
Abstract
Increased brain expression of vascular endothelial growth factor (VEGF) is associated with neurological disease, brain injury, and blood–brain barrier (BBB) dysfunction. However, the specific effect of VEGF on the efflux transporter P-glycoprotein, a critical component of the BBB, is not known. Using isolated rat brain capillaries and in situ rat brain perfusion, we determined the effect of VEGF exposure on P-glycoprotein activity in vitro and in vivo. In isolated capillaries, VEGF acutely and reversibly decreased P-glycoprotein transport activity without decreasing transporter protein expression or opening tight junctions. This effect was blocked by inhibitors of the VEGF receptor flk-1 and Src kinase, but not by inhibitors of phosphatidylinositol-3-kinase or protein kinase C. VEGF also increased Tyr-14 phosphorylation of caveolin-1, and this was blocked by the Src inhibitor PP2. Pharmacological activation of Src kinase activity mimicked the effects of VEGF on P-glycoprotein activity and Tyr-14 phosphorylation of caveolin-1. In vivo, intracerebroventricular injection of VEGF increased brain distribution of P-glycoprotein substrates morphine and verapamil, but not the tight junction marker, sucrose; this effect was blocked by PP2. These findings indicate that VEGF decreases P-glycoprotein activity via activation of flk-1 and Src, and suggest Src-mediated phosphorylation of caveolin-1 may play a role in downregulation of P-glycoprotein activity. These findings also imply that P-glycoprotein activity is acutely diminished in pathological conditions associated with increased brain VEGF expression and that BBB VEGF/Src signaling could be targeted to acutely modulate P-glycoprotein activity and thus improve brain drug delivery.
Introduction
Successful treatment of neurological disorders and brain injury requires delivery of therapeutics to the brain parenchyma, which is limited by the blood–brain barrier (BBB). The BBB restricts brain entry of neurotoxicants and of many therapeutic drugs via expression of high-resistance tight junctions that limit paracellular diffusion and multispecific, ATP-dependent xenobiotic efflux transporters that limit cellular accumulation (Hawkins and Davis, 2005; Miller et al., 2008). The most prominent of these BBB transporters is P-glycoprotein, which handles a large number of therapeutic drugs and is recognized as a major impediment to CNS pharmacotherapy (Miller et al., 2008). Modification of drugs to target them across the BBB has yielded little success in the clinic. Opening the paracellular pathway by osmotic disruption or the bradykinin agonist labradimil is limited in that such openings are nonselective, global, and transient. Moreover, there is evidence that P-glycoprotein can limit brain distribution of its substrates even under conditions in which the tight junctions of the BBB are disrupted (Spudich et al., 2006; Seelbach et al., 2007). Coadministration of a P-glycoprotein inhibitor with paclitaxel dramatically reduces tumor growth in brain, demonstrating that targeting P-glycoprotein can improve clinical outcome (Fellner et al., 2002). However, this approach increases distribution of paclitaxel throughout the brain, leaving healthy tissue vulnerable to neurotoxic side effects of the drug (Argyriou et al., 2008) as well as to environmental neurotoxicants and potentially neurotoxic therapeutics normally excluded from the brain by P-glycoprotein.
VEGF is a potent endothelial mitogen that promotes cell growth, survival, and angiogenesis (Ferrara et al., 2003). In brain, VEGF regulates neurogenesis as well as angiogenesis (Greenberg and Jin, 2005), and enhances learning and memory associated with hippocampal neurogenesis (During and Cao, 2006). Overexpression of VEGF in brain and/or brain microvasculature is associated with CNS pathologies including Alzheimer's disease, brain tumors, ischemia, and brain injury (Merrill and Oldfield, 2005; Thirumangalakudi et al., 2006). VEGF acts via receptor tyrosine kinases, which in turn initiate multiple intracellular signaling cascades regulating protein expression, cell growth, differentiation, migration, and vascular permeability (Cross et al., 2003). Signal transduction pathways downstream of the VEGF receptor flk-1 include known negative regulators of P-glycoprotein activity: protein kinase C (PKC) (Hartz et al., 2004, 2006) and Src kinase (Barakat et al., 2007). However, to date, the specific effect of VEGF on P-glycoprotein has not been demonstrated in any tissue.
Here we show that VEGF exposure acutely downregulates P-glycoprotein transport activity at the BBB in vitro and in vivo. Transporter downregulation was rapid and reversible and did not involve decreased P-glycoprotein protein expression. VEGF signaled to P-glycoprotein through flk-1 and Src kinase and is accompanied by Src-mediated phosphorylation of caveolin-1. Our experiments disclose a novel role for VEGF in the brain, and constitute the first known demonstration of VEGF and Src-dependent modulation of P-glycoprotein activity in vivo.
Materials and Methods
Reagents.
[N-ε(4-nitrobenzofurazan-7-yl)-d-Lys(8)]-cyclosporin A (NBD-CSA) was custom synthesized by R. Wenger (Basel, Switzerland). PSC833 was a gift from Novartis. Human recombinant VEGF165 was purchased from Sigma. 4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d] pyramidine (PP2), 4-amino-7-phenylpyrazol[3,4-d] pyrimidine (PP3), SU5416, 3-(3-thienyl)-6-(4-methoxyphenyl)pyrazolo[1,5-a] pyrimidine (TMPP), nocodazole, wortmannin, and lactacystin were purchased from Calbiochem. Staurosporine was purchased from Alomone Labs. The Src-activating phosphopeptide [YEEIP, Glu-Pro-Gln-Tyr(PO3H2)-Glu-Glu-Ile-Pro-Ile-Tyr-Leu] was purchased from Biomol. All isotopes were purchased from American Radiolabeled Chemicals. Mouse anti-P-glycoprotein (C219) was purchased from Abcam. Mouse anti-β-actin was purchased from Sigma. Mouse anti-caveolin-1 was purchased from BD Signal Transduction Laboratories. Rabbit anti-p14Y-caveolin-1 was purchased from Santa Cruz Biotechnology. All other reagents and chemicals were purchased primarily from Sigma.
Brain capillary isolation.
