
Abstract
Astrocytes release a variety of signaling molecules including glutamate, d-serine, and ATP in a regulated manner. Although the functions of these molecules, from regulating synaptic transmission to controlling specific behavior, are well documented, the identity of their cellular compartment(s) is still unclear. Here we set out to study vesicular exocytosis and glutamate release in mouse hippocampal astrocytes. We found that small vesicles and lysosomes coexisted in the same freshly isolated or cultured astrocytes. Both small vesicles and lysosome fused with the plasma membrane in the same astrocytes in a Ca2+-regulated manner, although small vesicles were exocytosed more efficiently than lysosomes. Blockade of the vesicle glutamate transporter or cleavage of synaptobrevin 2 and cellubrevin (both are vesicle-associated membrane proteins) with a clostridial toxin greatly inhibited glutamate release from astrocytes, while lysosome exocytosis remained intact. Thus, both small vesicles and lysosomes contribute to Ca2+-dependent vesicular exocytosis, and small vesicles support glutamate release from astrocytes.
Introduction
Ca2+-regulated exocytosis is a major pathway by which neurons and endocrine cells release neurotransmitters and hormones. In nerve terminals, synaptic vesicles, also termed small clear vesicles, are used for neurotransmission (Sudhof, 2004; Rizzoli and Betz, 2005; Chua et al., 2010). In contrast, neuroendocrine cells mainly use large dense-core vesicles to release neuropeptides and hormones (Burgoyne and Morgan, 2003; García et al., 2006). Small clear vesicles and large dense-core vesicles coexist in many types of neurons (De Camilli and Jahn, 1990), and both can undergo Ca2+-regulated exocytosis in the same nerve terminal (Klyachko and Jackson, 2002).
Astrocytes in the CNS regulate synaptic functions in an activity-dependent manner, through releasing gliotransmitters including glutamate, d-serine, and ATP (Yang et al., 2003; Gourine et al., 2010; Halassa and Haydon, 2010; Henneberger et al., 2010; Parpura and Zorec, 2010). Compared with the well characterized vesicular exocytosis in neurons and endocrine cells, the mechanisms of vesicle fusion in astrocytes are still under debate (Bezzi et al., 2004; Li et al., 2008). Using immunoelectron microscopy for vesicle glutamate transporter 1 (VGLUT1), Bezzi et al. (2004) identified a type of small vesicle ∼30 nm in diameter. These have characteristics similar to synaptic vesicles with expression of VGLUT1 and synaptobrevin 2 (Syb2)/vesicle-associated membrane protein 2 (Bezzi et al., 2004; Bowser and Khakh, 2007), and are exocytosed in a Ca2+-dependent manner (Bowser and Khakh, 2007; Marchaland et al., 2008). Cleavage of Syb2 and cellubrevin/vesicle-associated membrane protein 3 by Clostridium toxins greatly inhibits glutamate release from astrocytes (Bezzi et al., 1998; Araque et al., 2000; Pasti et al., 2001; Jourdain et al., 2007).
In addition to the specific labeling of vesicles with their resident proteins, uptake of styryl (FM) dyes is broadly used as an “unbiased” tool to study the dynamics of exo-endocytosis of synaptic vesicles and large dense-core vesicles (Cochilla et al., 1999; Aravanis et al., 2003; Rizzoli and Betz, 2004). FM1-43 loading in astrocytes only labels lysosomes, and Ca2+-dependent exocytosis of lysosomes has been revealed by confocal microscopy and total internal reflection fluorescence microscopy (TIRFM) (Zhang et al., 2007; Li et al., 2008). Based on these findings, it is proposed that the lysosome is the major type of vesicle for Ca2+-regulated exocytosis in astrocytes. Previously, the lysosome was identified as the main vesicle for Ca2+-dependent exocytosis in nonexcitable fibroblasts (Jaiswal et al., 2002, 2004). These reports presented views that were in conflict with those of small vesicle exocytosis in astrocytes.
In the present study, we investigated the types of vesicles supporting Ca2+-dependent exocytosis in astrocytes. Using double-staining with resident vesicular proteins, TIRFM of vesicle exocytosis, and sniffer current recording of glutamate release in cultured hippocampal astrocytes, we found that both small vesicles and lysosomes were Ca2+-regulated releasable vesicles in astrocytes. They fused with different efficiencies, and small vesicles were necessary for glutamate release. These results show that both small vesicles and lysosomes are components of vesicular exocytosis in astrocytes and may provide new insights into our understanding of the tripartite synapse.
Materials and Methods
Cell cultures.
The use and care of animals was approved and directed by the Institutional Animal Care and Use Committee of Peking University. Mouse hippocampal astrocytes were cultured as previously described (Chen et al., 2005). We used C57 mice (0–1 d old) without gender preference. Briefly, the hippocampus was dissected out in an ice-cold solution (containing, in mm: 137 NaCl, 5.4 KCl, 0.17 Na2HPO4, 0.22 KH2PO4, 10 HEPES, 22 sucrose, 17 glucose), cut into 3–4 pieces, and then trypsinized (0.25% trypsin in dissection solution) for 8–10 min. Cells were mechanically dissociated with a Pasteur pipette and plated in 75 cm2 flasks (Nuclon). They were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen). After reaching confluency (7–9 d), cells were trypsinized, resuspended, and plated on polyethyleneimine-coated glass coverslips in 35 mm dishes. Experiments were performed 24 h after the astrocytes regained normal morphology.
