
Abstract
For a motor unit to function, neurons and muscle cells need to adopt their correct cell fate, form appropriate cellular contacts, and assemble a specific repertoire of signaling proteins into presynaptic and postsynaptic structures. In the nematode Caenorhabditis elegans, a disruption of any of these steps causes uncoordinated locomotory behavior (unc phenotype). We report here the positional cloning of a new unc gene, unc-122, which we show by mosaic analysis and tissue-specific rescue experiments to act in muscle to affect locomotory behavior. unc-122 codes for a phylogenetically conserved type II transmembrane protein with collagen repeats and a cysteine-rich olfactomedin domain. Together with uncharacterized proteins in C. elegans, Drosophila, and vertebrates, UNC-122 defines a novel family of proteins that we term “Colmedins.” UNC-122 protein is expressed exclusively in muscle and coelomocytes and localizes to the postsynaptic surface of GABAergic and cholinergic neuromuscular junctions (NMJs). Presynaptic and postsynaptic structures are present and properly aligned in unc-122 mutant animals, yet the animals display neurotransmission defects characterized by an altered sensitivity toward drugs that interfere with cholinergic signaling. Moreover, unc-122 mutant animals display anatomical defects in motor axons that are likely a secondary consequence of neurotransmission defects. Both the neuroanatomical and locomotory defects worsen progressively during the life of an animal, consistent with a role of unc-122 in acute signaling at the NMJ. On the basis of motifs in the UNC-122 protein sequence that are characteristic of extracellular matrix proteins, we propose that UNC-122 is involved in maintaining a structural microenvironment that allows efficient neuromuscular signaling.
- motor axon
- neurotransmission
- neuromuscular junction
- genetics
- transmembrane protein
- C. elegans
Introduction
The ensemble of a muscle cell and its innervating motor neuron is called a motor unit. The development of a functional motor unit involves the appropriate specification of motor neuron and muscle cell fate, choice of neurotransmitter system, growth of the motor axon to its target muscle, functional innervation of the muscle target, and the assembly of presynaptic and postsynaptic signaling proteins. Various transcription factors that regulate motor neuron cell fate have been identified, one prominent class being LIM homeodomain transcription factors (Tsuchida et al., 1994; Jessell, 2000; Shirasaki and Pfaff, 2002). The involvement of LIM homeobox (Lhx) genes in motor neuron specification has been conserved across phylogeny from worms to flies and vertebrates (Hobert and Westphal, 2000; Thor and Thomas, 2002).
The nematode Lhx gene lim-6 is required for motor neuron development in the enteric motor units of Caenorhabditis elegans (Hobert et al., 1999a). The enteric motor unit is composed of two motor neurons, AVL and DVB, that innervate a group of three enteric muscle cells (Avery and Thomas, 1997). lim-6 is expressed in the AVL and DVB motor neurons, but rather than following the example of vertebrate Lhx genes in determining motor neuron identity and fate (Jessell, 2000; Shirasaki and Pfaff, 2002), lim-6 only affects subaspects of the motor neuron differentiation program (Hobert et al., 1999a). Specifically, loss of lim-6 has no impact on the generation of the DVB motor neuron, on the pattern of outgrowth of the main motor axon, or on the expression of several terminal differentiation markers. Loss of lim-6, however, causes an axon sprouting defect, characterized by the outgrowth of excessive axon sprouts from the cell body or main axon shaft of DVB (Hobert et al., 1999a). We have shown that axon sprouting of the DVB motor neurons is indicative of a defect in synaptic communication between DVB and its muscle target cells (Loria et al., 2003). Genetic ablation of the enteric muscle targets or their electric silencing also causes motor axon sprouting defects (Loria et al., 2003). Hence, one possible explanation for the motor axon sprouting defects of DVB and the apparent absence of developmental defects in lim-6 null mutants is that lim-6 affects aspects of neuromuscular signaling. Indeed, we found that lim-6 regulates the expression of the GABA synthesizing enzyme UNC-25 (Hobert et al., 1999a); however, the axonal defects of lim-6 mutants are more severe than those of unc-25 mutants, indicating that the function of lim-6 goes beyond the determination of neurotransmitter choice.
To better understand the DVB motor unit and hence the context and cellular process in which lim-6 exerts it function, we sought to identify and characterize genes that, when disrupted, cause lim-6-like phenotypic defects in the DVB motor neurons, i.e., the occurrence of ectopic axon sprouts (Loria et al., 2003; Mehta et al., 2004). We describe here a previously uncharacterized yet phylogenetically conserved transmembrane protein that is required in the muscle targets of many motor neurons to ensure appropriate signaling between the motor neuron and its muscle target.
Materials and Methods
General methods. General methods for the handling, maintenance, and genetic analysis of C. elegans are as described previously (Brenner, 1974). The wild-type parent for all strains was C. elegans var. Bristol (Brenner, 1974). DNA microinjection was performed as described previously (Mello et al., 1991). DNA was injected at different concentrations, depending on the experiment (see Figure legends). Transgenic lines were created in wild-type or unc-122(e2520) backgrounds using the injection markers ttx-3::gfp, ceh-22::gfp, or unc-122::gfp at 50 ng/μl as indicated.
Mutant strains. A listing of strains follows. CB4870: unc-122(e2520). This allele was extracted from the strain CB3337 (isolated by Ed Hedgecock, Johns Hopkins University, Baltimore, MD) that also contains unc-1(e2522) and dpy-3(e2521). MT7917: unc-122(n2916). This allele was isolated by Erik Jorgensen (University of Utah, Salt Lake City, UT) from a CB51 unc-13(e51) mutant background. EG79: unc-122(ox79). This allele was isolated in an N-ethyl-N-nitrosourea-mediated screen for synaptic transmission mutants and kindly provided by Erik Jorgensen. MT7915: dpy-5(e61)unc-122(n2916). SP24: dpy-5(e61)unc-54(e190). CB190: unc-54(e190). BC2776: eDf10/eDf24. CB2771: eDf5/eDf24. CB2777: eDf11/eDf24. CB2818: eDf13/eDf24. EG1285: lin-15(n765) oxIs12. CB156 unc-25(e156). OH271: unc-122(e2520); oxIs12. OH274: unc-122(e2520); unc-25(e156); oxIs12. OH2832: unc-25(e156); oxIs12. CB245 unc-17(e245). CB1072 unc-29(e1072). CB904: unc-38(e264). OH306: unc-122(e2520); unc-17(e245); oxIs12. OH211: unc-29(e1072) unc-122(e2520). GS2818: dpy-20(e1282); arIs37; arEx[cc::DpTx; rol-6]. GS1912: dpy-20(e1282); arIs37. OH810: unc-122(e2520); otIs127. OH1056: otIs127. EG1653: oxIs22. CZ333: juIs1. ZZ2004: unc-29(x568); xEx1. OH2290: otIs127; juIs1. OH2300: unc-122(e2520); oxIs22. OH2379: otIs127; unc-6(ev400). NW434: unc-6(ev400). OH2381: otIs127/+; jsIs42. OH2448: unc-29(x568)unc-122(e2520). AQ889: ljEx42. OH2452: unc-29(x568); otIs127; xEx1. OH2482: otIs127; ljEx42. OH368: unc-122(e2520); mgIs25. OH2833: unc-122(e2520); juIs1. OH2512: unc-122(e2520); ljEx42. OH2513: unc-29(x568)unc-122(e2520); xEx1.