All animal procedures were approved by the Animal Care and Use Committee of the National Institute of Environmental Health Sciences and conform to the guidelines and standards of the National Institutes of Health. Male Sprague Dawley retired breeder rats (Taconic) were killed by CO2 and decapitated, and their brains were removed for isolation of capillaries as previously described (Miller et al., 2000; Hartz et al., 2004). Briefly, brains from 5–10 rats were placed in ice-cold modified PBS (supplemented with glucose and pyruvate). Cerebral hemispheres were removed from the brainstem and cerebellum, and choroid plexuses, meninges, and white matter were dissected away from the gray matter. The gray matter was homogenized in a threefold volume of modified PBS and mixed with Ficoll solution to a final concentration of 20% Ficoll and spun for 20 min at 5800 × g. Pellets were resuspended in 1% BSA/modified PBS and washed on a column containing 4 ml/brain glass beads. Capillaries were collected from the beads by gentle agitation, washed three times with BSA-free modified PBS, and maintained at room temperature in modified PBS for all subsequent treatments. Enrichment of capillaries was confirmed by light microscopy; preparations were free of cellular contamination (neurons, astrocytes) and typically contained a minimum of cellular debris. Following isolation, capillaries were used for transport assays or protein extraction and Western blots.
P-glycoprotein transport assay.
In all transport experiments, capillaries were incubated at room temperature in modified PBS containing 2 μm NBD-CSA for 1 h. We previously demonstrated that 1 h incubation is sufficient to attain steady state with regard to NBD-CSA accumulation in the capillaries (Hartz et al., 2004). For treatments, two protocols were used. In one, capillaries were preincubated with transport or signaling inhibitors for 30 min before addition of NBD-CSA. In the other, VEGF or YEEIP was added after the 1 h incubation with NBD-CSA. Time-matched control groups from the same preparation were exposed to PBS alone during inhibitor and/or VEGF/YEEIP treatments. Except for time course studies, capillaries were imaged 1 h after addition of VEGF or YEEIP. In some experiments, 2 μm Texas Red (TR) replaced NBD-CSA during the incubation period; these were used to assess the effects of VEGF/YEEIP on paracellular (tight junction-restricted) permeability. For measurements, capillaries were visualized on a Zeiss 510 NLO confocal scanning microscope using an Ar (for NBD-CSA) or HeNe (for TR) laser, appropriate filter combinations, and a 40× 1.2 NA water-immersion objective. For each experiment, pinhole diameter, photomultiplier gain, and laser power were the same for all treatments. Each experiment was performed 2–4 times. A minimum of 10 capillaries per treatment were imaged in each experiment. Fluorescence intensity in the capillary lumen was quantified from stored images using Scion Image.
Protein extraction and Western blot.
To measure protein expression, capillaries were incubated at room temperature with VEGF or YEEIP in PBS. Following treatment, capillaries were pelleted at 5000 × g for 1 min, and the supernatant was aspirated and discarded. The tubes containing capillary pellets were frozen rapidly in liquid nitrogen and stored at −80°C for subsequent use. Pellets were resuspended in PBS containing protease and phosphatase inhibitors on ice and spun at 15,000 × g for 1 min. The supernatant (cytosolic fraction) was collected, and the pellet was washed with PBS and spun two more times. The washed final pellet was triturated in CelLytic MT mammalian tissue lysis buffer (Sigma) containing 10% PBS with protease and phosphatase inhibitors and sonicated for 20 s, then spun 10 min at 15,000 × g. The supernatant (membrane fraction) was collected and analyzed for protein content (Bradford method) and either stored at −80°C or used immediately for Western blot.
Western blots were performed on the NuPage electrophoresis system (Invitrogen). Proteins were electrophoresed on 4–12% Bis-Tris gels and blotted on Immobilon-FL polyvinylidene difluoride membranes (Millipore, Billerica, MA) per manufacturer's protocols. After blocking, membranes were incubated overnight with primary antibody. Blocking was done in LI-COR Odyssey Blocking Buffer (LI-COR Biosciences), and primary antibodies were diluted in the same buffer with 0.1% Tween 20. Membranes were then washed with PBS/Tween-20 and incubated with LI-COR IRDye-conjugated goat anti-mouse or anti-rabbit secondary antibodies for 1 h. After washing, bands were visualized on a LI-COR Odyssey Infrared Imager.
Immunoprecipitation.
Immunoprecipitation was performed based on previously described methods (Barakat et al., 2007). Briefly, capillaries were isolated and treated, pelleted, and stored at −80°C as described above. Pellets were resuspended in ice-cold immunoprecipitation buffer (50 mm Tris HCl, 150 mm NaCl, 0.1% SDS, 0.5% deoxycholate, 1% Tergitol-type NP-40, pH 7.5, protease and phosphatase inhibitors added), sonicated, and centrifuged for 10 min at 15,000 × g. Supernatants were analyzed for protein content by the bicinchoninic acid method. For each experiment, equal amounts of protein from each sample (50–100 μg) were precleared with 20 μl of protein G-Sepharose beads (50% in PBS, GE Healthcare) for 1 h on ice followed by centrifugation for 3 min at 1000 × g. Supernatants were incubated overnight with 1 μg of mouse anti-caveolin-1 antibody at 4°C with agitation. Protein G-Sepharose beads were added and incubated for 2 h at 4°C with agitation, followed centrifugation for 1 min at 3000 × g. After washing beads with immunoprecipitation buffer, immunoprecipitated proteins were solubilized in 30 μl of Laemmli buffer and heated 4 min at 95°C before gel loading, electrophoresis, and immunodetection of P-glycoprotein and caveolin-1 as described above.
In situ brain perfusion.