HEK293 cells were cultured as previously described (Yu et al., 2004).
Freshly isolated astrocytes.
Freshly isolated astrocytes were prepared as previously described (Zhou and Kimelberg, 2001; Zhang et al., 2004). Briefly, mouse hippocampus was dissected out in an ice-cold Ca2+-free solution (containing, in mm: 124 NaCl, 5 KCl, 2 MgSO4, 1 Na-pyruvate, 26 NaHCO3, 10 glucose, bubbled with 95% O2/5% CO2) and was treated with an enzyme solution [in 10 ml of normal EBSS buffer; 9.44 mg of papain (Worthington), 1.8 mg of l-cysteine (Fluka Biochemika), and 1.8 mg of EDTA)] for 15–20 min at 36°C. They were then washed twice with normal artificial cerebrospinal fluid (containing, in mm: 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.0 CaCl2, 1 Na-pyruvate, 26 NaHCO3, 1 NaH2PO4,11 glucose, bubbled with 95% O2/5% CO2). Cells were dissociated with a Pasteur pipette and plated on polyethyleneimine-coated glass coverslips. After 1 h incubation at 37°C, cells were fixed for immunocytochemistry.
Immunocytochemistry.
Astrocytes were fixed in 4% paraformaldehyde for 15 min at room temperature and permeabilized with 0.3% Triton X-100 for 5 min. Cells were blocked in PBS with 2% BSA for 0.5 h at room temperature. Primary antibodies were diluted in PBS with 2% BSA and incubated overnight at 4°C. After washing out the primary antibody with PBS and 0.1% saponin, cells were incubated with Alexa Fluor 488- or 594-conjugated secondary antibodies (in PBS with 2% BSA) (Invitrogen) for 2–3 h at room temperature. Antibodies against glial fibrillary acidic protein (GFAP) (1:1000; SYSY); Syb2 (1:1000; SYSY), cellubrevin (1:500; SYSY), and lysosome-associated membrane protein 1 (LAMP1) (1:200; Abcam) were used. Images were taken using an LSM 710 (Carl Zeiss) inverted confocal microscope with ZEN 2008 software (Carl Zeiss). For colocalization analysis, all puncta within a cell were selected and analyzed by the “Colocalization Analysis” plug-in of MBF ImageJ software (http://www.macbiophotonics.ca/imagej/).
Transfection.
Astrocytes were transfected with Lipofectamine 2000 (Invitrogen) 24 h after being plated on glass coverslips according to the manufacturer's suggestion. Each dish was transfected with 2 μl of Lipofectamine plus 1.5 μg of plasmid. Plasmids of synapto-pHluorin (Syb2-pHluorin) (a gift from Dr. Gero Miesenböck, Department of Physiology, Anatomy and Genetics, University of Oxford, UK), EGFP-Syb2, and Syb2-mOrange2 (both from Dr. Tao Xu, Institute of Biophysics, Beijing, China) were used to label small vesicles (Miesenböck et al., 1998; Zhao et al., 2009). Plasmid of EGFP-LAMP1 (a gift from Dr. Haoxing Xu, Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI) was used to label lysosomes (Dong et al., 2008). Plasmid of tetanus toxin (TeNT) was also from Dr. Tao Xu. Plasmid of EGFP-VGLUT1 was a gift from Dr. Jeffrey D. Erickson (Louisiana State University Health Sciences Center, New Orleans, LA). All experiments were performed 24–48 h after transfection except for the TeNT inhibition experiment (72 h).
TIRF imaging.
TRIFM was performed on an Olympus IX-81 inverted microscope with the Andor system, equipped with a 100× lens (Olympus; numerical aperture, 1.45; Li et al., 2004). Images were collected using Andor iQ software. Exposure time was 200 ms for all fluorescent proteins. For single-color imaging, sampling frequency was ∼4 Hz (250–260 ms per frame) and for dual-color imaging it was 2 Hz for each channel. All experiments were performed at room temperature (22–25°C). Based on previous reports (Zenisek et al., 2000; Bowser and Khakh, 2007), we defined a vesicle fusion event as when a sudden increase of synapto-pHluorin, syb2-mOrange2, or EGFP-LAMP1 signal after exposure to neutral pH extracellularly was followed by diffusion of the signal in the vicinity.
Sniffer current recording.
