
Abstract
Drugs that increase GABA levels in the brain reduce cocaine seeking in rodents and humans, suggesting that GABAergic inhibition regulates cocaine-seeking behavior. We previously reported that repeated cocaine exposure in vivo facilitates long-term potentiation by reducing the strength of GABAergic inhibition in dopamine neurons of the ventral tegmental area (VTA). Selective blockade of cocaine-induced reduction of GABAergic inhibition in the VTA might diminish cocaine-induced aberrant synaptic plasticity and addictive behavior. Here, we investigated the mechanism for cocaine-induced reduction of GABAergic inhibition. We show that a pathophysiologically relevant concentration of cocaine enables a normally ineffective stimulus to induce long-term depression (LTD) of IPSCs (I-LTD) in VTA dopamine neurons of midbrain slices. Activation of D2 dopamine receptors and group I metabotropic glutamate receptors and subsequent recruitment of endocannabinoid signaling are required for I-LTD induction. We further demonstrate that in vivo pretreatment with antagonists to these receptors blocks cocaine-induced reduction of GABAergic inhibition and that repeated cocaine exposure in vivo occludes the subsequent induction of I-LTD ex vivo. Together, these results suggest that repeated cocaine exposure reduces the strength of GABAergic inhibition in dopamine neurons by inducing I-LTD-like modification in vivo.
- endocannabinoid
- long-term depression
- synaptic plasticity
- GABA
- cocaine addiction
- dopamine
Introduction
There is an emerging consensus that associative learning plays an important role in drug addiction (Kauer, 2004; Kelley, 2004; Hyman et al., 2006). When environmental cues are repeatedly paired with drug use, an association between the cues and the drug is formed. As a result, drug-associated cues will acquire incentive salience (Robinson and Berridge, 1993), triggering drug craving and relapse. The mechanisms underlying this reinforcement learning remain poorly understood. Long-term potentiation (LTP) and long-term depression (LTD) are leading cellular mechanisms for learning and memory (Malenka and Bear, 2004). An attractive hypothesis for addictive behavior is that addictive drugs induce or alter long-term synaptic plasticity in the reward circuitry, which leads to the formation of drug-associated memories and ultimately, drug addiction. Not surprisingly, several studies have shown that in vivo cocaine exposure alters or induces LTP or LTD of excitatory synaptic transmission in the ventral tegmental area (VTA) (Ungless et al., 2001; Liu et al., 2005), nucleus accumbens (Thomas et al., 2000; Yao et al., 2004), prefrontal cortex (Huang et al., 2007), and amygdala (Goussakov et al., 2006), brain areas that are critically involved in drug addiction.
The dopamine neurons in the VTA of the midbrain are a primary target of many drugs of abuse including cocaine (Kauer, 2004). We have shown that LTP induction in VTA dopamine neurons is tightly controlled by GABAergic inhibition and repeated cocaine exposure in vivo for 5–7 d facilitates LTP induction by reducing the strength of GABAergic inhibition (Liu et al., 2005). This reduced inhibition might contribute to incentive sensitization by enabling cocaine-associated stimuli to induce persistent alteration of synaptic strength in a key component of the reward circuitry. Our study raises the possibility that blocking cocaine-induced reduction of GABAergic inhibition provide a means to diminish the aberrant synaptic plasticity and cocaine-seeking behavior. Indeed, drugs that enhance GABA levels in the brain reduce cocaine-seeking in rats (Gardner et al., 2002) and clinical trials (Brodie et al., 2003; Kampman et al., 2004). These findings demonstrate that manipulation of GABAergic inhibition is, in principle, a sound strategy for treating cocaine addiction. However, global enhancement of GABAergic inhibition by these drugs produces side effects (Brodie et al., 2003; Kampman et al., 2004). Selective blockade of cocaine-induced reduction of GABAergic inhibition in dopamine neurons could provide an alternative approach for therapeutic intervention of cocaine addiction.
We therefore sought to understand the mechanisms of cocaine-induced reduction of GABAergic inhibition. Because the latter is manifest as a persistent weakening of inhibitory synaptic transmission, we hypothesize that repeated cocaine exposure reduces GABAergic inhibition by inducing a long-term depression (LTD) of IPSCs (I-LTD)-like synaptic plasticity. To test this hypothesis, we determined whether (1) I-LTD could be induced under conditions that mimic in vivo cocaine exposure, (2) in vivo blockade of I-LTD prevented cocaine-induced reduction of GABAergic inhibition, and (3) repeated cocaine exposure in vivo occluded subsequent I-LTD induction ex vivo. Here, we provide evidence that endocannabinoid (eCB)-mediated I-LTD underlies the reduction of the strength of GABAergic inhibition induced by repeated cocaine exposure in vivo.
Materials and Methods
Cocaine treatment and slice preparation.
Experiments were performed using male Sprague Dawley rats [postnatal day 18 (P18)–P30], CB1-deficient mice, and wild-type mice of either sex (see below). In some experiments, rats were given daily intraperitoneal injections of either saline (0.9% NaCl, 1 ml/kg) or cocaine (15 mg/kg) for 1 d or 5–7 d. The effectiveness of the cocaine treatment was shown by the sensitization of locomotor activity (Liu et al., 2005). Subgroups of these rats were also given intraperitoneal injection of one of the following receptor antagonists 20 min before each cocaine injection (see Fig. 10). These include D1 receptor antagonist (R)-(+)-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine (SCH 23390; 2 mg/kg), D2 receptor antagonist sulpiride (5 mg/kg), metabotropic glutamate receptor 5 (mGluR5) antagonist 6-methyl-2-(phenylethynyl)-pyridine (MPEP; 10 mg/kg), and CB1 receptor antagonist N-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide (AM-251; 2 mg/kg). Rats or mice were anesthetized by isoflurane inhalation and decapitated, and horizontal midbrain slices (250 μm thick) were cut using a vibrating slicer (Leica, Nussloch, Germany) as described previously (Liu et al., 2005). Slices were prepared at 4–6°C in a solution containing (in mm) 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgSO4, 26 NaHCO3, 25 glucose, 11.6 sodium ascorbate, and 3.1 sodium pyruvate. For rats that received intraperitoneal saline or cocaine injection, midbrain slices were prepared 20–24 h after the last cocaine or saline injection. The slices were incubated in oxygenated artificial CSF (ACSF) containing (in mm) 125 NaCl, 3 KCl, 2.5 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. The ACSF was saturated with 95% O2 and 5% CO2. After recovery for at least 1 h at room temperature, a slice was transferred to the recording chamber and superfused with ACSF. In strontium (Sr2+) substitution experiments (see Figs. 8, 9), slices were superfused with ACSF in which 2 mm Sr2+/6 mm Mg2+ were substituted for 2.5 mm Ca2+/1 mm Mg2+ and NaCl was reduced to 120 mm to keep the osmolality of ACSF constant. All recordings were performed at 32 ± 1°C by using an automatic temperature controller (Warner Instruments, Hamden, CT).
CB 1-deficient mice.
CB1-deficient mice and their wild-type littermates (P16–P20) of either sex were used in this study. The mice were generated on a genetic background 129/SvJ (Ibrahim et al., 2003), and back-crossed unto an outbred ICR strain for at least nine generations. Genotyping of CB1 knock-out (CB1−/−), heterozygous (CB1+/−), and wild-type (CB1+/+) mice was performed by PCR technique on ear biopsies.
Electrophysiology.
Whole-cell recordings were made using a patch clamp amplifier (Multiclamp 700B) under infrared-differential interference contrast microscopy. Data acquisition and analysis were performed using a digitizer (DigiData 1440A) and analysis software pClamp 10 (Molecular Devices, Union City, CA) and Mini Analysis 6 (Synaptosoft, Decatur, GA). Signals were filtered at 2 Hz and sampled at 10 Hz. Dopamine neurons were identified by the presence of large Ih currents, rhythmic firing at low frequency, and prominent afterhyperpolarization (Johnson and North, 1992; Jones and Kauer, 1999; Liu et al., 2005). However, a previous study showed that these electrophysiological characteristics do not exclusively identify dopamine neurons (Margolis et al., 2006). Putative dopamine neurons in our study may contain a small number of nondopamine neurons, which should be randomly distributed in different experimental groups.
