

Abstract
Many Preterm-born children suffer from neurobehavioral disorders. Premature birth terminates the hypoxic in utero environment and supply of maternal hormones. As the production of interneurons continues until the end of pregnancy, we hypothesized that premature birth would disrupt interneuron production and that restoration of the hypoxic milieu or estrogen treatment might reverse interneuron generation. To test these hypotheses, we compared interneuronal progenitors in the medial ganglionic eminences (MGEs), lateral ganglionic eminences (LGEs), and caudal ganglionic eminences (CGEs) between preterm-born [born on embryonic day (E) 29; examined on postnatal day (D) 3 and D7] and term-born (born on E32; examined on D0 and D4) rabbits at equivalent postconceptional ages. We found that both total and cycling Nkx2.1+, Dlx2+, and Sox2+ cells were more abundant in the MGEs of preterm rabbits at D3 compared with term rabbits at D0, but not in D7 preterm relative to D4 term pups. Total Nkx2.1+ progenitors were also more numerous in the LGEs of preterm pups at D3 compared with term rabbits at D0. Dlx2+ cells in CGEs were comparable between preterm and term pups. Simulation of hypoxia by dimethyloxalylglycine treatment did not affect the number of interneuronal progenitors. However, estrogen treatment reduced the density of total and proliferating Nkx2.1+ and Dlx2+ cells in the MGEs and enhanced Ascl1 transcription factor. Estrogen treatment also reduced Ki67, c-Myc, and phosphorylation of retinoblastoma protein, suggesting inhibition of the G1-to-S phase transition. Hence, preterm birth disrupts interneuron neurogenesis in the MGE and estrogen treatment reverses interneuron neurogenesis in preterm newborns by cell-cycle inhibition and elevation of Ascl1. We speculate that estrogen replacement might partially restore neurogenesis in human premature infants.
SIGNIFICANCE STATEMENT Prematurity results in developmental delays and neurobehavioral disorders, which might be ascribed to disturbances in the development of cortical interneurons. Here, we show that preterm birth disrupts interneuron neurogenesis in the medial ganglionic eminence (MGE) and, more importantly, that estrogen treatment reverses this perturbation in the population of interneuron progenitors in the MGE. The estrogen seems to restore neurogenesis by inhibiting the cell cycle and elevating Ascl1 expression. As preterm birth causes plasma estrogen level to drop 100-fold, the estrogen replacement in preterm infants is physiological. We speculate that estrogen replacement might ameliorate disruption in production of interneurons in human premature infants.
Introduction
Approximately 15 million infants worldwide are born prematurely and approximately 1.9 million infants are born with very low birth weight (<1500 g) every year. These infants display significant diminution in cortical gray matter volume and continue to exhibit impaired cortical growth even in childhood and adolescence (de Kieviet et al., 2012). They manifest with moderate to severe neurodevelopmental disability at 1 year of age (Dyet et al., 2006; Thompson et al., 2007) and a number of them suffer neurobehavioral disorders during childhood and adolescence, including inattention, hyperactivity, anxiety disorders, reduced social skills, epilepsy, autism, and others (Botting et al., 1997; Anderson et al., 2003, 2004; Delobel-Ayoub et al., 2009; de Kieviet et al., 2012). The neurobehavioral disorders in childhood and adolescence are attributed to perturbations in the structure and function of cortical interneurons (Marín, 2012). As interneuron neurogenesis continues to the end of human pregnancy, preterm birth might influence production of cortical interneurons (Malik et al., 2013). Therefore, we asked whether preterm birth would influence generation of interneurons and what the underlying mechanism(s) were in determining this influence.
Interneurons, inhibitory neurons in the brain, play important and powerful roles in modulating the electrical activity of the excitatory pyramidal cells (Kelsom and Lu, 2013; Kepecs and Fishell, 2014). The interneurons are usually GABAergic and identified by the protein markers they express. The following subtypes of interneurons have been identified: parvalbumin (PV), somatostatin, calretinin, vasointestinal peptide, and neuropeptide Y. In rodents, somatostatin+ and PV+ interneurons originate from the medial ganglionic eminence (MGE), whereas calretinin+ and vasointestinal peptide+ interneurons derive from the caudal ganglionic eminence (CGE; Wonders and Anderson, 2006). Also in rodents, ∼70% of cortical interneurons are derived from the MGE, 30% from the CGE, and 5% from the preoptic area (Wonders and Anderson, 2006). In humans as well, most of the cortical interneurons are generated in the ventral ganglionic eminence and the production of cortical interneurons persists in the MGE and CGE during the third trimester of pregnancy (Arshad et al., 2016). Within MGEs, Nkx2.1 is the master regulator of interneuron production and activates a cascade of genes, including Lhx6, Sox6, and Satb2 (Wonders and Anderson, 2006). Other key transcription factors for interneuron neurogenesis are Dlx1/2, Ascl1 (Mash1), and GSX1/2. These genes are controlled by coordinated actions of sonic hedgehog (Shh) signaling and, thus, Shh critically regulates specification and differentiation of interneurons (Kelsom and Lu, 2013).
Preterm birth interrupts intrauterine life, terminates the hypoxic in utero environment, and disrupts the supply of placental and maternal hormones, as well as growth factors. Estrogen and progesterone are the major maternal hormones, and a drop in estrogen level in mice with ovariectomy reduces the density of PV+ interneurons, which are restored after treatment with 17β estradiol (E2), a form of estrogen (Wu et al., 2014). In addition, estrogen offers neuroprotection by antiapoptotic and anti-inflammatory activity, and modulates neuronal plasticity by regulating dendritic spine and synapse formation (Amantea et al., 2005; Brann et al., 2007; Brinton, 2009). Thus, estrogen might modulate the development of interneurons. Despite this evidence, the effect of prematurity and estrogen treatment on interneuron production has not been studied. Therefore, we hypothesized that premature birth would disrupt interneuron neurogenesis and that induction of hypoxia or estrogen treatment might restore production of interneurons.
To test these hypotheses, we used a preterm rabbit model in which we evaluated neurogenesis by quantifying total and cycling interneuron progenitors in the MGEs of preterm-born and term-born rabbits at equivalent postconceptional ages. We found that Nkx2.1+, Dlx2+, and Sox2+ progenitors were more abundant in the MGEs of preterm rabbits compared with term controls, and that estrogen treatment restored the population of progenitors, elevated Ascl1 transcription factor, and reduced c-Myc and phosphoretinoblastoma (p-Rb; serine 807/811) protein. The study proposes that estrogen replacement might ameliorate disruption in interneuron neurogenesis in premature newborns.
Materials and Methods
Animals.