Male Sprague Dawley rats (250–300 g, Charles River) were anesthetized by intraperitoneal injection (1 ml/kg) of ketamine cocktail (79 mg/ml ketamine, 3 mg/ml xylazine, 0.6 mg/ml acepromazine) and given heparin (10,000 U/kg). The common carotid arteries were exposed via ventral midline incision in the neck and cannulated with PE10 tubing connected to a perfusion circuit. Oxygenated Ringer solution (117 mm NaCl, 4.7 mm KCl, 0.8 mm MgSO4, 24.8 mm NaHCO3, 1.2 mm KH2PO4, 2.5 mm CaCl2, 10 mm d-glucose, 39 g/L 70 kDa dextran, 1 g/L bovine serum albumin, and 0.055 g/L Evans Blue, heated to 37°C) was delivered via the carotid cannulae at a rate of 3 ml/min with a peristaltic pump. In some experiments, [14C]-sucrose and [3H]-morphine (0.5 μCi each per ml of Ringer) were infused into the circuits with a syringe pump at 0.5 ml/min for 2.5, 5, 10, 15, or 20 min. In other experiments, [3H]-verapamil (0.1 μCi per ml of Ringer) was infused for 20 min. At the end of the perfusion, samples of perfusate were collected and the brain was removed. Cerebral hemispheres were stripped of meninges and choroid plexuses and minced by hand. Tissue and 100 μl samples of perfusate were thoroughly mixed with tissue solubilizer (Hyamine Hydroxide, MP Biomedicals) and digested for 2 d. Samples were prepared for scintillation counting by the addition of 100 μl of 30% acetic acid and 4 ml of liquid scintillation cocktail (CytoScint ES, MP Biomedicals), incubated in the dark overnight, and counted on a liquid scintillation counter. For experiments with [14C]-sucrose and [3H]-morphine, disintegrations per minute (dpm) were calculated from counts per minute (cpm) using standard quench correction methods. In pilot studies, samples from animals perfused simultaneously with [14C]-sucrose and [3H]-morphine yielded calculated brain distribution values for [14C]-sucrose and [3H]-morphine that were not significantly different from those determined following perfusion with each tracer alone using direct dpm determination with stored quench curves. Results are reported as the ratio of radioactivity in the brain to that in the perfusate (Rbr, μl/g):
 where C brain is the radioactivity measured in brain (dpm/g) and C perfusate that in the perfusate (dpm/μl). In all experiments involving intracerebroventricular injection (see Figs. 7B and 8), the reported Rbr was determined from the cerebral hemisphere ipsilateral to the injection site only. Unidirectional transfer coefficients (Kin) and initial volumes of distribution (V0) for [14C]-sucrose were estimated from least-squares linear regression of Rbr versus perfusion time (T) (Hawkins et al., 2007):
where C brain is the radioactivity measured in brain (dpm/g) and C perfusate that in the perfusate (dpm/μl). In all experiments involving intracerebroventricular injection (see Figs. 7B and 8), the reported Rbr was determined from the cerebral hemisphere ipsilateral to the injection site only. Unidirectional transfer coefficients (Kin) and initial volumes of distribution (V0) for [14C]-sucrose were estimated from least-squares linear regression of Rbr versus perfusion time (T) (Hawkins et al., 2007):
 [3H]-morphine uptake was best fit to a nonlinear model that accounts for efflux transport:
[3H]-morphine uptake was best fit to a nonlinear model that accounts for efflux transport:
 where kout is an efflux constant and Vbr is the effective distribution volume of [3H]-morphine at steady state (Seelbach et al., 2007). Curve fits and comparisons (F test) were performed with GraphPad Prism version 4.02. For perfusions with [3H]-verapamil, a single perfusion time (20 min) was used, dpm were determined directly using stored quench curves, and Rbr values were determined by Equation 1 above.
where kout is an efflux constant and Vbr is the effective distribution volume of [3H]-morphine at steady state (Seelbach et al., 2007). Curve fits and comparisons (F test) were performed with GraphPad Prism version 4.02. For perfusions with [3H]-verapamil, a single perfusion time (20 min) was used, dpm were determined directly using stored quench curves, and Rbr values were determined by Equation 1 above.
Intracerebroventricular injection.
Animals were anesthetized as described above and placed in a stereotactic frame. A 2 mm burr hole was drilled in the skull over the lateral ventricle (from bregma: 1.4 mm lateral, 0.8 mm caudal) and finished by hand with a 25 gauge needle. Injections were performed with a 26 gauge Hamilton syringe advanced 3.8 mm ventral from the surface of the dura. VEGF dissolved in sterile artificial CSF (aCSF) or aCSF alone was delivered slowly over the course of 30 s in a 2 μl volume. Injections were randomized between right and left hemispheres. The syringe was kept in place for 1 min following injection to prevent backflow. Following syringe removal, the injection site was covered with bone wax and the scalp incision closed with sutures. Animals were maintained under deep anesthesia for the duration of the experiments, with booster injections of anesthesia (typically half the initial dose) administered as needed. The in situ brain perfusion procedure was begun 30 min after the injection.
Results
VEGF reduces P-glycoprotein-mediated transport activity in isolated brain capillaries
We previously showed that isolated rat brain capillaries can be maintained in PBS at room temperature for up to 8 h without loss of viability or transport activity (Miller et al., 2000; Bauer et al., 2007). Isolated brain capillaries incubated in PBS containing NBD-CSA concentrate the fluorescent drug in the luminal space (Fig. 1A). In agreement with previous reports from this laboratory (Hartz et al., 2004), steady-state NBD-CSA fluorescence in capillary lumens was reduced 50–60% by the specific P-glycoprotein inhibitor PSC833 and by osmotic disruption of the tight junctions with 100 mm mannitol (see representative images in Fig. 1A). We previously showed that inhibitors of multidrug resistance-related proteins (MRPs) or breast cancer resistance-related protein (BCRP) did not affect luminal accumulation of NBD-CSA, and that the noninhibitable component of luminal NBD-CSA accumulation represents nonspecific binding and/or diffusion of the drug (Hartz et al., 2006). Thus, as before, here we have used PSC-sensitive luminal accumulation of NBD-CSA in isolated brain capillaries as a robust, quantitative indicator of P-glycoprotein-mediated transport activity in the capillaries.

VEGF reduces P-glycoprotein-mediated transport in isolated brain capillaries. A, Representative confocal micrographs showing steady-state (60 min) luminal fluorescence of the P-glycoprotein substrate NBD-CSA and the MRP-2 substrate Texas Red (TR). CON, Control; MAN, incubated with 100 mm mannitol to induce osmotic disruption of the tight junctions; PSC, incubated with P-glycoprotein inhibitor PSC833 (5 μm); LTC4, incubated with MRP2 inhibitor leukotriene C4 (0.3 μm); VEGF, incubated with 200 ng/ml VEGF. Scale bar, 5 μm. B, Dose–response curve for VEGF effect on P-glycoprotein-mediated transport. Data are mean luminal fluorescence of NBD-CSA ± SEM (PSC-insensitive fluorescence subtracted). VEGF significantly reduced luminal accumulation of NBD-CSA. Inset, Dose–response curve for VEGF effect on MRP2-mediated transport. Data are mean luminal fluorescence of TR ± SEM with LTC4-insensitive fluorescence subtracted. VEGF did not affect luminal accumulation of TR. Statistical comparison, **p < 0.01, ***p < 0.001 (vs NBD-CSA without VEGF), significance determined by one-way ANOVA with a Newman–Keuls multiple-comparison test. Representative experiments shown; data were collected from at least 10 capillaries per data point.