All recordings were made in the presence of glutamate receptor antagonists CNQX (20 μm) and AP-5 (50 μm). Neurons were voltage-clamped at −70 mV unless stated otherwise. For recording of evoked IPSCs, electrical stimulation was delivered by a bipolar tungsten stimulation electrode (World Precision Instruments, Sarasota, FL) that was placed ∼150 μm rostral to the recorded neuron, using square pulses with duration of 40 μs and frequency of 0.1 Hz. Unless stated otherwise, glass pipette was filled with a solution containing (in mm) 100 K-gluconate, 50 KCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 MgATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine at pH 7.2 (with KOH). The concentrations of K-gluconate and KCl were adjusted in some experiments (see below), whereas the remaining components were not changed. In experiments with postsynaptic Ca2+ buffer, 20 mm K-gluconate was replaced by 20 mm BAPTA (see Fig. 5C). For recording of maximal IPSCs, neurons were voltage-clamped at −20 mV, and K-gluconate (145 mm) and KCl (5 mm) were used in internal solution (see Fig. 10A). Maximal IPSCs were elicited by gradually increasing the stimulation intensity to recruit saturating IPSCs (Huang et al., 1999; Liu et al., 2005) and performed blind to the treatment history of the rat. For recording of miniature IPSCs (mIPSCs) in Figure 10, K-gluconate (0 mm) and KCl (150 mm) were used in internal solution, and tetrodotoxin (TTX) was added in the ACSF to block action potentials. For experiments in Figures 7, 8, and 9, K-gluconate (50 mm), KCl (100 mm), and QX-314 (lidocaine N-ethyl bromide; 1 mm) were used in internal solution. This solution allowed stable long-term recording (≥50 min) while offering high-resolution recording of mIPSCs in TTX and asynchronous events in strontium (Sr2+). Internal solution containing 150 mm KCl as used in Figure 10 did not allow stable long-term recording for I-LTD experiments, although it further improved the resolution of mIPSCs. Series resistance (15–30 MΩ) was monitored throughout the recordings and data were discarded if the resistance changed by >20%. Tetrahydrolipstatin (THL), cocaine hydrochloride, CNQX, and all other common chemicals were obtained from Sigma (St. Louis, MO), guanosine 5′-[β-thio]diphosphate trilithium salt (GDP-β-S) was from Biomol (Plymouth Meeting, PA) and all other drugs (e.g., agonists and antagonists) were from Tocris Bioscience (Ellisville, MO).
Statistics.
Data are presented as the mean ± SEM. For quantifying the magnitude of I-LTD, baseline IPSCs were averaged and compared with IPSCs averaged from the final 10 min of recording. Data sets were compared with either paired or unpaired Student's t test, or ANOVA followed by Tukey post hoc analysis. Results were considered to be significant at p < 0.05. The analysis of mIPSCs in TTX and asynchronous mIPSCs in Sr2+ was performed with cumulative probability plot (Van der Kloot, 1991) when two sets of data from the same neuron were compared. The cumulative probability of the amplitude was compared using the Kolmogorov–Smirnov test, which estimates the probability that two distributions are similar.
Results
Cocaine acutely depresses IPSCs in dopamine neurons
Our previous study has shown that daily intraperitoneal injection of cocaine (15 mg/kg) for 5–7 d reduces GABAergic inhibition in dopamine neurons, as detected in midbrain slices in vitro. The cocaine-induced reduction of the strength of GABAergic inhibition is manifest as a decrease in the mean amplitude of evoked maximal IPSCs, spontaneous mIPSCs and GABAA receptor agonist muscimol-induced currents and an increase in the probability of action potential firing in response to synaptic stimulation (Liu et al., 2005). It has been shown that a brief bath application of amphetamine produces a long-lasting depression of evoked EPSPs in rat amygdala slices (Huang et al., 2003). Because a primary action of both cocaine and amphetamine is to inhibit dopamine uptake (Elliott and Beveridge, 2005), we asked whether cocaine produced similar long-lasting depression of evoked IPSCs in VTA dopamine neurons in midbrain slices. We reasoned that this could account for the reduction of GABAergic inhibition induced by repeated cocaine exposure in vivo. However, we found that bath application of cocaine produced transient rather than long-lasting depression of evoked IPSCs in dopamine neurons in slices prepared from drug naive rats. This depression was concentration-dependent and reversible with washout of cocaine (Fig. 1A). Fitting the concentration-response data with a Boltzmann function yielded value for half-maximal inhibition (IC50) of 9.6 ± 1.0 μm (Fig. 1B). To determine whether dopamine receptor activation mediates this acute depression, we examined the effect of dopamine receptor antagonists on cocaine-induced depression of evoked IPSCs. Dopamine receptors consist of two subfamilies: D1-like (D1, D5) and D2-like (D2, D3, and D4) receptors (Pollack, 2004) (for simplicity, D1-like and D2-like dopamine receptors are termed as D1 and D2 receptors, respectively). We found that the D1 dopamine receptor antagonist SCH 23390 (10 μm) had no significant effect on the cocaine-induced depression (IC50, 9.1 ± 1.0 μm). In contrast, the D2 receptor antagonist sulpiride (10 μm) significantly attenuated the effect of cocaine on IPSCs and shifted the concentration-response curve for cocaine to the right (IC50, 18.5 ± 1.0 μm) (Fig. 1B). These results indicate that the activation of D2 receptors, but not D1 receptors, contributes to cocaine-induced depression of IPSCs.

D2 receptor activation contributes to cocaine-induced acute depression of evoked IPSCs. A, Bath application of cocaine produced reversible and concentration-dependent depression of IPSCs (n = 5–7 for each group). Sample IPSCs averaged from 20 consecutive trials before, during, and after application of 10 μm cocaine are shown on the top. Horizontal bar denotes time course of cocaine perfusion. B, D2 receptor antagonist sulpiride (10 μm) shifted concentration–response curve to the right, whereas D1 receptor antagonist SCH 23390 (10 μm) had no significant effect (n = 4–7 for each point).
Beside dopamine, cocaine inhibits the uptake of norepinephrine (NE) and serotonin (5-HT) (Elliott and Beveridge, 2005). The remaining depression is likely mediated by the activation of NE and/or 5-HT receptors. NE receptors are grouped into two major classes: α receptors (α1, α2) and β receptors (β1, β2, β3) (Philipp and Hein, 2004), whereas 5-HT receptors are grouped into seven major classes (5HT1–7) (Noda et al., 2004). Because of this complexity, separate studies are needed to determine which receptor subtypes mediate the remaining depression of IPSCs by cocaine.
In vivo microdialysis has revealed that the peak cocaine concentration in the mesolimbic dopamine system is ∼2–5 μm after intraperitoneal injection of cocaine (15 mg/kg) in the rat (Maisonneuve and Kreek, 1994). It is likely that intraperitoneal injection of cocaine at 15 mg/kg produces little, if any, acute depression of IPSCs in VTA dopamine neurons. Although cocaine at higher concentrations produces significant depression of IPSCs, the depression appears to be transient (Fig. 1A). Thus, cocaine-induced acute depression of IPSCs alone cannot explain the sustained reduction of GABAergic inhibition in VTA dopamine neurons induced by repeated cocaine exposure in vivo.
Cocaine enables I-LTD in dopamine neurons in midbrain slices
Dopamine neurons receive continuous synaptic bombardment in vivo; many synaptic connections are severed in the VTA slice preparation. We reasoned that pairing cocaine application with repetitive afferent stimulation to mimic in vivo synaptic activity could induce I-LTD, which is responsible for cocaine-induced reduction of GABAergic inhibition. VTA dopamine neurons receive GABAergic innervation mainly from medium spiny neurons in the nucleus accumbens (Kalivas et al., 1993), which fire action potentials up to 10–15 Hz in vivo (Carelli and Ijames, 2000; Chang et al., 2000). A moderate-frequency stimulation (10 Hz) mimicking this frequency was therefore chosen for I-LTD induction. However, we found that application of 10 Hz stimulation for 5 min had no significant effect on the amplitude of IPSCs in VTA dopamine neurons (94.8 ± 5.9% of baseline at 40–50 min; n = 8; p > 0.05) (Fig. 2A) (for the quantification of I-LTD magnitude, see Materials and Methods). Moderate-frequency stimulation (10–25 Hz) has been used successfully to induce LTD in the striatum and D2 dopamine receptor activation mediates or regulates this LTD induction (Kreitzer and Malenka, 2005; Ronesi and Lovinger, 2005). The striatum is richly innervated by dopamine axon terminals whereas dopamine is released at somatodendritic sites in the VTA (Beckstead et al., 2004). The 10 Hz stimulation likely induces more dopamine release in the striatum than in the VTA and the relative low dopamine activity in the VTA may account for the lack of I-LTD induction. We therefore determined whether inhibiting dopamine uptake with cocaine (3 μm) enabled I-LTD induction. Although cocaine at 3 μm by itself had no significant effect on evoked IPSCs (99.1 ± 7.2% of baseline, n = 7, p > 0.05) (Fig. 1A), the presence of cocaine (3 μm) during the 10 Hz stimulation induced robust I-LTD (67.3 ± 7.5% of baseline; n = 8; p < 0.01) (Fig. 2B). Thus, a pathophysiologically relevant concentration of cocaine enables a normally ineffective stimulus to induce I-LTD in VTA dopamine neurons.

Cocaine enables I-LTD induction in dopamine neurons in midbrain slices. A, Top, The 10 Hz, 5 min stimulation as indicated by the arrow had no significant effect on evoked IPSCs (n = 8). Bottom, Time course of normalized PPR during and after the 10 Hz stimulation. B, Top, The presence of cocaine (3 μm) 3 min before and during the 10 Hz stimulation led to I-LTD (n = 8). Bottom, The PPR was not significantly changed after I-LTD induction. C, D, The PPR (C) or CV (D) was measured under control conditions (PPR1 or CV1) and a test condition (PPR2 or CV2) in which either the holding potential (Vm) was changed from −70 to −90 mV (n = 7), the extracellular Ca2+ was changed from 2.5 to 1.5 mm(↓Ca2+, n = 7), or LTD was induced (n = 8). *p < 0.01 compared with corresponding baseline controls (paired Student's t test).