This study was performed after approval from the Institutional Animal Care and Use Committee of New York Medical College, Valhalla, New York. We used a preterm rabbit model that has been validated in our prior studies (Malik et al., 2013). The merits of using a rabbit model is that the rabbits are similar to humans in several ways: (1) the maximum growth of the brain occurs perinatally, (2) the brain is gyrencephalic, (3) the ganglionic eminences are relatively large, (4) the blood supply for the brain is from vertebral and internal carotid arteries, and (5) the maturation of lungs is complete before term, making them capable of survival with premature birth (Georgiadis et al., 2008; Muñoz-Moreno et al., 2013). More importantly, interneuron neurogenesis continues in pups born on embryonic day (E) 29 until postnatal day (D) 14, providing us with a unique opportunity to test the effect of prematurity on neurogenesis and study the underlying mechanisms. Timed-pregnant New Zealand rabbits were purchased from Charles River Laboratories. We performed Caesarean section to deliver the premature pups at E28.6 (rounded to E29 for simplicity) of gestational age (full term, 32 d). Newborn pups were reared in an infant incubator at a temperature of 35°C. We used rabbit milk replacer (Zoologic, PetAg) to gavage-feed the pups in a volume of ∼2 ml every 12 h (100 ml/kg/d) for the first 2 d, and feeds were advanced to 125, 150, and 200 ml/kg at D3, D5, and D7 respectively.
Estrogen and DMOG treatment.
The rabbit kits (E29) were treated sequentially with either intramuscular E2 or vehicle for 3 d and then killed. The treatment was initiated at birth and E2 was administered in a dose of 200 or 100 μg/kg, based on previous studies (Picazo et al., 2003; Twining et al., 2013; Corvino et al., 2015). DMSO was used as vehicle. To simulate hypoxia by stabilizing HIF-1, we treated E29 rabbit pups with dimethyloxalylglycine (DMOG; Cayman Chemical) starting immediately at birth. DMOG was administered in a dose of 100 mg/kg daily intramuscularly for 3 d, as was used previously to induce hypoxia in rabbit pups (Malik et al., 2013). The pups were killed at 72 h by using ketamine (35 mg/kg) and xylazine (5 mg/kg) anesthesia followed by decapitation. The comparison group received vehicle (15 μl of DMSO). Sex determination in the pups was done by performing quantitative real-time PCR for the SRY gene on the mRNA isolated from brain tissues.
Immunohistochemistry.
Immunohistochemical staining was performed as described previously (Ballabh et al., 2007). The primary antibodies used in experiments included the following: rabbit polyclonal Nkx2.1 (catalog #133638, Abcam), guinea pig polyclonal Dlx2 (provided by Dr. Kazuaki Yoshikawa, Osaka University, Japan), mouse monoclonal Ki67 (catalog #M7240, Dako), goat polyclonal Sox-2 (catalog #SC-6895, Santa Cruz Biotechnology). Secondary antibodies used were as follows: Alexa Fluor 488-donkey anti-mouse IgG (catalog #715-545-150), Alexa Fluor 488-AffiniPure donkey anti-rabbit IgG (catalog #711-165-152), Alexa Fluor 594-AffiniPure donkey anti-goat IgG (catalog #705-585-147) from Jackson ImmunoResearch Laboratories. Briefly, we hydrated the fixed sections in 0.1 m PBS, blocked the sections with normal donkey serum in PBS with 0.01% Triton-X, and then incubated with the primary antibodies diluted in PBS at 4°C overnight. After three washes in PBS, the sections were incubated with secondary antibody diluted in 1% normal goat serum in PBS at room temperature for 60 min. Finally, after washing in PBS, the processed sections were mounted with SlowFade Light Antifade reagent (Invitrogen) and were visualized under a Confocal microscope (Nikon Instruments). Stereology was performed using a fluorescent microscope (Axioskop 2 plus, Carl Zeiss) with a motorized specimen stage for automated sampling (Applied Scientific Instrumentation), CCD color video camera (Microfire, Optronics) and stereology software (Stereologer, Stereology Resource Center).
Stereological assessment of interneurons in the MGE and LGE.
We quantified a number of stereological parameters using a computerized software system (Stereologer, Stereology Resource Center), as described previously (Dummula et al., 2011). Briefly, 25-μm-thick coronal sections were cut using a cryostat with a section sampling interval of 90 μm to provide ≥5 sections at the level of midseptal nucleus. The sections were triple-labeled with Nkx2.1 or Dlx2 antibody with Ki67 and DAPI (nuclear stain) and were quantified as follows. The reference spaces (MGE and LGE) were outlined on the section under a 5× objective. The volume of the outlined area (reference space) was quantified using a point counting probe (frame, 25 × 25 μm; guard zone, 2 μm; interframe interval, 300 μm). Total and cycling (double-labeled with Ki67) Nkx2.1+ and Dlx2+ cells were counted using the optical dissector method (frame, 25 × 25 μm; guard zone, 2 μm; interframe interval, 280 μm). A coefficient of error of <0.10 was considered acceptable.
Western blot analyses.
We homogenized the frozen brain slice in a sample buffer (3% SDS, 10% glycerol, and 62.5 mm Tris-HCl, pH 7.4) using a mechanical homogenizer and then sonicated the lysate before centrifugation. Protein concentrations were measured using a BCA protein assay kit (Pierce Kit #23227, Thermo Fisher Scientific) and dilutions of BSA were used to create a standard curve. After boiling the samples in Laemmli buffer (catalog #161-0737, Bio-Rad), protein samples were separated by SDS-PAGE. Equal amounts of protein (20 μg) were loaded onto 4–15 or 4–20% gradient precast gels (Bio-Rad), based on the molecular weight of the target protein. Separated proteins were transferred onto polyvinylidene difluoride membranes by electrotransfer. Membranes were then incubated overnight in primary antibodies. We detected target proteins using a chemiluminescence ECL system (GE Healthcare) by using secondary antibodies conjugated with horseradish peroxidase (Jackson ImmunoResearch Laboratories). We stripped the membrane using stripping buffer (2.5% SDS, 0.7% 2-mercaptoethanol, 62.5 mm Tris-HCl, pH 6.8) and then incubated with β-actin antibody (catalog #A5316, Sigma-Aldrich) followed by secondary antibody and detection with chemiluminescence ECL system. As described previously (Ballabh et al., 2007; Vinukonda et al., 2016), the blots from each experiment were densitometrically analyzed using ImageJ, and optical density values for each protein of interest were normalized to β-actin. Antibodies used for Western blot analyses were rabbit polyclonal Sox6 (catalog #HPA001923, Sigma-Aldrich), mouse monoclonal Gad65 (Catalog# 26113, Abcam, Cambridge, MA, USA), mouse monoclonal Gad 67 (catalog #Mab5406, Millipore), mouse monoclonal Mash 1 (catalog #556604, BD Biosciences), mouse monoclonal Dlx1 (catalog #75-078, NeuroMab), mouse monoclonal Gli1 (catalog #49314, Abcam), rabbit p-Rb (serine 807/811; catalog #8516, Cell Signaling Technology), rabbit p-Rb (serine 780; catalog #9307, Cell Signaling Technology); rabbit cyclin-dependent kinase 6 (CDK6) polyclonal (SC7180, Santa Cruz Biotechnology), mouse monoclonal cyclin D2 (CCND2; Abcam), and mouse monoclonal β-actin (Sigma-Aldrich).