Mapping. Deficiency mapping with eDf13 and eDf10 placed unc-122 into a 1.7 map unit interval on chromosome I, which contains the unc-54 locus (see Fig. 3). Close linkage of unc-122 to unc-54 was observed during two- and three-factor mapping. A two-factor cross between unc-122(e2520) and unc-54(e190) produced 0 of 363 recombinants. Furthermore, to bias the analysis of recombinants, we performed a three-factor cross; 49 of 124 unc-122 homozygous animals produced from a three-factor cross between dyp-5(e61) and unc-54(e190) had recombined to pick up the dpy-5 locus, but none picked up the unc-54 locus. DNA fragments derived from the left of the unc-54 locus, covering the complete region of the eDf13 deficiency on the left side of unc-54, failed to rescue the unc-122 mutant phenotype (see Fig. 3). These results placed unc-122 close to the right of unc-54. The F11C3 and neighboring ZK270 cosmid were injected together at 20 ng/μl and fully rescued the coiling phenotype (13 of 13 lines). Primers were designed to amplify the F11C3.2 region spanning the predicted gene as well as 5′ and 3′ flanking regions up to the nearest genes (5′-gttttcccgctgataaattg-3′ and 5′-gaagttcgtcaggagacca-3′). The PCR fragment was injected at 20 ng/μl, and all lines obtained rescued the unc-122 locomotory defect (seven of seven lines).

UNC-122 is an OLF domain-containing protein. A, Map position of unc-122 and transformation rescue experiments. The approximate position of the eDf13 chromosomal deficiency is indicated as a gray bar. The right end extends to cosmid F11C3; the left end has not been mapped but must lie to the right of sur-2 (www.wormbase.org). B, Schematic sequence of UNC-122 with an alignment of its conserved motifs and indication of location of mutant alleles. Residues that are conserved in >50% of aligned sequences are boxed. Note that the e2520 allele harbors two mutations. The ox79 and n2916 alleles, independently isolated in two different laboratories (see Materials and Methods), are identical mutations. ClustalX was used as the alignment tool (Thompson et al., 1997). Accession numbers for previously unpublished proteins are as follows: cof-2, AY494975; unc-122, AY494976; Gliomedin, NP852047.
Reporter strains. A list of strains follows. juIs1: vesicle-associated membrane protein (VAMP)::green fluorescent protein (GFP) expressed under control of the unc-25 promoter in GABAergic neurons (Hallam and Jin, 1998) visualizes presynaptic specializations. oxIs12: GFP expressed under control of the unc-47 promoter in GABAergic neurons visualizes axon anatomy (McIntire et al., 1997). mgIs25: UNC-97::GFP visualizes dense body anatomy (Hobert et al., 1999b). oxIs22: UNC-49B::GFP visualizes postsynaptic GABA receptor clustering (Bamber et al., 1999). xEx1: extrachromosomal array [kindly provided by Jim Lewis (University of Texas, San Antonio, TX) and Jean-Louis Bessereau (Ecole Normale Superieure, Paris, France)] contains UNC29::GFP for visualization of acetylcholine receptor (AChR) clustering. ljEx42: extrachromosomal array (kindly provided by Alexander Gottschalk, University of California, San Diego, CA) that contains myc-tagged UNC-38 expressed under the unc-38 promoter visualizes postsynaptic acetylcholine receptors.
DNA constructs. The unc-122::gfp reporter gene construct contains an 800 bp fragment upstream of the ATG start codon of unc-122 that was amplified by PCR (5′-gttttcccgctgataaattg-3′ and 5′-gaagttcgtcaggagacca-3′) and subcloned into the gfp vector pPD95.75 (gift from A. Fire, Stanford University). The translational GFP reporter containing the promoter and all of the genomic region up to the stop codon fused to the N terminus of GFP was constructed by subcloning the genomic PCR fragment (5′-gttttcccgctgataaattg-3′ and 5′-tcaatatattctggtaagtcc-3′) into the gfp vector pPD95.75. Promoter subfragments (see Fig. 6) were created by PCR and subcloned in the same manner using the following primers: 5′-ggtaccataggagcagtt-3′, 5′-gttgacacgcagtttccctc-3′, 5′-ccccacggaaatgactatca-3′, 5′-ccactatattctggcaactagc-3′, and 5′-cgttgtgagcccaatgaagt-3′. Rescuing subfragments (see Fig. 5) were created by PCR using the following primers: 5′-ggctcacaatatggttgcga-3′, 5′-ggctcacaatAAGgttgcgacac-3′, 5′-ggacaagatactatcattccagtg-3′, 5′-gggcattgttgggccaatc-3′, 5′-ggttcctgtgccagtagtg-3′, 5′-gaacataagcactccaaaaccg-3′, 5′-ggaccaccaacactgggat-3′, and 5′-gaagttcgtcaggagacca-3′. The rescuing fragment containing 800 bp upstream and 1 kb downstream of the predicted F11C3.2 locus was subcloned into TOPO-XL. A unique NheI site was engineered into this construct by site-directed mutagenesis, and a GFP cassette was subcloned to create the N-terminal GFP fusion construct. GFP was also inserted in-frame within the unc-122 sequence by subcloning into the unique BglII site. The genomic-cDNA hybrid construct, which eliminates introns 3-7, was constructed by replacing the genomic BglII-NdeI fragment with the corresponding BglII-NdeI fragment from the cDNA. Truncations of this hybrid construct were generated by PCR using the following primers: 5′-ggctcacaatAAGgttgcgacac-3′, 5′-ggacaagatactatcattccagtg-3′ and 5′-gaagttcgtcaggagacca-3′.