To determine whether presynaptic or postsynaptic mechanisms mediate I-LTD, we compared the paired-pulse ratio (PPR) and the coefficient of variation (CV) of IPSCs before and after I-LTD induction. The PPR was calculated as the ratio of the mean amplitude of the second IPSCs over that of the first IPSCs (interpulse interval, 50 ms), whereas CV was calculated as σ/M, where σ and M are the SD and mean of the amplitude of first IPSCs, respectively. We found that I-LTD did not lead to significant changes in the PPR (baseline, 0.83 ± 0.07; I-LTD at 40–50 min, 0.82 ± 0.06; n = 8, p > 0.05) (Fig. 2B,C) and CV (baseline, 0.16 ± 0.06; I-LTD, 0.16 ± 0.05; p > 0.05) (Fig. 2D). A change in the PPR or CV suggests a presynaptic mechanism, whereas no change in the PPR or CV suggests a postsynaptic mechanism (Zucker and Regehr, 2002). Two experiments showed that this is indeed the case in dopamine neurons. First, holding the postsynaptic neurons at different membrane potentials (−70 and −90 mV) did not change the PPR (−70 mV, 0.73 ± 0.05; −90 mV, 0.67 ± 0.03; n = 7, p > 0.05) and CV (−70 mV, 0.16 ± 0.05; −90 mV, 0.16 ± 0.07; p > 0.05). Second, decreasing presynaptic GABA release by lowering extracellular Ca2+ from 2.5 to 1.5 mm significantly increased the PPR (2.5 mm Ca2+, 0.71 ± 0.06; 1.5 mm Ca2+, 0.82 ± 0.07; n = 7, p < 0.01, paired Student's t test) (Fig. 2C) and CV (2.5 mm Ca2+, 0.14 ± 0.05; 1.5 mm Ca2+, 0.21 ± 0.09; p < 0.01) (Fig. 2D). These results appear to suggest that I-LTD in VTA dopamine neurons is expressed postsynaptically (but see below).
Activation of D2 receptor and group I mGluRs is required for I-LTD induction
Cocaine at 3 μm is effective in inhibiting dopamine uptake in the VTA (Beckstead et al., 2004), whereas the repetitive synaptic stimulation (10 Hz, 5 min) likely activates dopaminergic and glutamatergic afferents. The activation of group I mGluRs leads to eCB-mediated I-LTD or LTD in the hippocampus, nucleus accumbens and cerebellum (Robbe et al., 2002; Chevaleyre and Castillo, 2003; Safo and Regehr, 2005; Edwards et al., 2006). In the dorsal striatum, the activation of D2 receptors mediates or regulates eCB-mediated LTD (Kreitzer and Malenka, 2005; Ronesi and Lovinger, 2005; Wang et al., 2006). We therefore determined the role of dopamine receptors and group I mGluRs in I-LTD induction in dopamine neurons.
We examined the effects of dopamine receptor antagonists on I-LTD induction. As shown in Figure 3A, the D2 receptor antagonist sulpiride (10 μm) significantly attenuated I-LTD (92.4 ± 4.7% of baseline; n = 8; p < 0.01 vs I-LTD control in Fig. 2B), whereas the D1 receptor antagonist SCH 23390 (10 μm) had no significant effect (68.4 ± 6.8% of baseline; n = 7; p > 0.05 vs I-LTD control in Fig. 2B). For the remainder of the Results, “I-LTD control” refers to Figure 2B unless stated otherwise. If cocaine enables I-LTD induction by enhancing D2 receptor activation, then the D2 receptor agonist quinpirole should mimic the effect of cocaine. We first examined the effect of quinpirole on evoked IPSCs. Quinpirole at 1 μm had no significant effect on evoked IPSCs (96.8 ± 5.8% of baseline; n = 5; p > 0.05), but induced reversible depression of evoked IPSCs at 10 μm (68.5 ± 8.1% of baseline; n = 6; p < 0.01) (Fig. 3B). Next, we determined whether quinpirole could replace cocaine to enable I-LTD induction. As shown in Figure 3C, the presence of quinpirole (1 μm) during the 10 Hz stimulation induced I-LTD (72.8 ± 6.8% of baseline; n = 8; p < 0.01 vs baseline), which was not significantly different from I-LTD induced in the presence of cocaine (Fig. 2B) (p > 0.05 vs I-LTD). These results suggest that D2 receptor activation is required for I-LTD in dopamine neurons. Finally, we determined whether quinpirole-induced depression of evoked IPSCs occluded subsequent induction of I-LTD. As shown in Figure 3D, in the continuous presence of quinpirole (10 μm), the same stimulation (10 Hz; 5 min; 3 cocaine) still induced I-LTD (77.0 ± 7.8% of baseline at 40–50 min; n = 6; p < 0.05 vs baseline), which was not significantly different from I-LTD induced in the presence of cocaine (Fig. 2B) (p > 0.05 vs I-LTD). Quinpirole alone induced short-term, but not long-term depression (Fig. 3B). The depression of IPSCs by quinpirole was not saturating. These results may explain why quinpirole did not occlude subsequent I-LTD induction.

Activation of D2 receptors is required for I-LTD induction. A, I-LTD was attenuated by D2 antagonist sulpiride (10 μm, n = 8), but not by D1 antagonist SCH 23390 (10 μm, n = 7). The arrows indicate the application of a 10 Hz, 5 min stimulation. B, The effect of bath application of quinpirole on evoked IPSCs (1 μm, n = 5; 10 μm, n = 6). C, The presence of quinpirole (1 μm) during the 10 Hz stimulation enabled I-LTD induction (n = 8). D, The presence of quinpirole (10 μm) throughout the experiment did not occlude I-LTD induction (n = 6).
To determine the involvement of group I mGluRs in I-LTD induction, we first examined whether group I mGluR antagonists blocked I-LTD induction. Group I mGluRs consist of mGluR1 and mGluR5, and both subtypes of receptors are known to be distributed on VTA dopamine neurons (Kane et al., 2005). Figure 4A shows that either the mGluR1 antagonist 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt; 80 μm) or the mGluR5 antagonist MPEP (10 μm) significantly attenuated I-LTD induction (Fig. 2B) (CPCCOEt, 86.5 ± 6.8%, n = 6, p < 0.01; MPEP, 91.9 ± 4.5%, n = 7, p < 0.01 vs I-LTD control). Next, we examined whether the group I mGluR agonist dihydroxyphenylglycine (DHPG) depressed evoked IPSCs in dopamine neurons. Bath application of DHPG (50 μm) for 10 min produced significant depression of evoked IPSCs that was barely reversible after washout (59.8 ± 10.3% of baseline at 40–50 min, n = 7, p < 0.01) (Fig. 4B). Finally, we examined whether the DHPG-induced long-lasting depression of evoked IPSCs occluded subsequent induction of I-LTD. In the continuous presence of DHPG (50 μm), combination of the 10 Hz stimulation with cocaine application failed to induce I-LTD (94.9 ± 9.7% of baseline at 40–50 min, n = 8, p > 0.05 vs baseline) (Fig. 4C). Together, the above results suggest that D2 dopamine receptor and group I mGluR activation are required for I-LTD induction in VTA dopamine neurons.

Activation of group I mGluRs is required for I-LTD induction in dopamine neurons. A, I-LTD was attenuated by mGluR1 antagonist CPCCOEt (80 μm, n = 6) or mGluR5 antagonist MPEP (10 μm, n = 7). B, Group I mGluR agonist DHPG (50 μm) induced long-lasting depression of evoked IPSCs (n = 7). C, The presence of DHPG throughout the experiment occluded subsequent I-LTD induction (n = 8). The arrows indicate the application of a 10 H2, 5 min stimulation.
Group I mGluR and D2 receptors could converge to activate the eCB signaling pathway to induce I-LTD. N-arachidonylethanolamine (AEA) and 2-arachidonoylglycerol (2-AG) are two major eCBs that activate CB1 receptors in the brain (Di Marzo et al., 1998; Piomelli, 2003). Group 1 mGluR activation preferentially leads to 2-AG production (Jung et al., 2005), whereas dopamine D2 receptor activation leads to the production of AEA, but not 2-AG (Giuffrida et al., 1999; Patel et al., 2003; Centonze et al., 2004). It is therefore of interest to know whether the disruption of AEA and 2-AG synthesis attenuates I-LTD induction. However, selective inhibitors of AEA synthesis have yet to be discovered. We examined the effect of blocking 2-AG synthesis on I-LTD induction.