Quantitative real-time PCR.
Gene expression was quantified by real-time PCR, as described previously (Ballabh et al., 2007; Vinukonda et al., 2016). Briefly, total RNA was isolated using a RNAeasy Mini kit (catalog #74104, Qiagen) from a coronal brain slice taken at the level of the midseptal nucleus. cDNA was synthesized using Superscript II RT enzyme (catalog #05081955001, Roche) followed by real-time quantitation using an ABI Prism 7900HT detection system. TaqMan probes were bought from Life Technologies. Their assay IDs were as follows: GAPDH (Oc03823402_g1), Ascl1 (AJD1UIG), Lhx6 (AIT975S), CCND2 (AID1VWY), Gad1 (Hs01065893-m1), Gad2 (Hs00609534_m1), Dlx5 (Oc03395679_m1), and rabbit SRY (NM_001171148.1).
Statistics and analysis.
Data are presented as means ± SEM. To compare the total number of interneuron progenitors and the number of proliferating interneuron progenitors (Nkx2.1 and Dlx2) in ganglionic eminences between two groups (term and preterm) at day 3 (D3 preterm vs D0 term) and day 7 (D7 preterm vs D4 term), we used two-way ANOVA. Maturity (term vs preterm) and postnatal age (D3 or D7 vs term equivalent) were two independent variables. Similarly, using two-way ANOVA, we compared the number of progenitors, quantitative real-time PCR, and Western blot analyses data between E2-treated and vehicle-treated pups at D3 and D7. All post hoc comparisons between means were done by Tukey's multiple-comparison test at 0.05 significance.
Results
Flattening and thinning of MGEs in E29 rabbits and progressive reduction in progenitors with age
In rodents, ganglionic eminences become thin and flat along the lateral ventricle wall at E15–E16 (Delgado and Lim, 2015). Likewise in rabbits, E29 pups did not display ganglionic eminences and Nkx2.1+ progenitors were mostly present along the lateral wall of the ventral domain with relatively few beside the medial wall. Accordingly, the proliferating Nkx2.1+ cells were abundant on the lateral wall of ventral subventricular zone (SVZ), but almost absent on the medial wall. Based on this morphology, the ventral SVZ along the lateral ventricular wall was considered as a remnant of the MGE, and the brain region above the MGE was considered as LGE (Fig. 1).

Thinning and flattening of MGEs and progressive diminution in the density of progenitors. A, Representative immunofluorescence of cryosections from the ventral telencephalon of E29 rabbit pups at D0 and D3 (as indicated) labeled with Nkx2.1 and Ki67 antibodies. The boxed area in cresyl violet image represents the brain region where the immunostained section was imaged under a confocal microscope. Bottom, High-magnification view of the boxed area in the top panel. Note abundance of Nkx2.1+ progenitors in the lateral wall and their relative paucity in the medial wall. There were fewer total Nkx2.1+ cells and proliferating Nkx2.1+ cells in the MGE at D3 compared with D0. Scale bar, as indicated. B, Data are mean ± SEM (n = 5 each group). Stereological quantification showed that the number of both total Nkx2.1+ cells and cycling Nkx2.1+ cells in the MGE declined as a function of postnatal age. C, Representative Western blot analyses for Nkx2.1 protein in brain homogenates from healthy preterm pups (E29) at D0, D3, and D7. Data are mean ± SEM (n = 5 each group). Nkx2.1 protein level declined with advancing in postnatal age.
We quantified Nkx2.1+ cells in the MGE of E29 rabbits at D0, D3, and D7 using unbiased stereology (Fig. 1). We found lower numbers of both total and cycling Nkx2.1+ cells as a function of postnatal age (P < 0.01, both). Western blot analyses confirmed that the expression of Nkx2.1 decreased with advancing postnatal age (p < 0.01). Together, these results show that Nkx2.1+ interneuronal precursors are abundantly present in the MGEs of E29 pups and diminish in number as the postnatal age increases.
Premature birth expands the population of interneuronal precursors in the MGE and LGE
We used our preterm rabbit model to assess the effect of prematurity on interneuron neurogenesis. To this end, we performed stereological quantification of the total number of progenitors and the number of proliferating progenitors in preterm (E29 pups at D3 and D7) versus term (E32 pups at D0 and D4) rabbit pups at an equivalent postconceptional age. The analyses of brain sections labeled with Nkx2.1-specific and Ki67-specific antibodies showed that the absolute number of both total and cycling Nkx2.1+ cells were higher in preterm pups at D3 compared with respective term controls at D0 (p = 0.016, 0.01, respectively; Fig. 2). The comparison between preterm pups at D7 and term pups at D4 showed a similar trend. However, the comparison was not significant. We next analyzed the density of progenitors as a function of postnatal age. We observed fewer total and cycling Nkx2.1+ cells at D7 compared with D3 in preterm rabbits (p = 0.007, 0.001). Likewise, there were fewer cycling Nkx2.1+ progenitors, but not total Nkx2.1+ cells, in term pups at D4 compared with D0 (p < 0.037).