Reporter gene analysis of cis-regulatory elements of the unc-122 and cof-2 genes. A, unc-122 constructs used to generate transgenic lines. Black bars indicate genomic sequence (5′ and 3′ of the gene); the white box indicates the heterologous unc-54 3′UTR. Expression pattern and rescue of the unc-122 mutant phenotype is indicated (expression is shown in C). For localization of presumptive cis-regulatory regions driving expression in muscle, see Figure 5. GFP and HA epitope coding sequences are not drawn to scale. Antibody staining to detect the HA-tagged protein is shown in Figure 8 in the context of subcellular localization of UNC-122. B, cof-2 promoter construct. The promoter contains cis-regulatory elements for expression in coelomocytes and muscles as shown in D-G. C-G, Transgenic worms expressing gfp reporter gene constructs. Stars indicate coelomocytes; arrows indicate body wall muscles in E and enteric muscle in all other panels. Most body wall muscles are not visible in D because of mosaicism. C, unc-122prom::gfp (the 800 bp promoter fusion from A). D-G, cof-2prom::gfp.

Functional dissection of the unc-122 locus: rescue analysis of fragments of the unc-122 locus. PCR products or subcloned DNA fragments were injected at 20 ng/μl into unc-122 mutant animals, and rescue of the coiling defect was scored. Rescue is defined as >50% of transgenic animals displaying wild-type (sinusoidal) movement patterns. Gray bars denote genomic regions 5′ and 3′ of the unc-122 locus. The deletion of the 5′ region without loss of rescuing activity argues for the presence of cryptic internal transcriptional and translational start sites. Potential alternative ATG start codons followed by cryptic potential signal sequences are indicated with gray arrows. Regions corresponding to the transmembrane domain (black), collagen repeats (blue), and OLF domain (red) are in color.
To generate the epitope-tagged rescuing transgene, a unique ClaI site was generated at the 3′ end of the gene by site-directed mutagenesis. A triple hemagglutinin (HA) tag was generated by PCR from pGTEP1 and subcloned into the ClaI site.
To generate the cof-2 reporter, the promoter was amplified by PCR and subcloned into the gfp vector pPD95.75 using the following primers: 5′-caacctacagtgcctttccgt-3′ and 5′-tttagttgaagaaactaggctcaatc-3′.
cDNA analysis. An unc-122 expressed sequence tag (EST) clone, yk410e3, was kindly provided by Y. Kohara (National Institute of Genetics, Mishima, Japan). Standard 5′ rapid amplification of cDNA ends (RACE) was used to confirm the 5′ end and construct a full-length cDNA. The accession number for unc-122 is AY494976. cof-2 was first incorrectly predicted to be two separate genes, C48E7.4 and C48E7.5. An alternative gene prediction by Intronerator (Kent and Zahler, 2000) predicted a gene structure with slightly different exons that fused C48E7.4 and C48E7.5 (and, incorrectly, also a third gene). On the basis of the Intronerator prediction for the fusion product of C48E7.4 and C48E7.5, we designed PCR primers (5′-atggaagaaaatttacattattgcaacttagc-3′ and 5′-ttatttagccagccaatgcaaaagt-3′) and obtained an RT-PCR product that was sequenced to confirm the correct gene structure. The accession number for cof-2 is AY494975.
Assay for coelomocyte function. The coelomocytes of C. elegans are scavenger cells that continuously and nonspecifically endocytose fluid from the pseudocoelom (body cavity) (Fares and Greenwald, 2001). When GFP is expressed in the muscle as a secreted form, it is released into the pseudocoelom and taken up by the coelomocytes (Fares and Greenwald, 2001). Using a reporter for coelomocyte endocytosis (arIs37; gift from Johnny Fares, University of Arizona, Tucson, AZ), we found no defect in uptake of GFP into coelomocytes in unc-122 mutant animals.
Generation of coelomocyte-deficient animals and exclusion of unc-122 activity in coelomocytes. We genetically ablated coelomocytes in unc-122 mutants using a transgenic construct expressing the diphtheria toxin A fragment (catalytic) under the control of the unc-122 promoter (pJF142; gift from Johnny Fares). We then scored for the ability of an unc-122 rescuing transgene on the same transgenic array to rescue the uncoordinated phenotype of unc-122. pJF142 and the rescuing transgene were injected at 10 and 20 ng/μl, respectively. ttx-3::gfp was used as an injection marker at 50 ng/μl. Animals in which the coelomocytes were removed, as evidenced by both the accumulation of GFP within the body cavity and the absence of visible coelomocyte cells by Nomarski optics, still retained the ability to be rescued by the unc-122 rescuing construct (36 of 38 animals).
Antibody staining. Worms were permeabilized using freeze fracture (Duerr et al., 1999) followed by fixation in ice-cold methanol and acetone. Slides were incubated for 1 hr in antibody buffer (0.5% Triton X-100, 1 mm EDTA, and 0.1% BSA in PBS with 0.05% sodium azide) containing 10% goat serum. Primary antibody incubations (1:100-1:300) were done overnight. Mouse monoclonal anti-UNC-17 antibodies (mAb1403) were kindly provided by Jim Rand (Oklahoma Medical Research Foundation, Oklahoma City, OK). MH46 antibodies were obtained from the Developmental Studies Hybridoma Bank (University of Iowa). Rabbit anti-GFP antibodies were obtained from Molecular Probes (Eugene, OR), and mouse monoclonal anti-GFP antibodies were obtained from Clontech (Cambridge, UK). Mouse monoclonal anti-myc antibodies (9E10) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rat anti-HA high-affinity antibodies (3F10) were obtained from Roche (Hertforshire, UK). After thorough washing with antibody buffer, slides were incubated in secondary antibody for 4 hr. (Cy3)- and (Cy2)-labeled secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA). After washing, slides were mounted in SlowFade reagent (Molecular Probes) and imaged by fluorescence microscopy.
Locomotory assays. Thrashing, body bends, and reversal were performed at room temperature as described previously (Tsalik and Hobert, 2003). Sensitivity to drugs was examined on nematode growth media plates containing either 1 mm aldicarb (ChemServices) or 100 μm levamisole as described previously (Lackner et al., 1999). Twenty to thirty worms per plate were monitored for paralysis over time. The criterion for paralysis was failure to respond to prodding with a platinum wire. Graphs are representative of at least two trials. Unless indicated otherwise, strains were grown at 20°C and scored as young adults.
