the responses of dural afferents. Naproxen treatment could have also inhibited the CSD-evoked meningeal afferent sensitization by targeting constitutively expressed meningeal cyclooxygenase in meningeal vascular and immune cells (Zhang et al., 2009) and the ensuing release of sensitizing prostanoids (Zhang et al., 2011a).
In summary, our findings point to a dissociation between the CSD-evoked cortical metabolic perturbations and the prolonged activation and mechanical sensitization of meningeal afferents. The findings that K(ATP) activation and cyclooxygenase inhibition have distinct effects on the CSD-evoked activation and sensitization of meningeal afferents may explain the ineffectiveness of cyclooxygenase inhibitors in many cases of migraine (Suthisisang et al., 2010). A combination therapy, which includes a cyclooxygenase inhibitor and a K(ATP) channel opener, may be explored for treating the headache in migraine with aura.
Footnotes
The study was supported by National Institutes of Health Grants NS-086830, NS-078263, and NS-101405 to D.L. We thank Dr. A. M. Strassman for helpful comments on the manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dan Levy, Department of Anesthesia, Critical Care and Pain Medicine, Beth Israel Deaconess Medical Center, DA717; 330 Brookline Avenue, Boston MA 02215. dlevy1{at}bidmc.harvard.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}