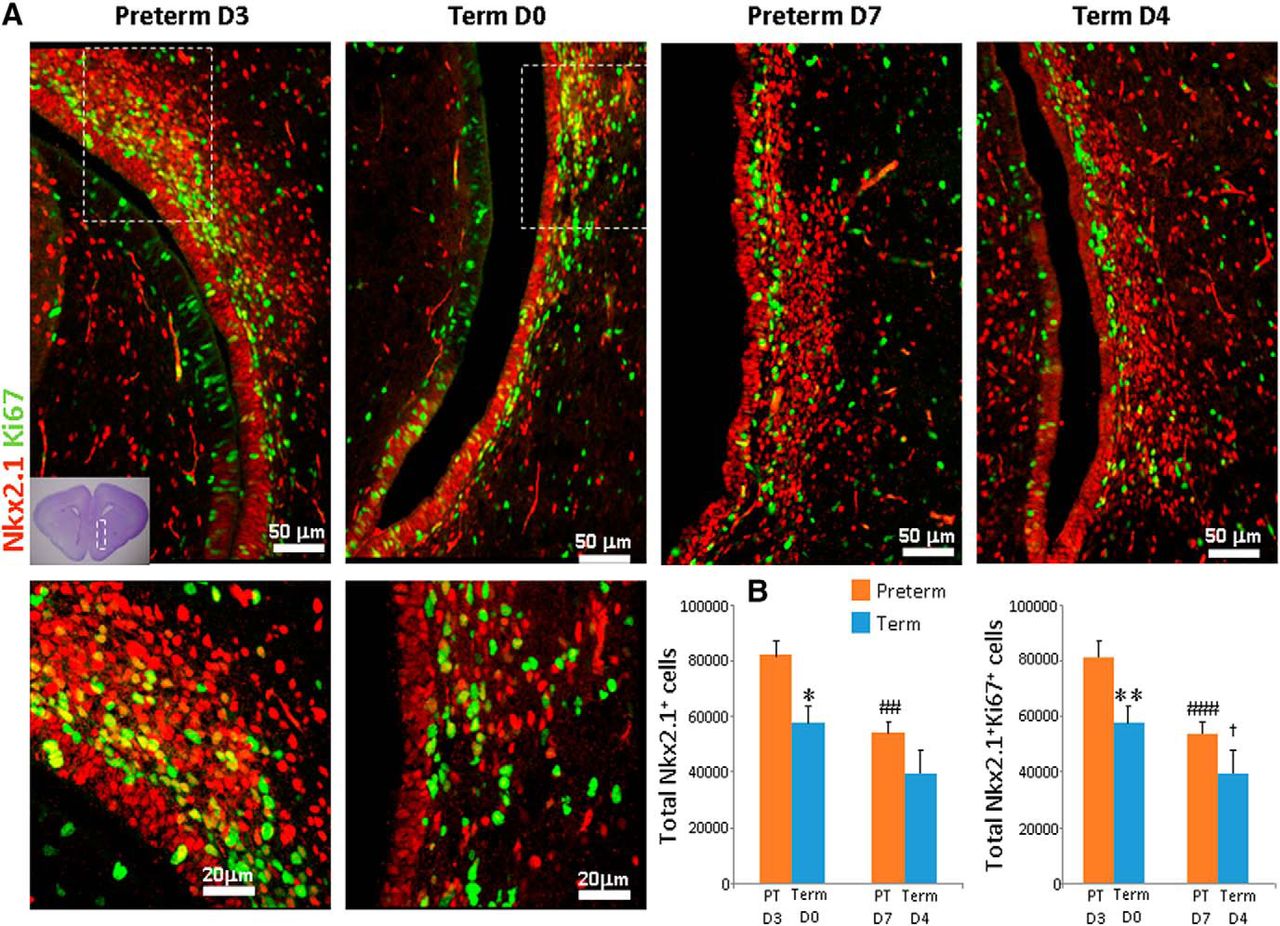
Total Nkx2.1+ cells and proliferating Nkx2.1+ cells were more abundant in preterm compared with term rabbits. A, Cryosections from term and preterm rabbits were labeled with Nkx2.1-specific and Ki67-specific antibodies. Bottom, High-magnification views of the boxed area in the top panel. The boxed area in the cresyl violet image (left lower corner) represents the brain region where the immunolabeled sections were imaged under a confocal microscope. Note greater abundance of total Nkx2.1+ cells and proliferating Nkx2.1+ cells in the MGEs of preterm rabbits (D3) compared with term controls (D0). Scale bar, as indicated. B, Data are mean ± SEM (n = 5 for each group). Stereological quantification showed that total Nk2.1+ cells and proliferating Nk2.1+ cells were more abundant in the MGEs of preterm (D3) compared with term (D0) rabbits. *p < 0.05 and **p < 0.01, preterm (D3) versus term (D0); ##P < 0.01, ###p < 0.001 for D3 preterm versus D7 preterm; and †p < 0.001 for term D0 versus term D7.
We next evaluated total number of Dlx2+ cells and the number of cycling Dlx2+ cells between term and preterm pups. Similar to Nkx2.1+ cells, both total Dlx2+ cells and proliferating Dlx2+ cells were more abundant in the MGEs of preterm pups at D3 compared with D0 term pups (p = 0.01, 0.04, respectively; Fig. 3). However, the number of Dlx2+ progenitors was comparable between D7 preterm and D4 term pups. Evaluation of Dlx2+ cells as a function of postnatal age revealed that both total Dlx2+ cells and cycling Dlx2+ cells were reduced at D7 relative to D3 in preterm pups in the MGE (both p's < 0.001). Similarly, the total Dlx2+ cells and cycling Dlx2+ cells were diminished at D4 compared with D0 term pups (both p's < 0.001).

The number of total Dlx2+ cells and the number of proliferating Dlx2+ cells were both higher in preterm compared with term rabbits. A, Representative immunofluorescence of cryosections from the MGEs of term and preterm rabbits labeled with Dlx2-specific and Ki67-specific antibodies. The boxed area in the cresyl violet image (top left corner) represents the brain region where the immunolabeled sections were imaged under a confocal microscope. Bottom, High-power view of the boxed area in the top panel. There were more total Dlx2+ cells and proliferating Dlx2+ cells in the MGEs of preterm rabbits (D3) compared with term controls (D0). Scale bar, as indicated. B, Data are mean ± SEM (n = 5 each group). The number of total Dlx2+ cells and the number of proliferating Dlx2+ cells were both higher in the MGEs of preterm (D3) compared with term (D0) rabbits. *p < 0.05 and **p < 0.01 for preterm versus term pups; ###p < 0.001 for D3 preterm versus D7 preterm pups; and ††† p < 0.05 for term D0 versus term D4 pups.
We next compared the population of Nkx2.1+ and Dlx2+ precursors in LGEs of term and preterm rabbits. We found more total Nkx2.1+ cells, but not the cycling Nkx2.1+ cells, in the preterm pups at D3 compared with term controls at D0 (p = 0.003, data not shown). The number of Nkx2.1+ cells was comparable between D7 preterm and D4 term pups. We next compared Nkx2.1+ cells as a function of postnatal age and found that there were fewer at D7 relative to D3 in preterm pups (p < 0.05, data not shown). Such reduction in the Nkx2.1 population was not noted in term pups. For Dlx2+ cells, there was no difference in their number between term and preterm pups at any of the two epochs.
Together, results show that premature birth results in an expanded pool of total and proliferating progenitors in the MGE and LGE. This might be because of greater production or delayed maturation of precursors, which might be attributed to early and untimely termination of the intrauterine environment.
Sox2+ cells are more abundant in preterm rabbits relative to term
Sox2+ cells produce neuronal progenitors and, indeed, conditional deletion of Sox2 impairs expression of Nkx2.1 in the MGE (Ferri et al., 2013). Since both cycling and total interneuronal progenitors (Nkx2.1+ and Dlx2+ cells) were elevated in the MGE of preterm pups at D3 relative to term controls, we postulated that Sox2+ cells might be more abundant in the MGEs of preterm rabbits relative to term controls. To this end, we labeled coronal brain sections from preterm rabbit brain with Sox2-specific and Ki67-specific antibodies and quantified them in the MGEs using unbiased stereology.