Results
unc-122 mutant animals display motor axon sprouting defects
We reasoned that genes required for the DVB motor neuron to develop and function appropriately may also have a function in the neuromuscular system that controls locomotory behavior. We therefore examined previously uncharacterized locomotory mutants, termed unc mutants, for defects in DVB motor neuron anatomy. In addition to unc-75 animals, which we described recently (Loria et al., 2003), we also find that unc-122 mutant animals display neuroanatomical defects that resemble those observed in lim-6 mutant animals (Fig. 1A). Similar to lim-6 mutants, these defects are strongly temperature sensitive (data not shown), but in contrast to lim-6 they show a lower penetrance (Fig. 1B) (Hobert et al., 1999a). Similar to other mutants that affect subtle aspects of AVL or DVB motor axon anatomy (Mehta et al., 2004), unc-122 mutant animals display no enteric muscle contraction defects (data not shown).

Motor axon anatomy defects of unc-122 mutant animals. A, Ectopic axon outgrowth (yellow arrowhead) from the DVB motor neuron (red arrow points to cell body), visualized with the unc-47::gfp reporter transgene oxIs12 in an adult animal. A schematic drawing of DVB motor neuron morphology is shown in the top panel, with synapses to its enteric muscle targets labeled schematically (blue arrows). B, Quantification of axon sprouting defects. Animals were raised at 25°C and scored as adults.
We examined axon morphology of other neuron classes in unc-122 mutants, including several motor neuron classes involved in locomotion, sensory neurons, and interneurons in head ganglia, and observed no obvious defects (data not shown).
The penetrance of the axon sprouting defects of unc-122 mutants in the enteric motor neurons can be enhanced significantly in a genetically sensitized background. Specifically, complete genetic elimination of GABA, the neurotransmitter of DVB, in unc-25-glutamic acid decarboxylase mutants (Jin et al., 1999) also yields lowly penetrant axon sprouting defects (Fig. 1B) (Loria et al., 2003). unc-25; unc-122 double null mutants display a synergistic axon sprouting phenotype, significantly stronger than each single mutant alone (Fig. 1B). A similar synergistic enhancement of unc-25 null mutants can be observed on removal of neuromuscular signaling pathways that act in parallel to unc-25, such as peptidergic signaling pathways (Loria et al., 2003; P. M. Loria and O. Hobert, unpublished observations). We conclude that unc-122 affects DVB motor axon anatomy, an effect that either could be caused indirectly by defects in neurotransmission or could be reflective of a more direct involvement of unc-122 in determining motor axon anatomy.
unc-122 affects different aspects of locomotory behavior
Because motor units controlling locomotory behavior display a more severe functional disruption than the motor units controlling defecation behavior, we sought to characterize the locomotory defects of unc-122 mutants in more detail. The impairment of locomotory behavior in unc-122 mutant animals is evident on several levels. First, although unc-122 mutant animals can move in a sinusoidal wave manner, they frequently coil up spontaneously (supplemental movies 1 and 2, available at www.jneurosci.org). Coiling is unbiased in the ventral or dorsal direction (data not shown). These coiling defects do not relate to defects in GABAergic signaling because removal of GABA (using unc-25 null mutant animals) does not affect the coiling behavior of unc-122 animals (supplemental movie 3, available at www.jneurosci.org, shows locomotory patterns of unc-25; unc-122 double null mutants). Second, the speed of straight sinusoidal locomotion is impaired in unc-122 mutant animals, as quantified by a measurement of body bends per time interval (Fig. 2A). Third, in liquid solution, unc-122 mutants display a decreased frequency of thrashing (Fig. 2B). Last, when moving forward or backward, unc-122 mutants display a decrease in reversal frequency (Fig. 2C).

Locomotory defects of unc-122(e2520) mutant animals. A, Body bends per time interval of adult animals moving on an agar plate at room temperature. B, Thrashing of adult animals at room temperature in a drop of M9 buffer. C, Reversal behavior of adult animals at room temperature on a nonseeded agar plate. D, Aldicarb (1 mm)-induced paralysis of adult animals cultivated at 20°C. E, Levamisole (100 mm)-induced paralysis of adult animals cultivated at 20°C.
We noted that unc-122 mutant animals show a significantly increased impairment of locomotory behavior in the adult stage compared with earlier larval stages (Fig. 2A,B). In addition, the motor axon defects in the DVB neuron are more severe in adult animals than in late larval stages (data not shown). The relatively low level of initial defects suggests that development of the motor circuit is affected minimally, yet the progressive worsening indicates that unc-122 is required continuously during the life of an animal. These observations provided us with the testable model that unc-122 may have a role in acute signaling rather than the initial development of the motor unit.
unc-122 mutants display a defective sensitivity to cholinergic agonist and antagonists
We tested more directly whether unc-122 mutants display defects in synaptic signaling by using two neuroactive drugs, aldicarb and levamisole, that are used to dissect neuromuscular signaling in C. elegans (Brenner, 1974; Hosono and Kamiya, 1991; Miller et al., 1996). Aldicarb inhibits acetyl cholinesterase (AChE), causing an accumulation of acetylcholine at the neuromuscular junction (NMJ), overstimulation of acetylcholine receptors, and eventual paralysis of wild-type animals. Defects in neurotransmission at the presynaptic site will lead to a less pronounced accumulation of acetylcholine, causing aldicarb resistance. In contrast, sensitivity to the acetylcholine receptor agonist levamisole will persist if the defect is on the presynaptic site; however, if postsynaptic signaling components are dysfunctional, animals will be resistant to the activity of levamisole. We find that unc-122 animals are not resistant but are hypersensitive to both aldicarb and levamisole (Fig. 2D,E). This observation indicates that unc-122 negatively regulates aspects of neurotransmission. The levamisole hypersensitivity of unc-122 is suppressed completely on removal of the cholinergic, levamisole-sensitive receptors unc-29 or unc-38 (Fleming et al., 1997) (Fig. 2E); however, unc-29; unc-122 and unc-38; unc-122 double mutants still coil, indicating that the coiling phenotype of unc-122 is not caused by an overstimulation of levamisole-sensitive nicotinic acetylcholine receptors in the body wall muscles.