Group 1 mGluRs are positively coupled to phospholipase C (PLC), which cleaves phosphatidylinositol 1,4,5-bisphosphate into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate, and DAG is subsequently converted into 2-AG by DAG lipase (Di Marzo et al., 1998; Piomelli, 2003). DAG lipase-α, the biosynthetic enzyme for 2-AG, is expressed at moderate to high levels on the plasma membrane of the dendrites of both dopamine and nondopamine neurons in the VTA (Matyas et al., 2008), suggesting that VTA neurons are capable of producing 2-AG. We first examined whether PLC inhibitor 1-[6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,5-dione (U73122) affected I-LTD induction in dopamine neurons. Slices were treated with PLC inhibitor U73122 (5 μm) for ≥1 h and U73122 was also bath applied throughout the experiment (Chevaleyre and Castillo, 2003; Edwards et al., 2006). As shown in Figure 5A, I-LTD was significantly attenuated in the presence of U73122 (Fig. 2B) (93.4 ± 9.1% of baseline, n = 6, p < 0.05 vs I-LTD control). Next, we examined whether DAG lipase inhibitors 1,6-bis(cyclohexyloximino-carbonylamino)hexane (RHC-80267) and THL affected I-LTD. RHC-80267 (30 μm) was bath applied throughout the experiment and THL was included in the internal solution and loaded into the recorded neuron via patch pipette (Safo and Regehr, 2005; Edwards et al., 2006). Figure 5B shows that both RHC-80267 and THL significantly attenuated I-LTD (RHC-80267, 91.9 ± 8.1%, n = 7, p < 0.05; THL, 96.0 ± 8.5%, n = 8, p < 0.05 vs I-LTD control). These results suggest that group I mGluRs activate PLC/DAG lipase pathway to induce I-LTD in dopamine neurons.

Signaling mechanisms for I-LTD induction in dopamine neurons. A, Disruption of 2-AG synthesis with PLC inhibitor U73122 (6 μm) attenuated I-LTD induction (n = 6). B, Disruption of 2-AG synthesis with DAG lipase inhibitor RHC 80267 (n = 7) or THL (n = 8) attenuated I-LTD induction. C, I-LTD could be induced in dopamine neurons loaded with Ca2+ chelator BAPTA (20 mm, n = 9) through patch pipette. D, I-LTD was blocked by intracellular loading of irreversible G-protein inhibitor GDP-β-S (2 mm, n = 6). The arrows indicate the application of a 10 Hz, 5 min stimulation.
Group I mGluR activation increases Ca2+ levels in dopamine neurons (Morikawa et al., 2003). Intracellular Ca2+ rise induces the release of AEA and 2-AG from hippocampal neurons (Di Marzo et al., 1994; Stella et al., 1997; Di Marzo et al., 1998). We therefore examined whether intracellular loading of Ca2+ chelator BAPTA (20 mm) blocked I-LTD induction. We found that I-LTD could be induced in the presence of BAPTA (79.8 ± 7.1% of baseline, n = 9, p < 0.01 vs baseline) (Fig. 5C). Although the magnitude of I-LTD appeared somewhat less than that of I-LTD control in Figure 2B, the difference was not statistically different (p > 0.05). Thus, intracellular Ca2+ rise is not the major factor that triggers 2-AG release during I-LTD induction.
Intracellular loading of dopamine neurons with DAG lipase inhibitor THL blocked I-LTD induction (Fig. 5B), suggesting that postsynaptic mGluRs are activated during I-LTD induction. To further test this idea, we examined the effect of postsynaptic blockade of mGluR signaling with irreversible G-protein inhibitor GDP-β-S on I-LTD induction. GDP-β-S (2 mm) was loaded into the dopamine neurons via the recording pipette, and the same stimulation (10 Hz, 5 min, 3 μm cocaine) was used for I-LTD induction. As shown in Figure 5D, I-LTD was blocked by GDP-β-S (96.9 ± 9.2% of baseline, n = 6, p < 0.05 vs I-LTD control). Thus, the activation of postsynaptic mGluRs is required for I-LTD induction.
The recruitment of eCB signaling in I-LTD induction
To determine whether eCB signaling mediates I-LTD in VTA dopamine neurons, we examined whether pharmacological blockade or genetic knock out of CB1 receptors blocked I-LTD in VTA dopamine neurons. We first examined the effect of the CB1 receptor antagonist AM-251 on I-LTD in dopamine neurons of rat midbrain slices. As shown in Figure 6A, bath application of AM-251 (2 μm) throughout the experiment blocked I-LTD (92.6 ± 5.5%, n = 8, p < 0.001 vs I-LTD control). However, when applied to the bath 10 min after I-LTD induction, AM-251 (4 μm) did not significantly affect the magnitude of I-LTD (72.3 ± 6.7%, n = 7, p > 0.05 vs I-LTD control). The requirement of CB1 receptor activation at similar time windows for I-LTD or LTD induction has also been observed in the hippocampus and dorsal striatum (Chevaleyre and Castillo, 2003; Ronesi and Lovinger, 2005). These results suggest that CB1 receptor activation is required for the induction, but not the maintenance, of I-LTD.

eCB signaling in inhibitory synaptic transmission and plasticity. A, Bath application of CB1 receptor antagonist AM-251 (2 μm) throughout the experiment blocked I-LTD induction in rat dopamine neurons (n = 8), whereas bath application of AM-251 (4 μm) starting 10 min after the 10 Hz stimulation had no significant effect on I-LTD expression (n = 7). The arrows indicate the application of a 10 Hz, 5 min stimulation. B, In the continuous presence of CB1 receptor agonist WIN 55,212-2, a combination of 10 Hz stimulation with cocaine application failed to induce I-LTD (n = 6). C, I-LTD was induced in dopamine neurons in slices from CB1+/+ mice (n = 6) or CB1+/− mice (n = 4), but was absent in slices from CB1−/− mice (n = 8). D, Bath application of CB1 receptor agonist WIN 55,212-2 (2 μm) decreased the amplitude of evoked IPSCs, and this depression was reversed by CB1 receptor antagonist AM-251 (4 μm, n = 7). E, The depression of evoked IPSCs by WIN 55,212-2 was accompanied by an increase in the PPR (*p < 0.05, paired Student's t test). F, DSI is absent in rat VTA dopamine neurons. Depolarization from −70 to 0 mV for 5 s did not induce significant DSI when CB1 receptor antagonist AM-251 (4 μm) was absent (n = 8) or present (n = 6).
If I-LTD is mediated by the activation of CB1 receptors, we expect that such I-LTD is occluded by CB1 receptor agonist R-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo- [1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone (WIN 55,212-2). WIN 55,212-2 (2 μm) was bath applied for at least 20 min and remained in the bath throughout the experiment. In the presence of WIN 55,212-2, the same stimulation (10 Hz, 5 min, 3 μm cocaine) did not induce significant I-LTD (99.5 ± 9.4% of baseline, n = 6, p > 0.05 vs baseline) (Fig. 6B). This result further confirms that CB1 receptor activation is required for I-LTD induction in dopamine neurons.
We next examined I-LTD induction in VTA dopamine neurons in slices prepared from CB1-deficient (CB1−/− and CB1+/−) and wild-type (CB1+/+) mice (Ibrahim et al., 2003). The same stimulation (10 Hz, 5 min, 3 μm cocaine) was used for I-LTD induction. We found that I-LTD was induced in dopamine neurons from CB1+/+ mice (70.1 ± 6.3% of baseline, n = 6 from 4 mice, p < 0.001 vs baseline control, paired Student's t test) and CB1+/− mice (68.3 ± 7.9% of baseline, n = 4 from 4 mice, p < 0.001 vs baseline control), but not from CB1−/− mice (96.2 ± 4.9%, n = 8 from 4 mice, p > 0.05 vs baseline control) (Fig. 6C). Together, these results provide strong evidence that CB1 receptor activation is required for I-LTD induction in VTA dopamine neurons.
We also examined the effect of CB1 receptor agonist WIN 55,212-2 on evoked IPSCs in rat VTA dopamine neurons. A previous study has shown that WIN 55,212-2 depressed evoked IPSCs in the VTA and this effect was attenuated by CB1 receptor antagonist N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methylpyrazole-3-carboxamide (SR141716) (Szabo et al., 2002). However, this study did not identify whether the recorded neurons were dopamine neurons or not. In our experiment, dopamine neurons were identified by their distinct firing characteristics and the presence of large Ih currents (Johnson and North, 1992; Jones and Kauer, 1999), as detailed in our previous study (Liu et al., 2005). We found that bath application of WIN 55,212-2 (2 μm) decreased the amplitude of evoked IPSCs (69.1 ± 4.9% of baseline, n = 7, p < 0.001) (Fig. 6D) and increased the PPR (baseline, 0.81 ± 0.09; WIN 55,212-2, 0.93 ± 0.08, p < 0.05, paired Student's t test) (Fig. 6E) in dopamine neurons, suggesting a presynaptic depression. This CB1 receptor-mediated depression of IPSCs was reversed by CB1 receptor antagonist AM-251 (4 μm) (Fig. 6D). CB1 ligands are highly lipophilic compounds that are not readily washed out from the slice, and AM-251 was therefore applied to block the effect of WIN 55,212-2. This result implies that the CB1 receptor activation is not responsible for the maintenance of I-LTD in dopamine neurons.