We demonstrated that the number of the total Sox2+ cells and the number of cycling Sox2+ cells were both higher in preterm rabbits at D3 compared with term controls at D0 (p = 0.02, 0.024 respectively; Fig. 4). This suggests that premature birth increases the proliferation and total pool of Sox2+ neuronal progenitors, in addition to Nkx2.1+ and Dlx2+ interneuronal progenitors.

The number of Sox2+ cells in the MGEs was higher in preterm pups than in term pups. A, Representative immunofluorescence of cryosections from the MGEs of term and preterm rabbits labeled with Sox2 and Ki67 antibodies. Bottom, High-magnification views of the boxed area in the top panel. Note higher density of Sox2+ cells in the MGEs of preterm pups relative to term controls. Scale bar, as indicated. B, Data are mean ± SEM (n = 5 each group). Stereological quantification showed that both total Sox2+ cells and cycling Sox2+ cells were more abundant in the MGEs of preterm (D3) compared with term (D0) rabbits. *p < 0.01, preterm (D3) versus term (D0).
Premature birth does not affect interneuronal precursors in the CGE
Approximately 30% of cortical interneurons originate from the CGE (Wonders and Anderson, 2006). Thus, we compared the population of Dlx2+ precursors in the CGE between preterm pups and term controls. To identify the CGE, we double-labeled coronal sections from preterm and term pups with Dlx2-specific, Coup-TFII (COUP transcription factor 2)-specific, and Ki67-specific antibodies. Coup-TFII is primarily expressed in CGEs (Kanatani et al., 2008). We found that proliferating Dlx2+ and CoupTFII+ cells were most prominent in the CGE at the level of posterolateral nucleus of thalamus. Hence, the comparisons for precursors in the CGE were done at this level between the groups.
We found D3 preterm pups, compared with D0 term pups, had 25% more total Dlx2+ cells. Similarly, D7 preterm pups, compared with D4 term controls, had 20% more Dlx2+ cells (Fig. 5). However, these comparisons were not significant. This suggests that preterm birth does not significantly affect the population of total and cycling interneuronal precursors in the CGE.

Dlx2+ cells in the CGEs were comparable between term and preterm pups. A, Representative immunofluorescence of cryosections from the CGEs of term and preterm rabbits labeled with Dlx2-specific and Ki67-specific antibodies. Bottom, High-magnification views of the boxed area in the top panel. Note similar abundance of Dlx2+ cells between two sets of term and preterm pups (as indicated). Scale bar, as shown. B, Data are mean ± SEM (n = 5 each group). Both total and proliferating Dlx2+ cells were comparable between term and preterm rabbits.
Cell-cycle drivers and transcription factors regulating neurogenesis in term versus preterm rabbits
Neurogenesis is linked to the proliferation and differentiation of neuronal progenitors and thus to the progenitors re-entering and exiting the cell cycle (Ohnuma, 2003). Therefore, using Western blot analyses. we quantified Ki67, p-Rb protein, CCND2, and CDK6. Cyclin D1 could not be assayed because of a lack of commercial antibody reactive to rabbit cyclin D1. Ki67, CCND2, CDK6, p-Rb (serine 807/811), and p-Rb protein (serine 780) levels were comparable between preterm pups and term controls at both D3 and D7 (Fig. 6A).

Ascl1 transcription factor was reduced in preterm relative to term rabbits, but molecules driving the cell cycle were comparable between term and preterm pups. A, Representative Western blot analyses for Ki67, p-Rb (serine 780), p-Rb (serine 807/811), CCND2, and CDK6 in brain homogenates from preterm (D3, D7) and term pups (D0, D4). Each lane represents lysate from a whole coronal slice taken at the level of midseptal nucleus of one brain. The second to last lane has a molecular weight marker. Data are mean ± SEM (n = 5 each group). For each lane, values normalized to β-actin levels. Ki67, CCND2, p-Rb (serine 807/811), p-Rb (serine 780), and CDK6 protein levels were comparable between preterm and term rabbits. B, Data are mean ± SEM (n = 5 each group). mRNA expression of Shh, Lhx6, Sox6, Dlx5, and Ascl1 were similar between preterm (D3) and term (D0) pups in both MGEs and CGEs. C, Western blot analysis for Dlx1, Gli1, Ascl1, and Sox6 was performed in brain homogenates from preterm (D3, D7) and term pups (D0, D4). Data are mean ± SEM (n = 5 each group). Values normalized to β-actin levels. Ascl1 protein levels were elevated in term pups at D4 compared with preterm pups at D7. Dlx1, Gli1, and Sox6 levels were comparable between term and preterm pups. *p < 0.05 for preterm (D7) versus term (D4) pups.
Production and maintenance of Nkx2.1+ progenitors are mediated by Shh signaling and the specification of interneuronal subtypes are regulated by transcription factors, including Dlx1/2, Ascl1, Lhx6/8, Gsh2, and Sox6 (Kelsom and Lu, 2013). Therefore, we quantified Dlx1, Ascl1, Sox6, and Gli1 in preterm rabbits (D3 and D7) compared with term controls. Dlx1 and Dlx2 have overlapping expressions in the ventricular zone and SVZ of the ventral telencephalon and have just subtle differences (Le et al., 2007). Dlx5 is coexpressed with Dlx1 and Dlx2 in the ventral SVZ and in the adjacent SVZ, which contains differentiating interneurons (Liu et al., 1997). We performed quantitative real-time PCR on tissues dissected from MGEs and CGEs and found that mRNA expression of Shh, Sox6, LHX6, Dlx5, and Ascl1 were comparable between preterm pups (D3) and term controls (D0) in both MGEs and CGEs (Fig. 6B). Western blot analyses on homogenates from coronal slices (midseptal nucleus level) showed that Ascl1 transcription factor was elevated in term pups at D4 compared with preterm pups at D7 (p = 0.046), but not in term pups at D0 versus preterm pups at D3. However, Dlx1, Sox6, and Gli1 proteins in preterm pups at D3 and D7 were comparable to the respective term controls (D0 and D4; Fig. 6C). Together, our results show that prematurity is associated with increased proliferation of interneuron progenitors and reduction in Ascl1 transcription factor; however, cell-cycle drivers and other transcription factors regulating interneuron neurogenesis are not affected.