Human recombinant VEGF (50–500 ng/ml) reduced luminal fluorescence of NBD-CSA in brain capillaries in a concentration-dependent manner (Fig. 1A,B). In contrast, exposing capillaries to the same range of VEGF concentrations had no effect on luminal accumulation of TR (Fig. 1B, inset), a fluorescent organic anion that is a substrate for MRP2; as shown previously, luminal accumulation of TR was substantially reduced by the MRP2 inhibitor LTC4 (Fig. 1A) (Bauer et al., 2008). These observations indicate that the effect of VEGF was specific for P-glycoprotein. Moreover, they show that VEGF did not reduce luminal NBD-CSA accumulation through increased tight junction leakage, since such an effect would have reduced accumulation of both substrates (see, e.g., the effects of hypertonic mannitol in Fig. 1A).
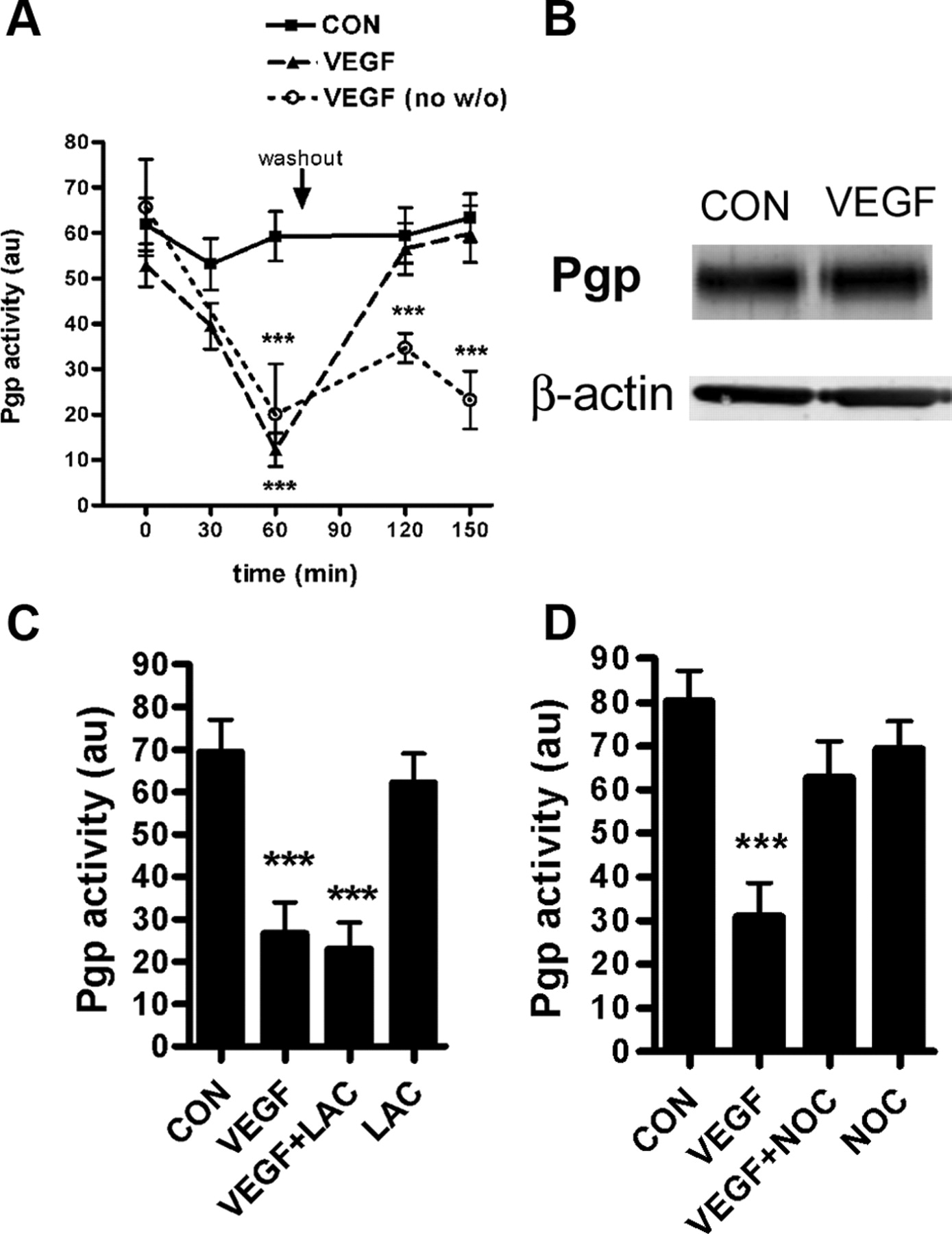
Figure 1 shows that a 1 h exposure to 200 ng/ml VEGF maximally reduced P-glycoprotein transport activity. We used this concentration for subsequent experiments investigating the mechanisms of VEGF action. To determine the time course of the VEGF effect, capillaries were incubated to steady state (1 h) with NBD-CSA and repeated measurements of P-glycoprotein transport activity were made at various times after addition of VEGF to the medium (Fig. 2A). VEGF rapidly reduced P-glycoprotein transport activity. In the continued presence of VEGF, transport activity remained at ∼60–75% below control levels for an additional 90 min. However, removing VEGF from the medium at 60 min returned transport activity to the control level 60 min later. Reversal of inhibitory effect suggests that the initial reduction in P-glycoprotein activity did not reflect degradation of the protein. This interpretation is supported by Western blots of protein extracted from capillaries, which show no change in P-glycoprotein protein expression following 60 min of VEGF treatment (Fig. 2B). Consistent with no change in transporter expression, preincubation of capillaries with the proteasome inhibitor lactacystin did not attenuate the effect of VEGF on P-glycoprotein activity, indicating that P-glycoprotein is not targeted to the proteasome in response to VEGF (Fig. 2C). In contrast, the microtubule polymerization inhibitor, nocodazole, blocked the effect of VEGF on P-glycoprotein activity, indicating a cytoskeleton-dependent mechanism, perhaps trafficking P-glycoprotein from the plasma membrane to an intracellular compartment (Fig. 2D).