We attempted to further corroborate the notion that unc-122 negatively regulates cholinergic signaling by asking whether removal of unc-122 can alleviate the effect of a partial disruption of presynaptic neurotransmitter release. Using a hypomorphic allele of the vesicular acetylcholine transporter unc-17, we find that the aldicarb resistance of unc-17 mutants is partly suppressed by removal of unc-122 (Fig. 2D). Because ACh and GABA act antagonistically to control locomotory behavior (Richmond and Jorgensen, 1999), this unc-122-mediated suppression of cholinergic defects could be explained by an impairment of GABA signaling. Although we cannot exclude such a possibility, we consider it unlikely because two pieces of genetic evidence point to unc-122 and GABA signaling acting in parallel and likely distinct pathways. First, unc-25; unc-122 double null mutants show additive locomotory defects; the animals coil and shrink (supplemental movie 3, available at www.jneurosci.org). Second, the aldicarb hypersensitivity observed during loss of GABA in unc-25 null mutants can be enhanced further by removal of unc-122 (Fig. 2D).
unc-122 encodes a phylogenetically conserved transmembrane protein
To gain a better understanding of the function of unc-122, we determined the molecular identity of the protein encoded by the unc-122 locus through positional cloning. Deficiency and multifactor mapping placed unc-122 in close proximity to the unc-54 locus (see Materials and Methods). Transformation rescue with cosmids adjacent to the unc-54 locus and smaller PCR fragments led to the identification of a 5 kb fragment that completely rescued the coiling phenotype of unc-122 mutants (Fig. 3A). This fragment contains a single predicted gene, F11C3.2. We confirmed the gene structure by sequencing an available EST clone and by 5′RACE analysis. All three available unc-122 alleles, which are phenotypically essentially indistinguishable, contain premature nonsense mutations in the open reading frame of F11C3.2 (Fig. 3B). We conclude that F11C3.2 is unc-122.
The premature stop codons are located in the C-terminal half of the protein and disrupt a phylogenetically conserved domain, the olfactomedin (OLF) domain (see below). We describe below experiments that show that all other, more N-terminally located domains of UNC-122 are not required for UNC-122 function. In conjunction with the observation that the placement of the e2520 allele over a chromosomal deficiency (eDf4) did not lead to increases in the severity of the mutant phenotype (data not shown), we conclude that all available alleles of unc-122 are likely genetic null alleles. The notion that our unc-122 alleles are loss-of-function alleles is supported by our observation that unc-122 alleles are not only completely recessive but are also phenocopied by reduction of unc-122 function through RNA interference (data not shown).
Both gene prediction and cDNA analysis reveal that UNC-122 is a 598 amino acid protein that contains a predicted transmembrane domain but no signal sequence, thus defining it as a type II transmembrane protein with a 19 amino acid long N-terminal intracellular domain (Figs. 3B, 4). In its extracellular domain, UNC-122 contains one copy of an OLF domain, a cysteine-rich domain first identified in the extracellular matrix protein olfactomedin and subsequently identified in several more proteins (Fig. 4). One of these, TIGR/myocilin, is mutated in human primary open angle glaucoma (Stone et al., 1997). Biochemical assays demonstrated that OLF domain-containing proteins can form disulfide-bonded multimers, suggesting that at least some OLF domain proteins may be structural proteins (Snyder et al., 1991; Hillier and Vacquier, 2003). Furthermore, a potential architectural role of UNC-122 is suggested by the presence of 16 copies of a collagen triple-helix repeat (Fig. 3B). These repeats are a defining feature of members of the collagen superfamily of extracellular matrix proteins (Mayne and Brewton, 1993). The sequence of the repeat unit is Gly-X-X, with the second and third positions frequently being proline and hydroxyproline. The presence of the collagen repeats sets UNC-122 apart from all previously reported OLF domain proteins (Fig. 4).

UNC-122 is phylogenetically conserved. A, Protein domain structure of OLF domain-containing proteins. Vertebrate genomes contain one other OLF domain-containing protein type, Latrophilin, a protein with various extracellular domains and seven transmembrane domains (Lelianova et al., 1997). Worms and flies each contain a single ortholog of Latrophilin that, however, contains no OLF domain. We named the family of collagen repeat-plus olfactomedin domain-containing proteins the Colmedins. B, Phylogenetic analysis of the OLF domain. A cladogram of a neighbor-joining tree was constructed using distance analysis in the PAUP program package (Swofford, 2000).
The only other protein in C. elegans that contains an OLF domain is also predicted to be a type II transmembrane protein with collagen triple-helix repeat (Fig. 4A). This gene was predicted previously to be two separate genes (one with an OLF domain, C48E7.4; the other with collagen repeats, C48E7.5), but our cDNA analysis reveals this previous prediction to be incorrect (Fig. 5B shows corrected gene structure). We have named this protein COF-2 (for Colmedin family member number 2; see below).
A putative vertebrate ortholog of UNC-122 has been identified recently and is termed Gliomedin (E. Peles, Weizmann Institute, Rehovot, Israel, personal communication; database accession number NP852047). Gliomedin is the only other non-nematode protein in the databases that exclusively contains the three structurally defining features of UNC-122: a type II transmembrane domain, collagen triple-helix repeats, and an OLF domain (Fig. 4A). Sequence similarity searches reveal that Drosophila melanogaster contains only one OLF domain protein in its genome, CG6867, the gene structure of which we confirmed by completely sequencing an available EST clone. Like UNC-122, COF-2, and Gliomedin, CG6867 is a type II transmembrane domain protein with collagen repeats, but curiously, it also contains two immunoglobulin domains not found in any other OLF domain protein (Fig. 4A). The clustering of UNC-122, COF-2, Gliomedin, and CG6867 into a common group on the basis of their characteristic assembly of specific domains is supported by a phylogenetic analysis of the sequence of the most conserved domain of the protein, the OLF domain (Fig. 4B). We name this family of proteins the “Colmedins” (for collagen and olfactomedin domains).
Functional mapping of relevant domains of the unc-122 locus
We tested the functional relevance of individual coding and noncoding regions of the unc-122 locus using transformation rescue assays. Surprisingly, we find that deletion of the intracellular domain, the transmembrane domain, and the collagen repeats from the original 5 kb rescuing fragment does not abolish its ability to rescue the locomotory defects of unc-122 mutants (Fig. 5). Moreover, we find that regions 5′ to the predicted start site of unc-122 are not required for rescuing activity (Fig. 5). Transcriptional regulatory elements must thus reside either in introns or 3′ to the gene. With the use of hybrid genomic-cDNA constructs, we narrowed these cis-regulatory regions to the last intron or the 3′UTR of the unc-122 locus (Fig. 5).