Estrogen treatment, but not DMOG treatment, reduces the population of total and cycling interneuronal precursors in the MGE
Since preterm birth terminates the in utero environment, causing the premature withdrawal of estrogen exposure to the preterm newborn, we hypothesized that estrogen treatment might restore the number of total and proliferating interneuronal precursors. To this end, we treated preterm rabbits with either estrogen (200 μg/kg) or vehicle for 3 d. The animals were killed at the end of D3, and total Nkx2.1, Dlx2+, and Sox2+ cells, as well as cycling Nkx2.1, Dlx2+, and Sox2+ cells, were stereologically quantified and compared between the two groups. The pups were balanced with respect to sex in the two groups, which was determined by performing quantitative real-time PCR for the SRY gene. We found that the number of total Nkx2.1+ cells and the number of proliferating Nkx2.1+ cells in MGEs were both significantly reduced in estrogen-treated pups relative to vehicle-treated controls (p = 0.015 and 0.03; Fig. 7A,B). We next compared Dlx2+ cells between the two groups. Consistent with Nkx2.1+ cells, the total number of Dlx2+ cells and the number of cycling Dlx2+ cells in the MGEs were both significantly diminished in estrogen-treated pups relative to controls (p = 0.005, 0.003; Fig. 7C,D). However, the total number of Sox2+ cells and the number of cycling Sox2+ cells in E2-treated rabbits were both similar to those in vehicle-treated rabbits. Since the effect of estrogen is dose-dependent (Strom et al., 2011), we next evaluated the effect of low-dose estrogen on interneuronal progenitors. To this end, we treated preterm rabbits with estrogen (100 μg/kg E2 for 3 d) and quantified total and cycling Nkx2.1+ and Dlx2+ cells in estrogen-treated and vehicle-treated pups. Similar to high-dose estrogen treatment, low-dose estrogen treatment reduced both total Nkx2.1+ cells and cycling Nkx2.1+ cells in the MGEs (p = 0.014, 0.001). Estrogen-treated pups, compared with vehicle-treated controls, had fewer total Dlx2+ cells (p < 0.01), but not fewer cycling Dlx2+ cells (p = 0.06). Together, these results show that estrogen treatment in both high and low doses reduces the population of interneuronal progenitors.

Estrogen treatment reduced total and cycling interneuronal progenitors. A, Representative immunofluorescence of cryosections from the MGEs of preterm rabbits labeled with Nkx2.1-specific and Ki67-specific antibodies. Bottom, High-magnification views of the boxed area in the top panel. Note reduced abundance of Nkx2.1+ and Nkx2.1+Ki67+ cells in estrogen-treated pups compared with vehicle-treated controls. Scale bar, as shown. B, Data are mean ± SEM (n = 5 each group). The number of total and proliferating Nkx2.1+ cells declined in estrogen-treated (both high-dose and low-dose groups) pups compared with vehicle-treated controls. C, Cryosections from the MGEs of preterm rabbits were labeled with Dlx2-specific and Ki67-specific antibodies. Bottom, High-magnification views of the boxed area in the top panel. Note diminished density of Dlx2+ and Dlx2+Ki67+ cells in estrogen-treated pups compared with vehicle-treated controls. Scale bar, as shown. D, Data are mean ± SEM (n = 5 each group). Total Dlx2+ cells were reduced in estrogen-treated (both high-dose and low-dose groups) pups compared with vehicle-treated controls. Dlx2+Ki67+ cells were reduced in high-dose estrogen pups relative to controls, but not in the low-dose estrogen pups. *p < 0.05, **p < 0.01 for estrogen-treated versus vehicle-treated pups.
Since estrogen can exert both proapoptotic or antiapoptotic effects on neural cells in a context-dependent manner (Nilsen et al., 2000; Zhang et al., 2017), we asked whether estrogen treatment would affect programmed cell death of interneuron progenitors. To this end, we labeled the coronal brain sections with Dlx2 antibody and then performed TUNEL. Stereological quantification showed that total number of TUNEL+ cells and Dlx2+ cells colabeled with TUNEL were comparable between estrogen-treated and vehicle-treated pups in the MGEs at D3 (Fig. 8A,B).

High-dose estrogen did not affect apoptotic cell death and DMOG treatment did not affect the population of interneuron progenitors. A, Cryosections from the MGEs of preterm rabbits (E29, D3) were labeled with Dlx2 antibody and then TUNEL staining was performed. Bottom, High-magnification views of the boxed area in the top panel. Note comparable density of TUNEL+ cells in the estrogen-treated and vehicle-treated pups. B, Data are mean ± SEM (n = 5 each group). Number of total TUNEL+ and Dlx2+TUNEL+ cells were comparable between estrogen-treated and vehicle-treated pups. C, Data are mean ± SEM (n = 5 each group). The total number of Nkx2.1+ cells and Dlx2+ cells and the number of cycling Nkx2.1+ cells and Dlx2+ cells in vehicle-treated pups were approximately the same as those in DMOG-treated pups. D, Representative Western blot analyses for VEGF in brain homogenates from preterm rabbits treated with DMOG and vehicle. Data are mean ± SEM (n = 5 each group). Adult rat brain was taken as control. Each lane values normalized to β-actin levels. VEGF protein levels were elevated in DMOG-treated rabbits compared with controls. *p < 0.05 for DMOG-treated versus vehicle-treated pups.
Oxygen is the key regulator of neurogenesis; and the intrauterine partial pressure of oxygen (pO2) level of the fetus is low compared with that of premature infants in extrauterine room air after birth. Therefore, to simulate the hypoxic environment, we treated E29 rabbits with intramuscular DMOG for 3 d and compared the population of Nkx2.1+ and Dlx2+ cells between DMOG-treated pups and vehicle-treated controls. We found that DMOG treatment did not significantly reduce the density of Nkx2.1+ and Dlx2+ cells in the MGEs of treated preterm rabbits compared with vehicle-treated controls (Fig. 8C). To determine whether DMOG treatment is inducing a hypoxic environment, we quantified VEGF protein by Western blot analyses (Fig. 8D). We found that DMOG treatment increased VEGF levels in rabbits treated with DMOG compared with vehicle-treated controls. This was consistent with our previous work (Malik et al., 2013), in which we found that DMOG treatment increases HIF1, VEGF, and erythropoietin (EPO) protein and gene expression in E29 premature pups 3 d old. Together, these results show that DMOG treatment did not significantly affect the population of interneuron progenitors despite effectively inducing a hypoxic environment.
Together, estrogen treatment reduced the number of both total and cycling interneuron progenitors, but induction of chemical hypoxia with DMOG treatment did not affect the population of progenitors.
Estrogen treatment reduces Ki67 and p-Rb levels, and elevates Ascl1 transcription factor
Neurogenesis is critically regulated by cell cycle; and phosphorylation of Rb protein by Cdk4/6-CyclinD (mid-G1-restriction) induces S-phase entry (Ross, 2011). Moreover, a number of transcription factors produced in ventral telencephalon affects proliferation and maturation of interneuronal progenitors (Wonders and Anderson, 2006), which can be influenced by estrogen. Therefore, we postulated that estrogen treatment might affect both the drivers of the cell cycle and the transcription factors orchestrating interneuron neurogenesis.