VEGF effects on P-glycoprotein transport activity and protein expression. A, Time course of VEGF action. Capillaries were incubated to steady state (1 h) with NBD-CSA, and P-glycoprotein activity was measured over multiple time points following addition of VEGF (200 ng/ml). After measurements at 60 min, the media were removed and replaced with fresh media, with one of the VEGF-treated groups getting VEGF-free media. Shown are pooled data from four experiments, each involving measurement from at least 10 capillaries per treatment. Statistical comparison, ***p < 0.001 versus control at the corresponding time point, significance determined by two-way ANOVA with a Bonferroni post hoc test. B, Western blot analysis of P-glycoprotein expression. Incubation of brain capillaries with VEGF (200 ng/ml, 1 h) does not change protein expression of P-glycoprotein in membrane fractions. C, The effect of VEGF on P-glycoprotein activity is not blocked by the proteasome inhibitor lactacystin (LAC, 10 μm). D, The effect of VEGF is attenuated by the microtubule polymerization inhibitor nocodazole (NOC, 20 μm). C, D, Representative experiments shown, measurements made in at least 10 capillaries per treatment. Statistical comparison, ***p < 0.001 versus control, significance determined by one-way ANOVA with a Newman–Keuls multiple-comparison test.
VEGF signals through flk-1 and Src
Preincubation of capillaries with SU5416, an inhibitor of the flk-1 receptor, blocked the effect of VEGF on P-glycoprotein transport activity (Fig. 3A). Thus, the effect of VEGF is receptor mediated. SU5416 is selective for flk-1 over other growth factor receptors (Fong et al., 1999), though interaction of SU5416 with the flt-1 cannot be ruled out (Itokawa et al., 2002). For this reason, TMPP, a drug that is selective for flk-1 over flt-1 (Fraley et al., 2002) was used to confirm the involvement of flk-1. TMPP blocked the effect of VEGF on P-glycoprotein activity, confirming that flt-1 is not likely involved (Fig. 3B).

VEGF signaling to P-glycoprotein. Shown are representative experiments; each experiment was repeated 2–4 times. Data are mean P-glycoprotein (Pgp) activity ± SEM, collected from at least 10 capillaries per condition. In all experiments shown, capillaries were preincubated with inhibitors 30 min before addition of NBD-CSA, then incubated to steady state (1 h) before addition of VEGF, and measurements were made 1 h later. Time-matched control groups from the same preparation were exposed to PBS during inhibitor and/or VEGF treatments. A, B, The effect of VEGF on P-glycoprotein activity is blocked by flk-1 inhibitors SU5416 (500 nm) and TMPP (50 nm). C, D, The effect of VEGF on P-glycoprotein activity is not blocked by the phosphatidylinositol-3-kinase inhibitor wortmannin (WORT, 100 nm), nor by the broad-spectrum PKC inhibitor staurosporine (STR, 100 nm). E, F, The Src family kinase inhibitor PP2 (1 μm) also blocks the effect of VEGF (E), but its nonfunctional analog PP3 (1 μm) does not (F). Statistical comparisons, ***p < 0.001 versus control, significance determined by one-way ANOVA with a Newman–Keuls multiple-comparison test.
We next examined signaling pathways downstream of VEGF/flk-1. Candidate pathways included phosphatidylinositol-3-kinase (PI3K)/Akt, PKC, and Src (Cross et al., 2003; Zachary, 2003). Wortmannin, the classic inhibitor of the PI3K/Akt pathway, and staurosporine, a broad-spectrum PKC inhibitor, both failed to block the effect of VEGF on transport (Fig. 3C,D), indicating that these pathways are not involved. In contrast to wortmannin and staurosporine, PP2, an inhibitor of Src family kinases, blocked the effect of VEGF (Fig. 3E), while the nonfunctional PP2 analog, PP3, was without effect (Fig. 3F). Consistent with a role for Src-kinase in VEGF signaling to P-glycoprotein, the Src kinase-activating peptide YEEIP (Liu et al., 1993) mimicked the effect of VEGF on P-glycoprotein transport activity in brain capillaries; the effect of YEEIP was blocked by PP2 but not PP3 (Fig. 4). As with VEGF, YEEIP did not affect luminal accumulation of TR and thus did not open tight junctions (data not shown). Previous studies from this laboratory demonstrated that tumor necrosis factor-α and endothelin-1 induced a similar rapid and reversible loss of P-glycoprotein activity without decreased protein expression (Hartz et al., 2006). Interestingly, tumor necrosis factor-α/endothelin-1 signaling to P-glycoprotein exhibited an absolute requirement for PKC activation. Thus, it appears that P-glycoprotein transport activity can be regulated by multiple signaling pathways.

Src-mediated modulation of P-glycoprotein transport activity. P-glycoprotein transport activity is decreased by the Src kinase activating peptide, YEEIP (100 μm, 1 h), an effect blocked by PP2 but not PP3. Statistical comparisons, ***p < 0.001 versus control, significance determined by one-way ANOVA with a Newman–Keuls multiple-comparison test.
VEGF causes Src kinase-dependent phosphorylation of caveolin-1
Caveolin-1 is a known substrate for Src kinase (Lee et al., 2000), and phosphorylation of caveolin-1 by Src in response to VEGF has been demonstrated in cultured endothelial cells (Labrecque et al., 2003). Barakat et al. (2007) recently demonstrated that Tyr-14 phosphorylation of caveolin-1 by Src increases the association of caveolin-1 with P-glycoprotein and negatively regulates P-glycoprotein transport activity in cultured brain endothelial cells. Using a Tyr-14 phospho-specific antibody, we found that exposing brain capillaries to VEGF or the Src activating peptide YEEIP increased Tyr-14 phosphorylation of caveolin-1 in brain capillaries (Fig. 5). Phosphorylation of caveolin-1 following VEGF treatment was abolished by preincubation of capillaries with the Src kinase inhibitor, PP2 (Fig. 5B). Neither VEGF nor PP2, alone or in combination, altered expression of P-glycoprotein or total caveolin-1 protein (Fig. 5).

VEGF increases Src-mediated Tyr-14 phosphorylation of caveolin-1. A, YEEIP (100 μm, 1 h) increases Tyr-14 phosphorylation of caveolin-1. B, Incubation of brain capillaries with VEGF (200 ng/ml, 1 h) and/or the Src kinase inhibitor PP2 (1 μm) is not associated with any change in protein expression of P-glycoprotein or total caveolin-1 (cav-1). Immunoreactivity for Tyr-14 phosphorylated caveolin-1 (p14Y-cav-1) is increased by VEGF; this effect is blocked by preincubation with PP2. Representative blots from a single experiment are shown; 10 μg of protein from membrane fractions were loaded per lane. All samples shown were from a single preparation of capillaries derived from 10 rat brains.