The functionality of engineered and secreted UNC-122 protein suggests that the endogenous unc-122 locus produces not only a transmembrane protein but also a secreted form of the protein. We indeed find by Western blot analysis that transgenic animals expressing an HA epitope-tagged, full-length UNC-122 protein produce two differently sized proteins (data not shown); however, at this point we have not determined whether these two protein products are the result of protein cleavage or products of two alternatively spliced forms of the gene.
unc-122 acts in muscle
To gain insights into the site of unc-122 action, we attempted to analyze its expression pattern using reporter gene fusion constructs. Reporter gene constructs in which gfp was inserted into various regions of the 5 kb rescuing unc-122 locus showed little to no gfp fluorescence or exclusive expression in coelomocytes, a scavenger cell type of C. elegans (Fig. 6). Smaller reporter gene constructs that contain only 5′ upstream regions of the unc-122 locus also yielded strong and consistent expression in coelomocytes (Fig. 6). Although this expression is strikingly similar to the coelomocyte expression of the sea urchin OLF protein amassin (Hillier and Vacquier, 2003), we were able to conclusively rule out the possibility that unc-122 acts in coelomocytes to control locomotory behavior. First, as we reported above, deletion of the 5′ region of the unc-122 locus, which contains the coelomocytic cis-regulatory elements, does not abolish rescuing activity (Fig. 5). Second, unc-122 cDNA driven by the coelomocyte regulatory elements is not able to rescue the unc-122 mutant phenotype (Fig. 7A). Third, if coelomocytes are genetically removed by expression of a cytotoxin (diphtheria toxin) under control of the coelomocyte regulatory element, no locomotory defects can be observed (see Materials and Methods). Finally, when coelomocytes are genetically removed in unc-122 mutant animals, these coelomocyte-deficient animals can still be rescued for their locomotory defects by injecting the wild-type 5 kb unc-122 locus (see Materials and Methods). Thus, coelomocytes are not necessary for unc-122 to fulfill its locomotory function. We also note that coelomocytes function normally in unc-122 mutant animals (see Materials and Methods).
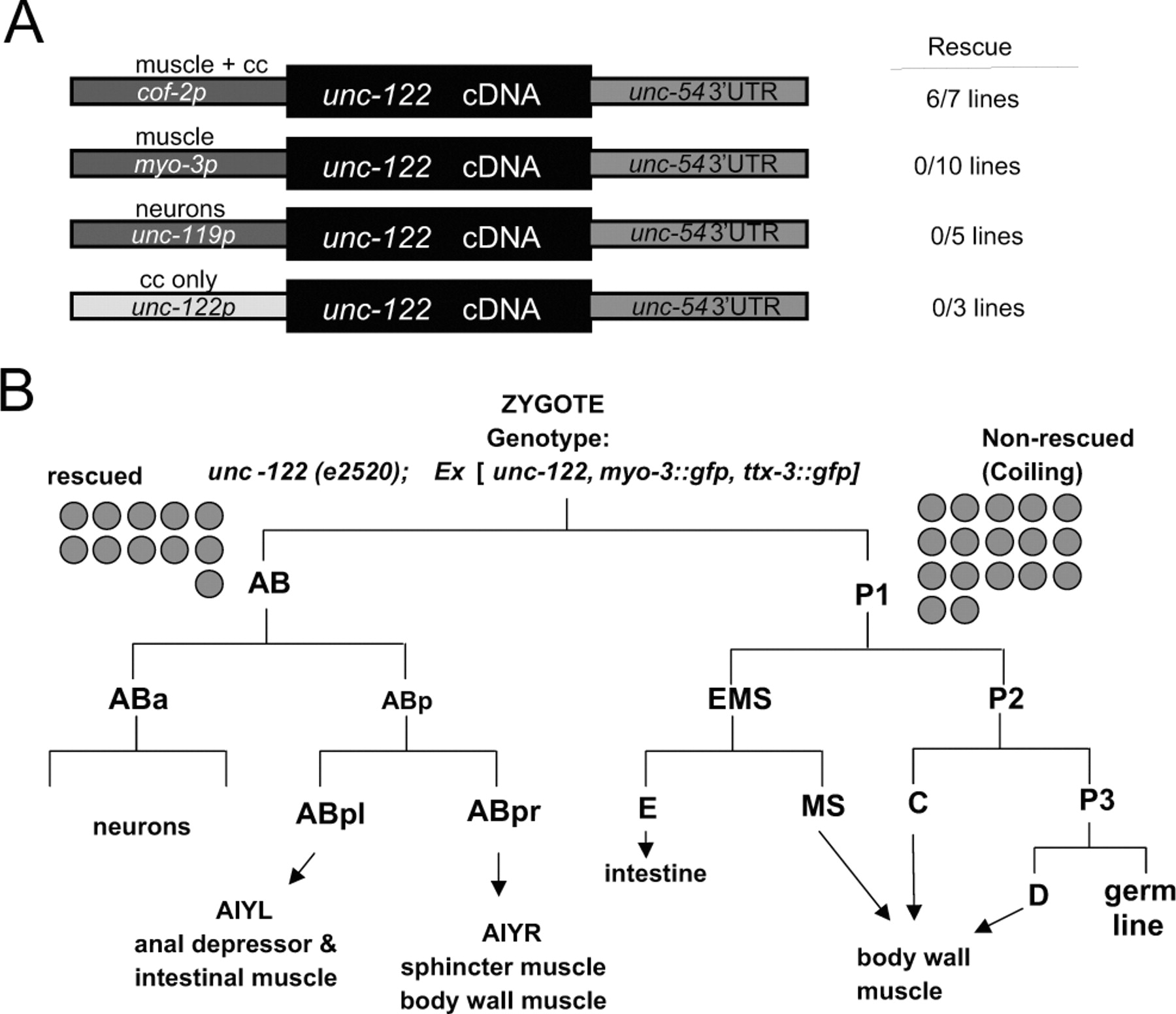
unc-122 acts in muscle. A, Tissue-specific rescue experiments. Constructs (not drawn to scale) were injected into unc-122(e2520) animals at 20 ng/μl. Rescue was scored as a line having >50% of animals that locomote without coiling. B, Mosaic analysis. The 5 kb unc-122 rescuing fragment (Fig. 2A) was injected into unc-122(e2520) animals (at 20 ng/μl) together with ttx-3::gfp and myo-3::gfp as lineage markers (at 50 ng/μl). Two classes of animals were scored for rescue of locomotory defects. First, animals in which AB cell descendants (ttx-3::gfp-expressing AIY cells and myo-3::gfp-expressing enteric muscle cells) express gfp but not P cell descendents (body wall muscle); second, animals in which body wall muscle, but not AIY or enteric muscles, expresses gfp. Gray circles indicate how many animals per class were found that had lost the array and were scored for rescue.