CB1 receptor activation often leads to short-term and long-term synaptic plasticity in the same synapses. In the hippocampus and cerebellum, high-frequency stimulation induces I-LTD and LTD, whereas postsynaptic depolarization induces transient suppression of inhibition (DSI) or excitation (DSE) (Chevaleyre and Castillo, 2003; Safo and Regehr, 2005). We examined whether DSI could be induced in VTA dopamine neurons in rat midbrain slices. Depolarization of dopamine neurons from −70 to 0 mV for 5 s induced minimal or no DSI in dopamine neurons (Fig. 6F). In contrast, the same depolarization induced robust DSI in CA1 pyramidal neurons in hippocampal slices, which was blocked by CB1 receptor antagonist AM-251 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results indicate that DSI and eCB-mediated I-LTD are dissociable in VTA dopamine neurons. Similar dissociation of short-term and long-term synaptic plasticity has been observed in the dorsal striatum, where eCB-mediated LTD is readily induced, but DSE is not (Kreitzer and Malenka, 2005).
Both presynaptic and postsynaptic mechanisms contribute to I-LTD expression
Our inability to detect significant change in the PPR and CV during I-LTD suggests that I-LTD is expressed postsynaptically (Fig. 2). However, electron microscopy studies have demonstrated that CB1 receptors are located at GABAergic axon terminals in the VTA (Matyas et al., 2008). In addition, the depression of IPSCs induced by CB1 receptor agonist WIN 55,212-2 is accompanied by a small, but statistically significant increase in the PPR (Fig. 6E). These results suggest that CB1 receptors are located at presynaptic sites in the VTA. These conflicting results prompted us to re-examine whether presynaptic or postsynaptic mechanisms mediate the expression of I-LTD. There is evidence that a presynaptic mechanism is not always accompanied by a change in the PPR and/or CV. For example, factors known to affect presynaptic transmitter release have no significant effect on the PPR of IPSCs in dentate gyrus of hippocampus (Kraushaar and Jonas, 2000). Analysis of quantal synaptic responses provides an alternative way to distinguish presynaptic and postsynaptic mechanisms of I-LTD. mIPSCs recorded in the presence of TTX represent quantal GABA release from presynaptic axonal terminals (Zhu and Lovinger, 2005). A change in quantal frequency indicates a presynaptic mechanism, whereas a change in quantal amplitude signifies a change in postsynaptic responsiveness. We set out to test whether I-LTD is associated with alteration of quantal frequency or amplitude.
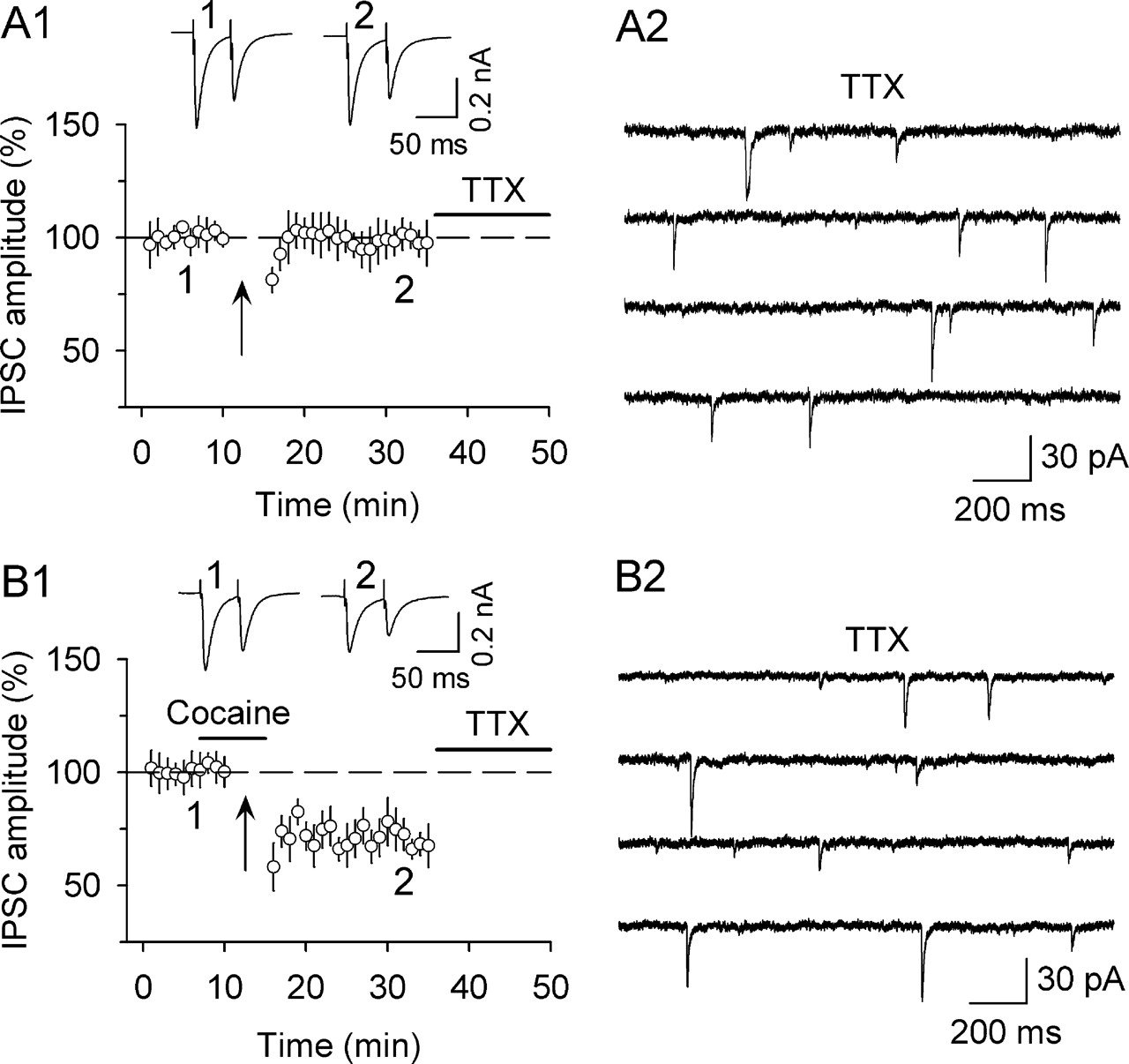
Because it is virtually impossible to compare mIPSCs from the same dopamine neurons before and after I-LTD induction, we compared mIPSCs from dopamine neurons that underwent I-LTD with mIPSCs from dopamine neurons that did not express I-LTD. After obtaining stable baseline evoked IPSCs, the 10 Hz stimulation was applied in the presence or absence of 3 μm cocaine in interleaved slices. As expected, I-LTD was induced when cocaine was present, and was not induced when cocaine was absent (Fig. 7A,B). Consistent with previous observations (Fig. 2), we found that I-LTD was not associated with any significant changes in the PPR (data not shown). Twenty minutes after I-LTD induction, TTX (1 μm) was bath applied to block action potential firing. Five minutes after the application of TTX, mIPSCs were recorded for 5–8 min from dopamine neurons in control (i.e., I-LTD was not induced) and I-LTD groups. We found that I-LTD did not lead to any significant changes in either the mean frequency (contro, 1.4 ± 0.2 Hz, n = 8; I-LTD, 1.3 + 0.3 Hz, n = 8; p > 0.05) or mean amplitude (control, 25.9 ± 4.5 pA; I-LTD, 23.8 ± 3.6 pA; p > 0.05) of mIPSCs

I-LTD did not lead to any significant changes in the amplitude or frequency of mIPSCs recorded in TTX. A1, A 10 Hz, 5 min stimulation (arrow) did not induce significant change in the amplitude of evoked IPSCs (n = 8). Twenty minutes after the 10 Hz stimulation, TTX was bath applied to block action potentials. A2, Sample mIPSCs recorded in A1. B1, The presence of cocaine (3 μm) during the 10 Hz stimulation (arrow) induced I-LTD (n = 8). B2, Sample mIPSCs recorded in B1.
Although mIPSCs originate from all inhibitory synapses contacting the dopamine neuron, eCB-mediated I-LTD is largely limited to the stimulated synapses (Chevaleyre and Castillo, 2004), which are only a small subset of the synapses on the recorded neuron. This may explain why we did not detect any significant changes in the amplitude or frequency of mIPSCs during I-LTD. To record quantal synaptic responses selectively from the stimulated synapses, we induced asynchronous quantal responses by substituting Ca2+ with strontium (Sr2+) in the ACSF (Goda and Stevens, 1996; Oliet et al., 1996; Choi and Lovinger, 1997). When Sr2+ is substituted for Ca2+, the fast synchronous component of evoked IPSCs is markedly reduced, but the slow asynchronous release of quanta is greatly enhanced. Analysis of Sr2+-induced asynchronous quantal events has revealed that LTP/LTD in the hippocampus is expressed postsynaptically (Oliet et al., 1996), whereas LTD in the dorsal striatum is expressed presynaptically (Choi and Lovinger, 1997). A distinct advantage of this approach is that it permits us to analyze quantal events from the stimulated synapses.