To investigate the possibility that Src-dependent phosphorylation of caveolin-1 increased its association with P-glycoprotein in brain capillaries, we immunoprecipitated caveolin-1 as previously described (Barakat et al., 2007). P-glycoprotein was detected in immunoprecipitated samples, but not in samples in which either the antibody or capillary protein had been omitted, confirming that the immunoreactivity for P-glycoprotein observed was due to coprecipitation with caveolin-1 rather than nonspecific binding to the Sepharose beads (Fig. 6). Caveolin-1 was similarly detected in only the immunoprecipitated samples, confirming that it had been precipitated (Fig. 6). Both proteins were well resolved from heavy and light chains of IgG, as confirmed by comparison with capillary lysate (Fig. 6). Though a modest (∼20%) increase in coprecipitated P-glycoprotein was observed with VEGF in some experiments, this was not consistent across all experiments performed. Moreover, in those experiments where increased P-glycoprotein signal was seem, the Src inhibitor PP2 was without effect. Thus, in rat brain capillaries, we found no evidence for VEGF- or Src-induced association of P-glycoprotein with caveolin-1.

Coimmunoprecipitation of caveolin-1 and P-glycoprotein. Caveolin-1 was immunoprecipitated from 100 μg of brain capillary protein. Representative experiment shown. CON, VEGF, and VEGF + PP2 represent samples from a single capillary preparation treated as indicated. Ab, Caveolin-1 antibody carried through the IP procedure without capillary protein. +, Five micrograms of brain capillary protein (positive control). −, One hundred micrograms of brain capillary protein carried through the IP procedure without antibody (negative control). IgG heavy chain (hc) and light chains (lc) were well resolved from detected bands for P-glycoprotein and caveolin-1. No consistent changes were observed in P-glycoprotein precipitated with caveolin-1 in any condition.
Intracerebroventricular VEGF increases brain uptake ofP-glycoprotein substrates in vivo
In situ brain perfusion is a well established method for measuring net transport of molecules across the BBB under near-physiological conditions while controlling for confounding variables such as blood flow, blood pressure, tracer concentration, and metabolism (Smith and Allen, 2003). We used [3H]-morphine and [3H]-verapamil, both substrates for P-glycoprotein (Dagenais et al., 2001; Seelbach et al., 2007), together with [14C]-sucrose, a polar, nontransported molecule with limited brain distribution (Hawkins et al., 2007), to assess BBB P-glycoprotein function and paracellular (tight junction-limited) permeability of the BBB, respectively. In mice, complete knock-out of the mdr1a/b genes (coding for P-glycoprotein) only increases in vivo brain uptake of morphine by ∼30% (Dagenais et al., 2001), indicating that this drug is at best a weak substrate for P-glycoprotein. Morphine is nonetheless an attractive substrate for use in dual-label transport experiments with a tight junction marker such as sucrose. Since the morphine distribution volume in brain is only ∼3 times higher than that of sucrose, any signal bleed of 3H into the 14C counting window is negligible. While verapamil is a much more sensitive indicator of P-glycoprotein activity (Dagenais et al., 2001), its steady-state distribution volume in rats is nearly 100 times that of sucrose under control conditions, meaning that even a ∼1% spill of 3H signal into the 14C counting window could lead to artifacts (i.e., an overestimated Rbr for sucrose), an effect we observed in pilot studies. For this reason, morphine was infused together with sucrose to assess P-glycoprotein activity and paracellular permeability of the BBB simultaneously, while verapamil was infused alone to confirm the effect of VEGF on P-glycoprotein activity and to investigate the role of Src kinase in this effect.
We compared curve fits of the morphine uptake data to both a linear model (Eq. 2) that assumes unidirectional uptake and a nonlinear model that accounts for efflux (Eq. 3) using the Akaike's Information Criteria test. In every case except the group treated with the P-glycoprotein inhibitor cyclosporine-A (CSA, 8 μm), morphine uptake was better fit to the nonlinear model, as we would expect if significant brain efflux of morphine was driven by P-glycoprotein. The fact that morphine uptake in the presence of CSA was best described by a linear model confirmed that morphine is indeed a substrate for P-glycoprotein. However, to facilitate comparison of effective distribution volumes (Vbr) for morphine between groups, the nonlinear model was used for all morphine uptake data reported.
CSA increased the [3H]-morphine Vbr from 41.9 ± 3.9 μl · g−1 in controls to 53.0 ± 6.4 μl · g−1 (F(2,53) = 3.976, p = 0.0246) without changing the [14C]-sucrose influx rate (Kin = 0.3 ± 0.2 μl · g−1 · min−1 in control vs 0.4 ± 0.2 μl · g−1 · min−1 in CSA, F(2,53) = 0.2923, p = 0.7477). This indicates inhibition of P-glycoprotein without disruption of the tight junctions (Fig. 7A). In contrast, osmotic disruption of the tight junctions by introduction of a 1.3 m mannitol bolus into the perfusion circuit immediately before isotope infusion significantly increased both the [14C]-sucrose influx rate (Kin = 1.8 ± 5.2 μl · g−1 · min−1, F(2,48) = 26.80, p < 0.0001) and the [3H]-morphine effective distribution volume (Vbr = 169.9 ± 90.9 μl · g−1, F(2,48) = 34.42, p < 0.0001).

Effect of VEGF on in vivo brain distribution of [14C]-sucrose and [3H]-morphine. A, Sucrose and morphine distribution in control and CSA-treated rats (8 μm in perfusate). Rats were perfused with [14C]-sucrose and [3H]-morphine simultaneously, n = 4–11 per time point per condition. Sucrose distribution versus perfusion time was best fit to a unidirectional uptake model (Eq. 2). Best fit lines were not significantly different between groups (F(2,53) = 0.2923, p = 0.7477). Morphine distribution versus perfusion time was best fit to a model with an efflux component (Eq. 3). Comparison of curve fits showed that the curves were not equivalent (F(2,53) = 3.976, p = 0.0246), reflecting increased brain distribution of morphine in the CSA group. Curve fits and comparisons were performed with GraphPad Prism version 4.02. B, VEGF (500 ng in 2 μl of aCSF) or 2 μl of aCSF alone was injected into the lateral ventricle 30 min before in situ brain perfusion. VEGF significantly increased brain distribution of morphine (F(2,52) = 4.935, p = 0.0109) but not sucrose (F(2,52) = 1.792, p = 0.1768) in the cerebral hemisphere ipsilateral to the injection.