To this end, we assayed Ki67, Rb phosphorylation, CCND2, c-Myc, and CDK6 in E2 versus vehicle-treated preterm pups. Western blot analyses revealed that Ki67 and c-Myc expression were reduced in E2-treated rabbits relative to vehicle-treated controls at D3 (p = 0.049, 0.04), but not at D7. p-Rb (serine 807/811) and CCND2 levels were significantly reduced in E2-treated preterm pups compared with vehicle-treated controls at D7 (p = 0.005, 0.04, respectively), but not at D3 (Fig. 9). Cdk6 and p-Rb (serine 780) level was comparable between E2-treated and vehicle-treated pups at both epochs (CDK6 data not shown). Quantitative real-time PCR showed that CDK6 and CCND2 were comparable between term and preterm pups at both epochs (data not shown). The discrepancy between CCND2 protein expression and mRNA expression can be attributed to other levels of regulation between transcript and protein product.

Estrogen treatment reduced Ki67, CCND2, and p-Rb (serine 780) expression. A, Representative Western blot analyses for Ki67, p-Rb (serine 780), p-Rb (serine 807/811), CCND2, and CDK6 in brain homogenates from preterm pups treated with estrogen and vehicle. Data are mean ± SEM (n = 5 each group). Each lane represents lysate from a whole coronal slice taken at the level of midseptal nucleus of one brain. Each lane values were normalized to β-actin levels. B, Data are mean ± SEM (n = 5 each group). Ki67 and c-Myc expression were reduced at D3, and p-Rb (serine 807/811) and CCND2 levels were diminished at D7 in estrogen-treated pups relative to controls. p-Rb (serine 780) expression was comparable between estrogen and vehicle-treated pups. *p < 0.05 for estrogen-treated versus vehicle-treated pups.
We next compared transcription factors and enzymes regulating interneuron neurogenesis between E2-treated and vehicle-treated pups at both D3 and D7. Real-time PCR using Taqman probes revealed that mRNA expression of Ascl1 and Gad1 was elevated in E2-treated pups relative to vehicle-treated controls at D7 (p = 0.012, 0.008, respectively), but not at D3 (Fig. 10A). Gad2 and Lhx6 mRNA accumulation was similar between E2-treated and vehicle-treated pups. Consistent with these findings, Western blot analyses demonstrated that Ascl1 protein levels was higher in E2-treated pups relative to vehicle-treated controls at D7 (p = 0.001), but not at D3 (Fig. 10B). Gli1, Sox6, and Dlx1 transcription factors were comparable between E2-treated and vehicle-treated pups at both epochs (data on Sox6 not shown). To understand how E2 may activate Ascl1 expression, we searched for transcription factor binding motifs in the upstream region of the rabbit Ascl1 gene and found an estrogen response element (ERE) located ∼9.2 kb from the transcription start site. This ERE is highly conserved and located ∼8 kb upstream of the human Ascl1 gene (Fig. 10C). Moreover, by mining previously published genome-wide transcription factor binding data (Schmidt et al., 2010), we found that this ERE was very likely functional as it was bound by an estrogen receptor α in the human MCF (Michigan Cancer Foundation) breast cancer cell line, in which estrogen was active.

Estrogen treatment elevated Ascl1 and GAD1 expression. A, Gene expression was quantified by quantitative real-time PCR. Data are mean ± SEM (n = 5 each group). mRNA expression of Ascl1 and Gad1 was elevated in estrogen-treated pups relative to controls. However, Gad2 and Lhx6 mRNA accumulation were comparable between estrogen-treated and vehicle-treated pups. B, Western blot analyses for Ascl1, Dlx1, and Gli1 were performed in brain homogenates of estrogen-treated and vehicle-treated pups at D3 and D7. Data are mean ± SEM (n = 5 each group). Values normalized to β-actin levels. Ascl1 was elevated in estrogen-treated preterm pups at D7 compared with vehicle controls. Dlx1, Gli1, and Sox6 were comparable between estrogen-treated and vehicle-treated preterm pups. *p < 0.05 for estrogen-treated versus vehicle-treated pups. C, A distal ERE is in the upstream region of Ascl1 gene. Note estrogen receptor ChIP-seq peak in human cells (top), the sequence alignment of the ERE, and its comparison to known estrogen receptor binding motif (bottom). The conserved ERE is ∼8 and ∼9.2 kb upstream to the transcription start sites of the human and rabbit Ascl1 genes, respectively.
Together, estrogen treatment enhanced Ascl1 and reduced p-Rb (serine 780), CCND2, and c-Myc levels, which might be contributing to suppression in the density of interneuron progenitors.
Discussion
Inattention, hyperactivity, autism, emotional disturbance, poor social skills, epilepsy, and lower intellectual abilities are found in ∼50% of preterm-born children (Potijk et al., 2016). Recent studies have shown that the cellular and molecular basis of these developmental and neuropsychiatric disorders are specific defects in the development and function of interneurons (Marín, 2012). Hence, we evaluated production of interneurons in preterm versus term rabbits at an equivalent postconceptional age and determined whether estrogen replacement in premature rabbits reverses the prematurity-induced disruption in neurogenesis. We demonstrated that premature birth resulted in increased proliferation and expansion of interneuron progenitors in the MGE. In addition, estrogen treatment reduced proliferation and diminished the population of progenitors. This effect of estrogen can be attributed to an elevation in Ascl1 transcription factor and inhibition of the G1/S cell-cycle phase, as reflected by reduced Ki67, CCND2, and p-Rb (serine 780) levels in estrogen-treated premature rabbits relative to controls. The study highlights the disturbance of neurogenesis in preterm rabbits possibly related to withdrawal of estrogen and restoration in production of interneurons with estrogen replacement.
The novel and most important finding in the present study was the demonstration of increased proliferation and population of interneuron progenitors—Nkx2.1+, Dlx2+, and Sox2+ cells—in the MGEs of premature rabbits compared with term-equivalent controls. To our knowledge, this is the first demonstration of the effect of prematurity on interneuron neurogenesis. Increased proliferation and number of interneuron progenitors in preterm rabbits indicate augmented generation of interneurons. However, arrested maturation of progenitors might also contribute to increased abundance of progenitors in preterm newborns. An arrest or delay in the maturation of Nkx2.1+ and Dlx2+ cells might reduce the interneuron population in the cerebral cortex. Even though interneurons constitute only 20–30% of all cortical neurons, arrested maturation and integration of interneurons in the cerebral cortex would contribute to reduced brain growth in premature infants, which has been demonstrated in a number of MRI studies in preterm-born children and adolescents (Monson et al., 2016). Moreover, this disruption in neurogenesis might affect development of cortical circuitry, cause imbalance between excitatory and inhibitory cortical neurons, and lead to neurobehavioral disorders. Together, these results show how prematurity disrupts interneuron neurogenesis, which can potentially contribute to impaired cortical growth in preterm-born children.