We first determined whether this approach can distinguish presynaptic and postsynaptic changes in VTA dopamine neurons. A critical requirement is that Sr2+ -induced asynchronous events are quantal. We found that simple substitution of Ca2+ with Sr2+ was not satisfactory. The fast synchronous component of evoked IPSCs was smaller but still predominant, whereas asynchronous events were few. After testing several Sr2+/Mg2+ ratios, we found that that substituting 2.5 mm Ca2+/1 mm Mg2+ with 2 mm Sr2+/6 mm Mg2+ in the ACSF led to a significant decrease in the amplitude of synchronous IPSCs and to the appearance of many asynchronous synaptic events during the 500 ms period after the synaptic stimulation (Fig. 8A1). In five of six neurons tested, the amplitude distribution of asynchronous events was not significantly different from that of spontaneous mIPSCs recorded from the same dopamine neuron after TTX (1 μm) was added to block action potentials (Fig. 8A2). Consistent with previous studies (Oliet et al., 1996; Choi and Lovinger, 1997), our result suggests that Sr2+- induced asynchronous synaptic events are quantal. Another important consideration is whether Sr2+- induced asynchronous mIPSCs are sensitive to presynaptic and postsynaptic manipulations. Applying paired-pulse stimulation (interpulse interval of 30 ms), which is known to increase the probability of neurotransmitter release, increased the frequency of asynchronous mIPSCs to 214.7 ± 24.8% of that induced by single stimulation (p < 0.001, n = 4) (Fig. 8B1), but had no significant effect on the cumulative amplitude distribution of mIPSCs (Fig. 8B2). However, changing the holding potential from −70 mV to −90 mV led to a significant increase in the amplitude of asynchronous mIPSCs (Fig. 8C2), but had no significant effect on the frequency of the mIPSCs (102.8 ± 6.9% of control at −70 mV, n = 5).
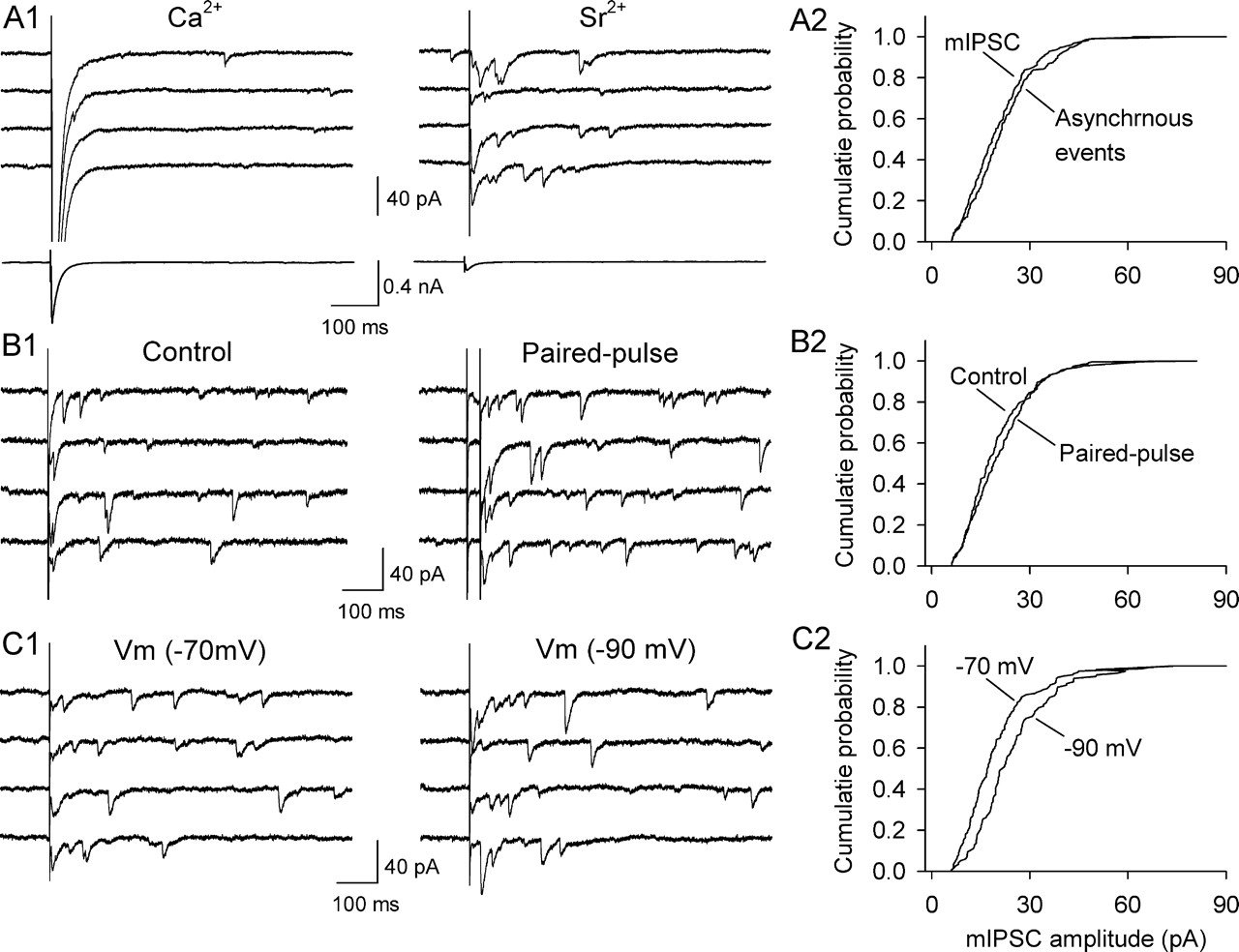
Sr2+-induced asynchronous IPSCs are quantal and are sensitive to presynaptic and postsynaptic manipulations. A1, Top, Sample IPSCs evoked in the presence of Ca2+ and Sr2+. Bottom, Evoked IPSCs averaged from 30 trials are shown at a different scale. A2, Comparison of cumulative amplitude distribution of Sr2+-induced asynchronous events with that of mIPSCs recorded in TTX in the same dopamine neuron (p > 0.05). B1, Paired-pulse stimulation enhanced the frequency of Sr2+-induced asynchronous mIPSCs. B2, Comparison of cumulative amplitude distribution of Sr2+-induced asynchronous mIPSCs evoked by single stimulation with that evoked by paired-pulse stimulation in the same dopamine neuron (p > 0.05). C1, Changing the holding potential from −70 to −90 mV increased the amplitude of Sr2+-induced asynchronous mIPSCs. C2, Comparison of cumulative amplitude distribution of Sr2+-induced asynchronous mIPSCs at −70 mV with that at −90 mV in the same dopamine neuron (p < 0.001, Kolmogorov–Smirnov test).
Having established the validity of this approach, we next examined whether I-LTD is associated with any changes in the frequency or amplitude of Sr2+-induced asynchronous mIPSCs. Three sets of I-LTD experiments were first performed in Ca2+-containing ACSF and switched into Sr2+-containing ACSF later. In the first set of experiments, the 10 Hz stimulation was applied in the absence of 3 μm cocaine. As expected, I-LTD was not induced (Fig. 9A1, control group). In the second set of experiments, the presence of cocaine (3 μm) during the 10 Hz stimulation enabled I-LTD induction (Fig. 9B1, I-LTD group). In the third set of experiments, the presence of CB1 receptor antagonist AM-251 (2 μm) blocked I-LTD induction (Fig. 9C1, AM-251 group). Twenty minutes after the 10 Hz stimulation, Sr2+-containing ACSF was washed in while the synaptic stimulation was continued. Sr2+ has been shown to substitute for Ca2+ in inducing and maintaining LTD in cultured hippocampal neurons and striatal slices (Goda and Stevens, 1996; Choi and Lovinger, 1997). Asynchronous mIPSCs recorded 10 min after Sr2+ wash-in from these three groups were analyzed and compared (Fig. 9A2,B2,C2). We found that I-LTD was accompanied by a significant decrease in both mean frequency (control, 8.6 ± 0.7 Hz, n = 7; I-LTD, 6.3 ± 0.5 Hz, n = 8; p < 0.05) and amplitude (control, 23.8 ± 2.6 pA; I-LTD, 15.7 ± 1.8; p < 0.01) of Sr2+-induced asynchronous mIPSCs. Both effects were blocked by AM-251 (mean mIPSC frequency, 8.3 ± 0.7 Hz; p < 0.05 vs I-LTD; mean mIPSC amplitude, 22.3 ± 2.2 pA; p < 0.05 vs I-LTD; n = 8) (Fig. 9D,E). Together, these results suggest that both presynaptic and postsynaptic mechanisms contribute to I-LTD expression or maintenance in dopamine neurons.