A previous distribution study showed that ∼42% of a VEGF bolus was retained in brain 1 h after intracerebroventricular injection, and autoradiography indicated that VEGF was concentrated primarily (but not entirely) in the ipsilateral hemisphere (Storkebaum et al., 2005). Thus, intracerebroventricular injection is an effective means to deliver an acute bolus of VEGF to the cerebral hemisphere ipsilateral to the injection site. In preliminary studies, we found that intracerebroventricular injection of aCSF (2 μl) had no effect on [14C]-sucrose distribution to the ipsilateral cerebral hemisphere (Rbr measured by a 20 min perfusion) measured within 1 h of injection (13.5 ± 0.8 μl · g−1 vs 15.7 ± 1.0 μl · g−1 control). A range of VEGF doses (100–1000 ng in 2 μl of aCSF) were then tested; only at 1000 ng of VEGF did [14C]-sucrose distribution tend to increase (Rbr = 18.8 ± 2.5 μl · g−1, p > 0.05). We therefore used a 500 ng bolus of VEGF, a dose that did not alter tight junction permeability, for all subsequent intracerebroventricular injection studies.
Intracerebroventricular injection of VEGF significantly increased brain distribution of morphine compared to aCSF-injected controls (Fig. 7B). Vbr of [3H]-morphine increased from 38.7 ± 2.2 μl · g−1 in the aCSF group to 49.8 ± 3.0 in the VEGF group (F(2,52) = 4.935, p = 0.0109). In the same animals, the [14C]-sucrose influx rate was unchanged from aCSF-injected controls (Kin = 0.4 ± 0.1 μl · g−1 · min−1 for aCSF, 0.4 ± 0.1 μl · g−1 · min−1 for VEGF, F(2,52) = 1.792, p = 0.1768) (Fig. 7B). Thus, using morphine as an indicator, VEGF diminished P-glycoprotein transport activity in vivo without affecting tight junction permeability.
Inhibition of P-glycoprotein with CSA increased brain distribution of [3H]-verapamil 4.3-fold relative to naive controls (n = 3 per group, p < 0.05), comparable to the 5.1-fold increase reported with transporter knock-out in mice (Dagenais et al., 2001). Verapamil thus provides a more sensitive in vivo indicator of P-glycoprotein transport activity. Figure 8 shows that intracerebroventricular injection of VEGF significantly increased brain distribution of [3H]-verapamil. This effect was blocked by intraperitoneal injection of the Src kinase inhibitor PP2 immediately before intracerebroventricular injection of VEGF (Fig. 8). Thus, these in vivo experiments confirmed both the reduction of P-glycoprotein transport activity with VEGF exposure and the involvement of Src kinase in VEGF signaling.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of VEGF and Src inhibition on in vivo brain distribution of [3H]-verapamil. Intracerebroventricular injection of VEGF significantly increases brain distribution of [3H]-verapamil in the cerebral hemisphere ipsilateral to the injection site during a 20-min brain perfusion (*p < 0.05). Intraperitoneal injection of PP2 (1 mg/kg body weight, immediately before intracerebroventricular VEGF/aCSF injection) abolishes the effect of intracerebroventricular VEGF on brain verapamil distribution (n = 4–5 per group).
Discussion
In the present study, we found a rapid, specific, and concentration-dependent reduction of P-glycoprotein transport activity in isolated brain capillaries exposed to VEGF. Tight junctional permeability was not altered. The VEGF effect on transport was fully reversible and did not involve reduced protein expression of P-glycoprotein. VEGF acted through the flk-1 receptor and Src kinase, but not through PI3K/Akt or PKC. Direct activation of Src kinase by YEEIP mimicked the effects of VEGF on P-glycoprotein activity in brain capillaries. Finally, intracerebroventricular injection of VEGF increased brain distribution of the P-glycoprotein substrates morphine and verapamil without increasing BBB permeability to sucrose. This effect was blocked by peripheral administration of the Src kinase inhibitor PP2. Thus, inhibition of P-glycoprotein transport activity by VEGF occurred both in isolated brain capillaries in vitro and the intact BBB in vivo.
Recent experiments with a rat brain endothelial cell line showed increased interaction of caveolin-1 with P-glycoprotein is associated with decreased transporter activity, and that Tyr-14 phosphorylation of caveolin-1 by transfected Src kinase promotes this interaction (Barakat et al., 2007). We show here that exposing intact brain capillaries to either VEGF or the Src kinase activating peptide YEEIP increased specific Tyr-14 phosphorylation of caveolin-1; VEGF-induced caveolin phosphorylation was blocked by the Src kinase inhibitor, PP2. However, in coimmunoprecipitation experiments, we did not see increased association of caveolin-1 with P-glycoprotein following VEGF treatment. Several possibilities could explain this discrepancy. First, immortalization and culture of brain capillary endothelial cells causes changes in protein expression and function; indeed, expression and function P-glycoprotein is especially sensitive to culture conditions (Gaillard et al., 2000). Second, induced association of these proteins may be more dynamic and/or less strong than their constitutive association (note that P-glycoprotein was pulled down with caveolin-1 in all conditions). The induced association may not survive tissue lysis and protein extraction. Finally, phosphorylation of caveolin-1 may occur independently of P-glycoprotein inhibition and not be necessary for VEGF signaling to P-glycoprotein. Further, the effects of PP2 could be explained by direct inhibition of flk-1 tyrosine kinase activity by PP2. Though we are not aware of any interaction between PP2 and flk-1 that does not involve inhibition of Src activity, we cannot rule out this possibility. However, the inhibitory effect of PP2 together with mimicry of VEGF effects by the Src activator YEEIP strongly suggest that VEGF acts on P-glycoprotein via Src, and implies that other factors associated with increased Src signaling may acutely regulate P-glycoprotein.
Loss of P-glycoprotein transport activity could reflect decreased expression of the transporter protein, removal of the protein from the cell surface, a shift of the protein to a different plasma membrane microdomain, or allosteric modulation, possibly via protein-protein interactions. For VEGF-induced loss of P-glycoprotein activity, we found no reduction in transporter protein expression nor any effect of a proteosome inhibitor, ruling out reduced expression as an underlying mechanism. At present, we cannot distinguish with certainty among the remaining possibilities. Nevertheless, loss of activity was fully reversible when VEGF was removed from the medium and was blocked when capillaries were pretreated with nocodazole, a microtubule polymerization inhibitor, in the present study. These findings suggest involvement of cytoskeletal-dependent removal of the protein from the cell surface or protein movement to a different plasma membrane microdomain where it is inactive. In preliminary experiments, VEGF exposure decreased proteolysis of P-glycoprotein in brain capillaries following brain perfusion with a protease solution (Hawkins et al., unpublished data), suggesting that VEGF stimulates internalization of P-glycoprotein. Confirmation of this finding will begin to illuminate the mechanism by which VEGF regulates P-glycoprotein.