Because reporter gene analysis yielded no insights into the site of unc-122 function, we sought to identify the cells in which unc-122 acts to control locomotory behavior by using rescue assays with heterologous expression constructs, mosaic analysis, and epitope tagging of the locus. For the first approach, we expressed the unc-122 cDNA under control of neuron- or muscle-specific promoters but observed no rescue (Fig. 7A). Because of preliminary evidence that overexpression of unc-122 can cause locomotory defects (data not shown), we were concerned that a failure to observe rescue was caused by too high a level of unc-122 expression in these transgenic lines. To circumvent this problem, we made use of the observation that the 5′ regulatory region of the cof-2 gene contains cis-regulatory elements driving moderate reporter expression in muscle (Fig. 6) (expression is also observed in coelomocytes; their involvement in unc-122 function was excluded as described above). We found that unc-122 cDNA expressed under control of the cof-2 cis-regulatory elements is capable of rescuing the unc-122 coiling phenotype (Fig. 7A). We take these rescue experiments as the first of a total of three pieces of evidence that unc-122 acts in muscle to affect locomotory behavior.
The next piece of evidence about the focus of unc-122 action was obtained by mosaic analysis. We concentrated specifically on neuronal and muscle lineages for this analysis because locomotory defects are usually caused by defects in either lineage. Using gfp markers that label neuronal and muscle cell lineages, we find that loss of unc-122 in neuronal lineages has no impact on locomotory behavior, whereas loss of unc-122 in muscle cell lineages affects locomotory behavior (Fig. 7B). Hence, unc-122 acts in muscle rather than neurons to affect locomotory behavior.
Last, we tagged the unc-122 locus with the HA epitope tag (Fig. 6A). This construct is able to fully rescue the mutant phenotype of unc-122 (five of five lines show rescue). We chromosomally integrated one unc-122::HA-expressing array and performed anti-HA antibody staining. We observed expression of UNC-122 in body wall muscle and coelomocytes (Fig. 8) (data not shown). We note that this expression pattern is identical to the reporter gene-inferred expression pattern of the unc-122-like gene cof-2. In summary, based on cell-specific heterologous expression, mosaic analysis, and antibody staining, we conclude that unc-122 acts in muscle to affect locomotory behavior.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
UNC-122 is enriched at muscle postsynaptic zones but does not affect their development. A, Schematic drawing of individual muscle cells and their muscle arms that extend into the VNC to make neuromuscular junctions (circled) with individual motor axons (red). Only a subset of cells and NMJs are shown. Note that individual motor neurons make multiple en passant synapses with many muscle arms and that muscle arm and endplate morphology can be very complex, stretching over a substantial area; this is shown in more detail in supplemental Figure 1. B, UNC-122 is enriched at NMJs. All animals contain an unc-122::HA-expressing, chromosomally integrated array (otIs127) that rescues the mutant unc-122 phenotype. To visualize UNC-38 localization, the ljEx42 array was used. Colocalization of UNC-122 to muscle endplates was observed not only with UNC-38 but also with UNC-29 (data not shown). In unc-6 mutants, neurons and consequently muscle arms are mislocalized, causing the generation of aberrantly localized NMJs. C, Presynaptic and postsynaptic differentiation of NMJs appears normal in unc-122 mutants. All panels show unc-122 mutant animals. In the top panel, cholinergic presynaptic specialization (visualized with anti-UNC-17) and postsynaptic GABAergic NMJs (visualized with antibody staining against UNC-49::GFP; note the visualization and intactness of muscle arms) are shown; in the bottom panel, presynaptic and postsynaptic cholinergic specializations are shown. The animals in the bottom panel contain the unc-29(x568) allele in the background, which is necessary for correct localization of the unc-29::gfp reporter expressed from the xEx1 array (Fleming et al., 1997). Wild-type controls appear indistinguishable (data not shown).
UNC-122 localizes to neuromuscular junctions
Epitope-tagged UNC-122 shows a punctate localization along the ventral nerve cord (VNC) (Fig. 8). These sites do not coincide with muscle-hypodermis attachment sites, as observed by antibody costaining against an attachment marker protein (data not shown). In C. elegans, muscle cells extend thin extensions (“muscle arms”) into the VNC, where they make neuromuscular junctions with motor axons (Fig. 8A). Contact points between muscle arms and motor axons are not focal in nature but distribute longitudinally along motor axons (supplemental Fig. 1), hence giving the muscle endplate an extended appearance (schematically shown in Fig. 8A). To determine whether the localization of UNC-122 indeed coincides with the muscle endplate of NMJs, we performed costaining experiments, using presynaptic and postsynaptic markers that are specific for individual motor neuron classes. We examined cholinergic presynaptic and postsynaptic specializations using antibody staining against the vesicular acetylcholine transporters UNC-17 (Alfonso et al., 1993) and epitope-tagged acetylcholine receptors (UNC-29 and UNC-38) (Fleming et al., 1997). We found that UNC-122 clusters precisely oppose UNC-17 clusters, consistent with the localization of UNC-122 to muscle endplates (Fig. 8B). UNC-122 clusters do not precisely overlap but rather interdigitate with clusters of UNC-38 AChR (Fig. 8B) or UNC-29 (data not shown); the relatively large size of muscle endplates suggests that UNC-122 and UNC-38 may localize to distinct subdomains within muscle endplates. Conceptually similar results can be found at GABAergic NMJs, which were visualized with antibody staining against the tagged GABA receptor UNC-49 (Bamber et al., 1999) and tagged VAMP protein, driven specifically in GABAergic neurons (data not shown).
It has been shown that muscle arm extension can respond flexibly to perturbations of the motor neurons (Hedgecock et al., 1990). In the absence of the C. elegans netrin homolog UNC-6, axons show general defasciculation, with axons that normally run in the dorsal nerve cord often being displaced ventrally. In unc-6 mutants, the dorsal muscles send their muscle arms ventrally across the lateral epidermis to locate the misplaced axon (Hedgecock et al., 1990). We find that in unc-6 mutants, UNC-122 is enriched in NMJs that form onto the misplaced motor neurons (Fig. 8B).