I-LTD is associated with a decrease in the amplitude and frequency of Sr2+-induced asynchronous mIPSCs. A1, The 10 Hz stimulation (arrow) did not induce I-LTD in the absence of cocaine. Twenty minutes after the stimulation, Sr2+ was washed in. (A2) Sample asynchronous mIPSCs recorded in A1. B1, The presence of cocaine during the 10 Hz stimulation (arrow) induced I-LTD. B2, Sample asynchronous mIPSCs recorded in B1. C1, The presence of AM 251 (2 μm) throughout the experiment blocked I-LTD. C2, Sample asynchronous mIPSCs recorded in C1. D, Comparison of mean amplitude of asynchronous mIPSCs among control, I-LTD, and AM-251 groups (*p < 0.05 compared with the other two groups, ANOVA). E, Comparison of mean frequency of asynchronous mIPSCs among control, I-LTD, and AM-251 groups (*p < 0.05 compared with the other two groups, ANOVA; n = 7–8).
eCB-mediated I-LTD has been shown to be specific to the stimulated pathway in the hippocampus (Chevaleyre and Castillo, 2004). To determine whether I-LTD in VTA dopamine neurons is limited to the stimulated synapses, we examined whether I-LTD was associated with any changes in the spontaneous mIPSCs that occurred outside of the 500 ms time window of stimulation-induced asynchronous mIPSCs in Sr2+. These spontaneous mIPSCs likely originate from all synapses contacting the recorded neuron. We compared the spontaneous mIPSCs from control group and I-LTD group and found that there was no significant difference in either mean amplitude (control, 23.7 ± 2.7 pA, n = 7; I-LTD, 21.4 ± 2.2 pA, n = 8; p > 0.05) or mean frequency (control, 2.3 ± 0.5 Hz; I-LTD, 2.1 ± 0.4 Hz; p > 0.05) of the spontaneous mIPSCs. These data are consistent with the finding that I-LTD did not lead to any significant changes in either the frequency or amplitude of mIPSCs recorded in TTX (Fig. 7). Thus, our data suggest that I-LTD in dopamine neurons is specific to the stimulated pathway.
In vivo application of antagonists to the receptors involved in I-LTD prevented cocaine-induced reduction of GABAergic inhibition
Our previous study has shown that daily intraperitoneal injection of cocaine for 5–7 d reduces the mean amplitude of maximal IPSCs and mIPSCs in VTA dopamine neurons of midbrain slices, and the frequency of mIPSCs remains unchanged (Liu et al., 2005). Maximal IPSCs were elicited by gradually increasing the stimulation intensity to recruit saturating IPSCs (Huang et al., 1999; Liu et al., 2005), which indicate a good estimate of total inhibitory inputs to the recorded neuron. mIPSCs recorded in the presence of TTX (0.5 μm) represent quantal GABA release from presynaptic axonal terminals (Zhu and Lovinger, 2005). Together, maximal IPSCs and mIPSCs provide a good estimate of the strength of GABAergic inhibition.
If repeated cocaine exposure in vivo for 5–7 d reduces GABAergic inhibition by inducing I-LTD in vivo, we expect that in vivo application of inhibitors of I-LTD will prevent cocaine-induced reduction of GABAergic inhibition. Indeed, we found that pretreatment (i.p.) of rats with D2 receptor antagonist sulpiride (5 mg/kg), mGluR5 antagonist MPEP (10 mg/kg), or CB1 receptor antagonist AM-251 (2 mg/kg) 20 min before each intraperitoneal cocaine injection (15 mg/kg) blocked cocaine-induced reduction of the mean amplitude of maximal IPSCs and mIPSCs, as detected in midbrain slices prepared 20–24 h after the last cocaine injection. In contrast, pretreatment with D1 receptor antagonist SCH 23390 (2 mg/kg) had no significant effect (Fig. 10A,B). Pretreatment with one of these antagonists before each saline injection had no significant effect on the mean amplitude of maximal IPSCs, compared with that of saline injection alone (Fig. 10A). The doses for these antagonists are based on published studies (Zhou et al., 2001; Rutkowska, 2004; Hodge et al., 2006). Because mGluR1 blockade or knock out causes ataxia by disrupting cerebellar function (Aiba et al., 1994; Coesmans et al., 2003; Bear, 2005), we did not test whether mGluR1 antagonist CPCCOEt affected cocaine-induced reduction of the mean amplitude of maximal IPSCs and mIPSCs. The above results indicate that prevention of in vivo I-LTD induction blocks cocaine-induced reduction of GABAergic inhibition.

Effects of in vivo pretreatment (i.p.) with antagonists to the receptors involved in I-LTD on cocaine-induced decrease in the amplitude of maximal IPSCs and mIPSCs in dopamine neurons in midbrain slices, which were prepared 20–24 h after the last saline or cocaine injection. A, Top, Sample IPSCs in response to stimuli of incremental intensities (20–140 μA). Calibration: 0.2 nA, 50 ms. The neurons were voltage-clamped at −20 mV. Bottom, Intraperitoneal injection of cocaine for 5–7 d reduces the amplitude of maximal IPSCs compared with saline injection (no pretreatment). Pretreatment (i.p.) with CB1 antagonist AM-251, D2 antagonist sulpiride (Sulp), or mGluR5 antagonist (MPEP) 20 min before each intraperitoneal cocaine injection blocked cocaine-induced reduction of the mean amplitude of maximal IPSCs (*p < 0.05 compared with cocaine alone, ANOVA followed by Tukey test), whereas pretreatment with D1 antagonist SCH 23390 (SCH) had no significant effect (n = 9–12 for each black bar). Pretreatment with one of these antagonists before saline injection had no significant effect on maximal IPSC amplitude, compared with that of saline injection alone (n = 8–12 for each clear bar). B, Same as in A, except that mIPSCs were recorded at a holding potential of −70 mV (n = 10–12 for each bar). Calibration: 40 pA, 0.4 s. In this figure, saline-treated groups are represented by clear bars, and cocaine-treated groups by black bars.
To ensure an accurate measurement of maximal IPSCs, we placed the stimulation electrode at the fixed distance (∼150 μm) to the soma of the recorded dopamine neuron. Furthermore, electrophysiological recordings were performed blind to the treatment history of the rat. Pilot experiments indicated that the positioning of the stimulation electrode in a certain range (150–300 μm to the soma of the recorded neuron) had little impact on the amplitude of maximal IPSCs, as increasing the stimulation intensity can overcome the distance between the recording and stimulation electrodes. As a result, the amplitude of maximal IPSCs remains unchanged. Nevertheless, the two precautions were followed throughout this study.
Repeated cocaine exposure in vivo occludes the subsequent induction of I-LTD ex vivo
As mentioned above, our previous study has shown that intraperitoneal cocaine injection for 5–7 d reduced GABAergic inhibition in dopamine neurons, whereas a single cocaine injection or saline injection had no significant effect (Liu et al., 2005). If I-LTD underlies cocaine-induced reduction of GABAergic inhibition, the magnitude of I-LTD would be reduced in dopamine neurons in midbrain slices prepared from 5 to 7 d cocaine-treated rats because such I-LTD had occurred in vivo. We therefore compared I-LTD in dopamine neurons in slices prepared from 1 d and 5–7 d saline- and cocaine-treated rats. The same conditioning stimulation (10 Hz, 5 min, 3 μm cocaine) was used for the induction of I-LTD. As shown in Figure 11, I-LTD with comparable magnitude was induced in dopamine neurons in slices from 1 d saline and cocaine-injected rats (1 d saline, 67.1 ± 5.2%, n = 6; 1 d cocaine, 68.9 ± 5.5%, n = 8; p > 0.05 vs I-LTD control). In contrast, I-LTD was diminished in slices from 5 to 7 d cocaine-treated rats (92.9 ± 7.3%, n = 6; p < 0.01 vs I-LTD control), but not in slices from 5 to 7 d saline-treated rats (63.5 ± 7.2%, n = 7; p < 0.01 vs 5–7 d cocaine-treated group; p > 0.05 vs I-LTD control). These data suggest that repeated cocaine exposure in vivo occludes the subsequent induction of I-LTD ex vivo.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Repeated, but not single, cocaine exposure occludes the subsequent induction of I-LTD induction ex vivo. A, Summary of the effects of single intraperitoneal saline (n = 6) or cocaine (n = 8) injection on subsequent I-LTD induction in midbrain slices. B, Summary of the effects of repeated daily intraperitoneal saline (n = 7) or cocaine (n = 6) injection for 5–7 d on I-LTD induction in midbrain slices. In all experiments, slices were prepared 20–24 h after the last saline or cocaine injection. The arrows indicate the application of a 10 H2, 5 min stimulation.
Discussion
In this study we investigated the mechanism by which repeated cocaine exposure in vivo reduces the strength of GABAergic inhibition in VTA dopamine neurons. We first characterized in detail the properties of eCB-mediated I-LTD in dopamine neurons and then tested whether eCB-mediated I-LTD is responsible for cocaine-induced reduction of GABAergic inhibition.
eCB-mediated I-LTD in dopamine neurons
We find that the activation of group I mGluRs and D2 receptors leads to eCB-mediated I-LTD in dopamine neurons, and the presence of cocaine during the 10 Hz stimulation is required for I-LTD induction. These features are in contrast to I-LTD or LTD induction in other brain areas. For example, the activation of group I mGluRs alone triggers eCB-mediated I-LTD in the hippocampus (Chevaleyre and Castillo, 2003; Edwards et al., 2006) and LTD in the cerebellum (Safo and Regehr, 2005). High-frequency stimulation used likely causes strong activation of group I mGluRs, which leads to substantial eCB release. On the other hand, moderate-frequency stimulation (10–25 Hz) activates group I mGluRs and/or D2 receptors to induce eCB-mediated LTD in the striatum (Robbe et al., 2002; Kreitzer and Malenka, 2005; Ronesi and Lovinger, 2005). The striatum is richly innervated by dopamine terminals and the heightened D2 receptor activity may preclude the requirement of cocaine in LTD induction. These studies suggest that a threshold level of CB1 receptor activity controls I-LTD or LTD induction.