VEGF has long been associated with increased vascular permeability, and thought to contribute to increased BBB permeability in hypoxia and some brain tumors (Vogel et al., 2007). As such, it is critical that we distinguish any apparent decrease in P-glycoprotein transport activity from nonspecific opening of the BBB. In the present study, we found no effect of VEGF on paracellular (tight junction-restricted) permeability to TR in vitro nor to sucrose in vivo at VEGF concentrations and exposure times that significantly reduced P-glycoprotein transport activity. This is in agreement with previous studies that reported VEGF to have no effect on BBB permeability when injected directly into a nondiseased brain, a finding that lead investigators to conclude that “… tumor-derived angiogenic vessels and normal cutaneous vessels respond to permeability mediators in a way that normal brain vessels do not” (Criscuolo et al., 1990). It is also important to use caution in comparing data from in vivo models and freshly isolated brain capillaries to data from cell culture studies, as critical changes in brain endothelial cell phenotype do occur in culture. For example, mRNA for the flt-1 receptor is decreased 21-fold in primary brain endothelial cell culture compared to freshly isolated capillaries (Calabria and Shusta, 2008). Flt-1 is thought to act as a “scavenger” for VEGF, binding it with higher affinity than the flk-1 receptor and thus negatively regulating the biological effects of VEGF mediated by flk-1 (Hiratsuka et al., 1998). This may explain why developmentally mature, nondiseased brain capillaries (freshly isolated or in situ) are relatively unresponsive to the permeabilizing effect of VEGF compared to endothelial cell cultures, as suggested by our results and others (Criscuolo et al., 1990; Harrigan et al., 2002).
Given the association of increased brain VEGF with CNS disease (Greenberg and Jin, 2005), the present results suggest that BBB P-glycoprotein activity may be altered by VEGF in certain disease states. Indeed, there is clinical evidence for P-glycoprotein activity being depressed in injured portions of the brain. Ederoth et al. (2004) used microdialysis to measure brain concentrations of morphine, a P-glycoprotein substrate, in traumatic brain injury (TBI) patients. They found a higher concentration in injured versus noninjured portions of the brain and attributed this finding to impaired drug efflux. The implications of diminished P-glycoprotein transport activity are twofold. First, loss of P-glycoprotein function could render TBI patients vulnerable to central toxicity and or/side effects of peripherally acting drugs, such as ion channel blockers (i.e., verapamil) and immunosuppressants (i.e., CSA). Indeed, the altered brain pharmacokinetics for morphine, a weak P-glycoprotein substrate, argue that this is the case. Thus, the potential for adverse drug reactions may be increased. Conversely, delivery of therapeutics to treat brain injury may be enhanced at the site of injury. Note that agents showing considerable promise as neuroprotective therapeutics in TBI based on animal studies include FK-506 (Singleton et al., 2001) and CSA (Hatton et al., 2008), both of which are P-glycoprotein substrates. The neuroprotective efficacy of these agents in TBI may benefit in part from improved distribution to the site of injury as a result of VEGF-induced acute downregulation of P-glycoprotein activity.
It is becoming increasingly clear that functional changes in brain microvascular physiology play a significant role in the pathogenesis and progression of neurodegenerative disease, including Alzheimer's disease. Both aberrant angiogenic signaling (including increased levels of VEGF) (Kalaria et al., 1998; Tarkowski et al., 2002; Thirumangalakudi et al., 2006; Desai et al., 2009) and diminished expression/activity of P-glycoprotein (Vogelgesang et al., 2002, 2004) have been observed in the brains of Alzheimer's disease patients. Low levels of the homeobox gene MEOX2 (encoding the protein GAX) have been reported in brain endothelial cells from Alzheimer's patients (Wu et al., 2005). GAX is a regulator of vascular differentiation, and in transgenics with one copy of the MEOX2 gene deleted, clearance of Aβ by low-density lipoprotein receptor-related protein 1 (LRP) are impaired. Interestingly, angiogenesis in response to hypoxia was also impaired in these animals, despite expected elevations of VEGF in response to hypoxia (Wu et al., 2005). In other words, the microvasculature in Alzheimer's disease may be exposed to high levels of VEGF, but cannot respond normally to it. In another study, VEGF was found to colocalize with β-amyloid plaques (Yang et al., 2004). Endothelial deposition of β-amyloid is inversely correlated with P-glycoprotein expression (Vogelgesang et al., 2002), and there is evidence that P-glycoprotein also contributes to the brain clearance of Aβ (Cirrito et al., 2005; Kuhnke et al., 2007), likely in coordination with LRP and the receptor for advanced glycosylation end products (RAGE) (Zlokovic et al., 2000). At this point, it is not known whether VEGF is related to the diminished function of BBB P-glycoprotein in Alzheimer's, though the data presented herein suggest that anti-VEGF therapy could present a novel strategy for improving brain clearance of Aβ.
In summary, our data show that VEGF acutely and reversibly depresses the transport activity of P-glycoprotein, a key BBB drug efflux transporter. VEGF signaled through the flk-1 receptor and Src, and depression of P-glycoprotein activity by VEGF is associated with Src-dependent phosphorylation of caveolin-1, a signal known to trigger caveolin-dependent endocytosis. To our knowledge, this is the first demonstration of VEGF regulating P-glycoprotein in any tissue. Our findings disclose a novel role for VEGF in brain. These findings also imply that brain pharmacokinetics of xenobiotics may be altered via modulation of P-glycoprotein in conditions associated with increased levels of VEGF in the brain including TBI and Alzheimer's disease.
Footnotes
- Received October 13, 2009.
- Revision received November 25, 2009.
- Accepted December 2, 2009.
-
This work was supported by the Intramural Research Program of the National Institutes of Health, Project #Z01 ES080048-18 LP.
- Correspondence should be addressed to Dr. David S. Miller, National Institute of Environmental Health Sciences, MD F1-03, P.O. Box 12233, Research Triangle Park, NC 27709. miller28{at}niehs.nih.gov
- Copyright © 2010 the authors 0270-6474/10/301417-09$15.00/0