Together with the demonstration of a muscular focus of action of unc-122, we conclude from these localization experiments that UNC-122 localizes to the postsynaptic side of NMJs.
UNC-122 is not required to determine gross aspects of muscle ultrastructure
The focus of unc-122 gene action in muscle prompted us to assess muscle fate and ultrastructure in unc-122 mutant animals. Using gfp markers that label various aspects of muscle anatomy, we found first that all markers are appropriately expressed, indicating that muscle cell fate is appropriately executed. Second, overall muscle shape, muscle filament attachment to the membrane (via dense bodies), and muscle arm extension are intact (data not shown; see Materials and Methods for gfp reporters). Moreover, electron microscopy analysis of cross sections of unc-122 mutant animals showed no defects in overall muscle arm morphology, sarcomere structure, or dense body structure (D. H. Hall, personal communication).
The localization of UNC-122 at NMJs prompted us to ask whether presynaptic and postsynaptic specializations form appropriately in unc-122 mutant animals. We visualized presynaptic specialization of GABAergic motor neurons using VAMP::GFP and of cholinergic motor neurons using anti-UNC-17 antibodies and found each to be unaffected (Fig. 8C) (data not shown). Moreover, we analyzed the localization of the three postsynaptic neurotransmitter receptors mentioned above, unc-49, unc-29, and unc-38, and found them to localize normally in unc-122 mutant animals (Fig. 8C) (data not shown). Taken together, the intact muscle arm targeting and correct clustering of several presynaptic and postsynaptic components indicate that UNC-122 is not required to determine gross aspects of muscle innervation by motor axons.
Discussion
Starting with the anatomy of the enteric motor neuron DVB as an indicator of motor unit development and function, we have characterized the unc-122 gene and found it to code for a postsynaptic protein required for neuromuscular signaling. Although the characterization of unc-122 does not reveal additional insights into the mechanism of lim-6 homeobox gene function in DVB, our study reveals a novel muscular component required for motor unit function. Our study furthermore underscores the fact that motor axon morphology is exquisitely sensitive to disruption in neurotransmission, a notion made previously in various experimental systems (Brown et al., 1981; Zhao and Nonet, 2000; Loria et al., 2003).
We infer from the following observations that postsynaptically localized UNC-122 acts in neuromuscular signaling rather than in the development of the motor unit. First, we have been unable to observe any abnormalities in the execution of muscle cell fate or muscle cell morphology. Morphological analysis included the visualization of presynaptic and postsynaptic proteins, the localization of which we found to be unaffected by loss of unc-122. Second, the abnormal drug response profile of unc-122 mutants is a commonly used indicator of neuromuscular signaling defects and in the past has revealed a multitude of components required for efficient neurotransmission (Brenner, 1974; Hosono and Kamiya, 1991; Miller et al., 1996). Third, the motor axon defects as well as locomotory defects worsen significantly during the life of an animal. In other words, the initially limited defects are indicative of the muscle structure and initial function being little affected, whereas the worsening indicates that the normally occurring execution of neuromuscular signaling is disrupted, causing a progressive inability to maintain the structural and functional integrity of the motor unit.
The extracellular motifs of UNC-122, collagen repeats, and the cysteine-rich OLF domain are characteristic units of extracellular matrix proteins. Indeed, several OLF domain proteins have been localized to the extracellular matrix and shown to physically interact with components of it (Snyder et al., 1991; Yokoe and Anholt, 1993; Filla et al., 2002; Hillier and Vacquier, 2003). Moreover, OLF domain proteins can form higher-order, disulfide-bonded multimers, further underscoring their function as structural proteins (Snyder et al., 1991; Hillier and Vacquier, 2003). In addition, the recently uncovered function of the OLF domain protein amassin as a cell adhesion protein (Hillier and Vacquier, 2003) indicates that UNC-122 may act as a structural and perhaps adhesive component of the NMJ.
The vertebrate NMJ is composed of a plethora of extracellular matrix proteins, such as specific laminins and collagens (Sanes and Lichtman, 1999; Patton, 2000). These proteins have highly specialized functions in creating individual structural NMJ microdomains required for efficient neuromuscular signaling, and they target specific proteins to these microdomains. For example, collagen Q is a component of the AChE complex and is required to localize this enzyme to the synaptic cleft (Feng et al., 1999). The collagen repeats of UNC-122 may serve a similar role in localizing C. elegans signaling molecules, such as AChE and other factors, to the NMJ. Alternatively, UNC-122 may create a structural microenvironment that limits the cell surface clustering of postsynaptic receptors. Although we observe no difference in localization of three known postsynaptic receptors, it needs to be emphasized that we have used overexpressed reporter constructs for this analysis, which does not allow us to make inferences about quantitative aspects of endogenous receptor levels at the NMJ in unc-122 mutants. Last, it can be envisioned that UNC-122 is a synaptic adhesion molecule that stabilizes synaptic contacts. Loss of unc-122 may result in a disruption of a tight contact between presynaptic and postsynaptic zones, thus indirectly causing synaptic signaling defects.
Footnotes
- Received December 11, 2003.
- Revision received January 8, 2004.
- Accepted January 13, 2004.
This work was funded by grants from the National Institutes of Health, the Rita Allen Foundation, the Muscular Dystrophy Association, and the Christopher Reeve Paralysis Foundation. We are indebted to Erik Jorgensen for pointing out the existence of the unc-122 mutation in the CB51 strain and for providing us with another unc-122 allele, and David Hall, Martin Wakeland, and Ken Nguyen for EM analysis. We thank Elior Peles for communicating results on Gliomedin, Martine Forrester and Wendy Yip for conducting behavioral assays, Adam Wenick for recording muscle arm structure with C50F7.1::gfp, and members of the worm community, specifically Erik Jorgensen, Jim Lewis, Jean Louis Bessereau, Alexander Gottschalk, Johnny Fares, and Jim Rand, for providing transgenic strains and reagents.
Correspondence should be addressed to Oliver Hobert, Department of Biochemistry and Molecular Biophysics, Center for Neurobiology and Behavior, Columbia University, College of Physicians and Surgeons, 701 West 168th Street, New York, NY 10032. E-mail: or38{at}columbia.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/242191-11$15.00/0