Different eCB release pathways can act synergistically to enhance the presynaptic depression (Alger, 2005). For example, muscarinic receptor and group I mGluR agonists enhance DSI in the hippocampus (Hashimotodani et al., 2005; Edwards et al., 2006) and a D2 receptor agonist enhances DSE in the VTA (Melis et al., 2004). Furthermore, D2 receptors and group I mGluRs act in concert to induce eCB-mediated frequency-dependent depression and LTD in the striatum (Kreitzer and Malenka, 2005; Yin and Lovinger, 2006). Similar synergistic action may also occur in dopamine neurons during I-LTD induction. It would be easier to reach the threshold for I-LTD by activating both receptors than by activating either receptor alone. As a result, I-LTD could be readily induced after in vivo cocaine exposure (see below).
Intracellular loading of DAG lipase inhibitor THL or G-protein inhibitor GDP-β-S into dopamine neurons blocked I-LTD (Fig. 5), suggesting that I-LTD is induced postsynaptically. However, investigation of the mechanisms for I-LTD expression yields mixed results. Analysis of the PPR and CV suggests that I-LTD is expressed postsynaptically, whereas analysis of Sr2+-induced asynchronous mIPSCs indicates that both presynaptic and postsynaptic mechanisms contribute to the expression of I-LTD. Because a presynaptic mechanism is not always accompanied by a change in the PPR (Kraushaar and Jonas, 2000), analysis of the PPR may fail to detect a presynaptic component of I-LTD expression. Thus, it is most likely that both presynaptic and postsynaptic mechanisms contribute to the expression of I-LTD in the VTA. We speculate that the D2 receptor activation contributes to both presynaptic and postsynaptic expression of I-LTD. D2 receptors are known to exist on somatodendritic sites of midbrain dopamine neurons (Beckstead et al., 2004). The D2 receptor activation could lead to a retrograde depression of GABA release via AEA production (Giuffrida et al., 1999; Centonze et al., 2004), as well as a decrease in postsynaptic GABA responsiveness (Trantham-Davidson et al., 2004). Alternatively, after presynaptic CB1 receptor activation, both presynaptic and postsynaptic mechanisms are recruited for I-LTD expression and maintenance. We find that CB1 receptor antagonist AM-251 did not affect I-LTD when applied 10 min after the 10 Hz stimulation, consistent with previous findings that CB1 receptor activation is not responsible for the maintenance of I-LTD or LTD (Chevaleyre and Castillo, 2003; Ronesi and Lovinger, 2005; Edwards et al., 2006). These two possibilities are not mutually exclusive and could happen in sequence. Future studies are needed to clarify the signaling mechanisms for I-LTD expression and maintenance.
We could induce eCB-mediated I-LTD, but not DSI, in VTA dopamine neurons. In the hippocampus, I-LTD depends on the activation of PLC/DAG lipase and cAMP/protein kinase A pathways. However, DSI, which although thought to be mediated by 2-AG (Kim and Alger, 2004), was unaffected by inhibitors to either pathway (Chevaleyre and Castillo, 2003; Edwards et al., 2006; Chevaleyre et al., 2007). Furthermore, repetitive DSI failed to induce I-LTD (Edwards et al., 2006). Thus, DSI and I-LTD are mechanistically distinct and could be independently induced. In the hippocampus, DSI requires the presence of cholecystokinin-expressing interneurons (Wilson et al., 2001; Foldy et al., 2006; Glickfeld and Scanziani, 2006), which express high levels of CB1 receptors on their axonal terminals (Katona et al., 1999; Tsou et al., 1999). The lack of DSI in the VTA could be attributable to the absence or rarity of such interneurons.
eCB-mediated I-LTD as an underlying mechanism for cocaine-induced reduction of GABAergic inhibition
eCB-mediated LTD and I-LTD have been induced in slices from many brain areas. However, whether they represent a mechanism that underlies synaptic plasticity in vivo remains poorly understood. Our results provide evidence for eCB-mediated I-LTD as a mechanism for the reduction of GABAergic inhibition in VTA dopamine neurons induced by repeated cocaine exposure in vivo. First, I-LTD can be induced under conditions that mimic in vivo cocaine exposure. We find that pairing cocaine application (3 μm) with repetitive afferent stimulation (10 Hz, 5 min) induces I-LTD in VTA dopamine neurons in midbrain slices, whereas either cocaine or the 10 Hz stimulation alone is ineffective. This concentration of cocaine approximates the level in the brain after in vivo cocaine exposure (Maisonneuve and Kreek, 1994), whereas the 10 Hz stimulation mimics the natural frequency of synaptic activity in vivo (Carelli and Ijames, 2000). These results strongly suggest that I-LTD is induced in vivo after cocaine exposure. Second, pretreatment with antagonists to the receptors involved in I-LTD induction prevented cocaine-induced reduction of the amplitude of maximal IPSCs and mIPSCs, indicating that I-LTD and cocaine-induced reduction of GABAergic inhibition share common mechanisms. Finally, repeated cocaine exposure in vivo for 5–7 d occludes the subsequent induction of I-LTD ex vivo, suggesting that such I-LTD had occurred in vivo. Together, these data strongly support the hypothesis that I-LTD underlies cocaine-induced reduction of reduction of GABAergic inhibition.
We hasten to point out that I-LTD is only a beginning step that initiates cocaine-induced reduction of GABAergic inhibition. Although both I-LTD and the reduced inhibition are expressed as long-lasting depression of IPSCs, the duration for the depression is different: I-LTD presumably lasts a few hours, whereas cocaine-induced reduction of GABAergic inhibition lasts at least 2 weeks after withdrawal from cocaine treatment (Liu et al., 2005). Newly established LTP in vivo can be depotentiated by spontaneous neuronal activity (Zhou et al., 2003). Analogous to this depotentiation, in vivo I-LTD induced by a single cocaine exposure is likely vulnerable to “de-depression”. However, repeated cocaine exposure for 5–7 d may consolidate and reconsolidate I-LTD, leading to sustained reduction of GABAergic inhibition. This may explain why repeated, but not single, cocaine exposure occludes the subsequent induction of I-LTD ex vivo. The mechanism underlying the transition from the early I-LTD to the sustained reduction of GABAergic inhibition is not yet known. Synaptic glutamate receptors are removed via endocytosis after LTD induction (Luscher et al., 1999; Luthi et al., 1999; Beattie et al., 2000), and similar mechanisms may operate at inhibitory synapses on dopamine neurons after I-LTD induction.
The implication of I-LTD in cocaine-seeking behavior
An important consequence of eCB-mediated DSI and I-LTD is to prime excitatory synapses for LTP induction and to increase excitability (Carlson et al., 2002; Chevaleyre and Castillo, 2003, 2004; Zhu and Lovinger, 2007). I-LTD shown here may underlie cocaine-induced facilitation of LTP induction and increase in excitability in VTA dopamine neurons (Liu et al., 2005). Thus, blocking I-LTD induction provides a means to block cocaine-induced aberrant synaptic plasticity and addictive behavior. Consistent with this idea, growing evidence suggests that CB1 receptor and mGluR5 antagonists have considerable therapeutic potential in drug addiction (Lea and Faden, 2006; Maldonado et al., 2006). CB1 receptor antagonist SR141716 attenuates relapse induced by environmental cues or cocaine re-exposure (De Vries et al., 2001; Xi et al., 2006), whereas mGluR5−/− mice do not self-administer cocaine (Chiamulera et al., 2001) and mGluR5 antagonist MPEP decreases cocaine self-administration in rats and monkeys (Lee et al., 2005; Paterson and Markou, 2005). These remarkable results suggest that CB1 receptors and mGluR5 play an important role in cocaine addiction. However, mGluR5 and CB1 receptors are not a direct target of cocaine and the mechanisms by which these antagonists impair cocaine-seeking behavior are virtually unknown. We have shown that mGluR5 and CB1 receptor activation is required for I-LTD and cocaine-induced reduction of GABAergic inhibition in dopamine neurons. Given the important role of GABAergic inhibition in controlling cocaine-seeking behavior (Brodie et al., 2003; Kampman et al., 2004), we believe that blocking cocaine-induced reduction of GABAergic inhibition may contribute to the effectiveness of these antagonists in diminishing cocaine seeking. As such, drugs that block cocaine-induced reduction of GABAergic inhibition in dopamine neurons could be promising candidates for treating cocaine addiction.
Footnotes
- Received September 4, 2007.
- Revision received December 15, 2007.
- Accepted December 20, 2007.
-
This work was supported by the National Alliance for Research on Schizophrenia and Depression (Q.-s.L.) and National Institutes of Health Grant R01 DA09155 (C.J.H.). We thank Debra Cockayne (Roche, Palo Alto, CA) for providing CB1-deficient mice and Stephanie Balthazor for genotyping of the CB1-deficient mice. We also thank Qiang Zhou (Mount Sinai School of Medicine, New York, NY) for helpful comments on this manuscript.
- Correspondence should be addressed to Qing-song Liu, Department of Pharmacology and Toxicology, Medical College of Wisconsin, 8701 Watertown Plank Road, Milwaukee, WI 53226. qsliu{at}mcw.edu
- Copyright © 2008 Society for Neuroscience 0270-6474/08/281385-14$15.00/0