Prematurity affects not only the interneuron neurogenesis, but it suppresses glutamatergic neurogenesis as well (Malik et al., 2013). Since glutamatergic neurogenesis continues until the 28th gestational week and interneuron neurogenesis continues until the end of pregnancy, preterm birth is more likely to affect GABAergic than glutamatergic neurogenesis (Malik et al., 2013; Arshad et al., 2016). Preterm birth terminates the intrauterine life prematurely and interneuron neurogenesis is disrupted in extrauterine environment. The intrauterine environment is relatively hypoxic and receives enrichment from maternal and placental sources. Intrauterine fetal arterial oxygen tension (PO2 = ∼25 mmHg) is lower compared with levels in premature infants (PO2 = ∼60 mmHg) in NICUs. Low oxygen tension keeps the neural and other stem cells in an undifferentiated state (Mohyeldin et al., 2010). However, contrary to this notion, we found that term rabbits with a longer in utero sojourn exhibited fewer total and cycling interneuron progenitors compared with preterm rabbits. Preterm birth is an interruption in the maternal and placental supply of estrogen, progesterone, and several growth factors, as well as nutrients, which can potentially affect neurodevelopmental processes. Together, the effects on neurogenesis in preterm-born infants can be attributed to premature withdrawal of the intrauterine environment and drops in hormonal levels and supply of growth factors.
Our other key finding in this study was that estrogen treatment reduced total and proliferating interneuron progenitors (Nkx2.1+ or Dlx2+ cells) in the MGE. Consistent with our studies, several reports have shown that estrogen alters proliferation and maturation of neuronal progenitors in both culture studies and in vivo experiments. In culture experiments, estrogen treatment in appropriate doses significantly increases proliferation of neural stem cells (NSCs) and enhances their differentiation into neurons (Li et al., 2017). In another study, E2 increased the proliferation of NSCs and the ratio of neurons to glia cells (Brännvall et al., 2002). In female rats, estrogen treatment affects hippocampal dentate gyrus granule cell proliferation in a dose-dependent and timing-dependent manner (Tanapat et al., 2005). Hence, it seems that the effect of estrogen on proliferation versus differentiation of NSCs during embryonic aging is dose and context dependent. In models of brain injury, estrogen exhibits neuroprotection and promotes maturation of interneurons. For example, in a rat model of trimethyltin-induced hippocampal degeneration, estrogen treatment increased the GAD67 and PV expression (Corvino et al., 2015). Likewise, estrogen minimizes brain injury in models of traumatic and ischemic brain injury (Tskitishvili et al., 2014; Day et al., 2017). Together, these studies reinforce the notion that estrogen treatment has a wide range of effects, including proliferation, differentiation, and neuroprotection, and that the impact is dose and context dependent.
Ascl1 is the principal regulator of the transcriptional program in the ventral telencephalon, by which it controls temporal progression in the proliferation and maturation of interneuronal progenitors (Vasconcelos and Castro, 2014). Genetic ablation of Ascl1 reduces neurogenesis and causes neurodevelopmental defects, whereas its overexpression promotes cell-cycle exit and induces neuronal differentiation (Casarosa et al., 1999; Castro et al., 2011; Kelsom and Lu, 2013). In the present study, we found that estrogen treatment upregulated expression of Ascl1 and Gad67, as well as phosphorylation of the retinoblastoma protein. Hence, estrogen-induced elevation in Ascl1 might have increased Gad1 expression and inhibited G1-to-S phase transition of the cell cycle, thereby promoting maturation of interneuronal progenitors (Liu et al., 2017). Our genomic data analyses revealed that a distal ERE in the Ascl1 gene is highly conserved between rabbits and humans, and the ERE is bound by estrogen receptor in human cells, suggesting that the ERE can be a potential enhancer by which estrogen regulates Ascl1 transcription. To our knowledge, this is the first study proposing a novel mechanism that estrogen-induced differentiation of interneuron precursors is mediated by an elevation in Ascl1.
Neurogenesis involves both neuronal proliferation and differentiation. Proliferation is orchestrated by cell-cycle entry and differentiation by cell-cycle exit. In the present study, estrogen treatment reduced expression of CCND2 and diminished phosphorylation of Rb. CCND2 knock-out experiments have revealed that CCND2 promotes SVZ proliferation (Glickstein et al., 2007). Rb phosphorylation inactivates Rb and releases EF2, which drives G1 phase into S phase (Giacinti and Giordano, 2006). Indeed, reduced p-RB level inhibits the cell cycle (G1-to-S phase progression), resulting in reduced cell proliferation. Therefore, estrogen-induced downregulation of CCND2 and reduced phosphorylation of Rb protein would promote cell-cycle exit, reduce proliferation, and enhance maturation of interneuronal progenitors. Although estrogen treatment decreased Ki67 and c-Myc levels at day 3, a significant reduction in p-RB was noted at D7 with a downward trend at D3 in our studies. Together, estrogen-induced reduction in proliferation of interneuron progenitors is related to its substantial impact on the cell cycle.
Preterm birth reduces the level of plasma estrogen 100-fold (Trotter and Pohlandt, 2000). Indeed, the use of replacement estrogen and progesterone treatment has undergone a clinical trial in premature infants and has shown marginal benefit in improving bone mineral accretion rate and reducing the incidence of chronic lung disease (Trotter et al., 1999; Hunt et al., 2004). Estrogen treatment in pregnant rabbits increases surfactant production and improves lung maturation in fetuses (Khosla et al., 1981). In addition, estrogen treatment abrogates white matter injury in animal models of hypoxia-ischemia and hyperoxia, protects immature neurons and astroglia, and promotes synapse formation (Hilton et al., 2006; Gerstner et al., 2007, 2009; Nunez et al., 2007; McCarthy, 2008). These links underscore the importance of estrogen treatment in promoting both neural and physical growth and development of premature infants. Importantly, no significant adverse effect of estrogen replacement has been reported in the clinical trial on preterm infants (Hunt et al., 2004). We also did not notice any apparent adverse effect of estrogen treatment in preterm rabbits. However, adverse effects of estrogen treatment reported in adult humans include nausea, vomiting, loss of appetite, increased thirst, muscle weakness, confusion, tiredness, abdominal tenderness, and vaginal bleeding. Estrogen might also suppress the hypothalamic–pituitary axis.
In conclusion, prematurity disrupts neurogenesis and estrogen treatment reverses this disturbance in interneuron production. Estrogen treatment seems to mediate the effect of prematurity by inhibiting the cell cycle and elevating expression of Ascl1 transcription factor. Given that estrogen level drops prematurely in preterm infants, estrogen restores interneuron production, and given that estrogen confers neuroprotection in a number of animal models, we speculate that estrogen replacement might restore neurogenesis and improve the neonatal outcome of extremely premature infants.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grants R21NS085508 (P.B.) and R01NS083947 (P.B.). The authors thank Joanne Abrahams for the assistance with images.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Praveen Ballabh, Department of Pediatrics and Neuroscience, Albert Einstein College of Medicine–Children's Hospital at Montefiore, Kennedy Center, 1410 Pelham Parkway, Bronx, NY 10461. Pballabh{at}msn.com





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}