AsRed2 (Clontech), alone as a control, or with plasmid of glutamate receptor (GluR)-L497Y (Stern-Bach et al., 1998; Lee et al., 2007), was transfected into HEK293 cells. GluR-L497Y was a gift from Dr. Stephen F. Traynelis (Department of Pharmacology, Emory University School of Medicine, Atlanta, GA). Briefly, 1.5 μg of GluR-L497Y plasmid and 0.3 μg of AsRed2 were cotransfected into HEK293 cells with 2 μl of Lipofectamine. On the next day, HEK293 cells were trypsinized and plated on top of astrocytes (on 8 × 8 mm glass coverslips). Electrophysiology was performed 4–8 h after plating. Cells were bathed in standard extracellular solution (containing, in mm: 150 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH adjusted to 7.4 by NaOH) at room temperature. Voltage clamp in the whole-cell configuration was used to record mechanical stimulation-induced glutamate release. Intracellular solution contained (in mm): 140 CsCl, 5 EGTA, 10 HEPES, 2 Mg2+ATP, pH adjusted to 7.4 with CsOH. GluR-L497Y-positive cells were first selected by perfusion of 200 μm glutamate with a fast electronic switching perfusion system (MPS-2, INBIO Inc.). Red cells were first perfused with glutamate (200 μm, 10 s) to test whether they also expressed GluR-L497Y. Among these, 95% responded to glutamate perfusion. Only GluR-positive cells were used to record mechanical stimulation-induced glutamate release from astrocytes. Current was sampled by Pulse software and an EPC-10/2 amplifier (HEKA Elektronik) at 2 kHz. A micromanipulator (Burleigh Instruments) was used for patch clamp and for mechanical stimulation.
To block VGLUT function in cultured astrocytes, we treated astrocytes with trypan blue (1 μm) or rose bengal (0.5 μm) in standard extracellular solution for 30 min at 37°C as previously reported (Ogita et al., 2001; Montana et al., 2004).
Fluorescent dyes and Ca2+ imaging.
FM1-43 (Invitrogen) and LysoTracker (Invitrogen) were used to label vesicles in cultured astrocytes (Li et al., 2008). Astrocytes were loaded with FM1-43 (10 μm, 30 min) or LysoTracker (50 nm, 30 min) in standard extracellular solution. They were then washed 3 times (5 min each) before experiments. FM1-43 and LysoTracker images were captured with an LSM 710 (Carl Zeiss) inverted confocal microscope. Intracellular Ca2+ was measured with the low-affinity Ca2+ indicator Fluo-4 AM (Invitrogen). Briefly, astrocytes were loaded with 5 μm dye for 5 min at 37°C and washed 3 times with normal external solution (5 min each) before experiments. [Ca2+]i was measured by epifluorescence imaging using an Olympus IX-70 inverted microscope equipped with a monochromator-based system (TILL Photonics). X-chart software (HEKA Elektronik) was used to collect imaging data at 1 Hz.
Drugs and reagents.
All drugs and reagents were from Sigma, unless specified otherwise.
Statistics.
All electrophysiological data were analyzed with IGOR software (Wavemetrics). Imaging data were analyzed with ImageJ (NIH) and Photoshop CS3 (Adobe Systems). Data are presented as mean ± SEM. Significance of difference was tested using Student's t test (*p < 0.05; **p < 0.01; ***p < 0.001; n.s. = not significant, p > 0.05).
Results
Small vesicles and lysosomes coexisted in astrocytes
Both small vesicles and lysosomes have been identified as secretory vesicles in astrocytes (Bezzi et al., 2004; Chen et al., 2005; Bowser and Khakh, 2007; Jaiswal et al., 2007; Li et al., 2008). We visualized small vesicles and lysosomes in cultured astrocytes by antibody labeling with Syb2 and LAMP1, a lysosome marker (Dong et al., 2008), respectively. Our cell cultures were almost purely astrocytes (Fig. 1A). We found no evidence of colocalization of Syb2 and LAMP1 proteins (n = 20 cells; Fig. 1B), indicating that small vesicles and lysosomes are molecularly distinct (Schoch et al., 2001; Jaiswal et al., 2002; Sudhof, 2004; Dong et al., 2008). Bezzi et al. (2004) reported that astrocytes in situ express cellubrevin but not Syb2. However, Zhang et al. (2004) suggested that freshly isolated astrocytes do express Syb2. We found 75 ± 1% (n = 33 cells) of Syb2 puncta colocalized with cellubrevin signals (Fig. 1C). These data showed that Syb2 and cellubrevin colocalized in small vesicles in astrocytes, so we used Syb2 as a marker for these vesicles.

Small vesicles and lysosomes coexisted in hippocampal astrocytes. A, Representative images of mouse cultured hippocampal astrocytes stained with antibody against GFAP (red) and the nuclear stain DAPI (blue). Scale bars: (left), 100 μm; (right), 20 μm. B, Double immunostaining with Syb2 (left; green) and LAMP1 (middle; red) in a cultured astrocyte. The merged image is shown on the right. C, Double staining with Syb2 (left; green) and cellubrevin (middle; red) in another astrocyte. Merged image is shown on the right. The boxed areas in B and C are magnified in the insets. Scale bars, 10 μm.
Since astrocytes may have a different protein expression profile under culture conditions (Cahoy et al., 2008; Fiacco et al., 2009), we determined whether small vesicles and lysosomes still coexisted in freshly isolated astrocytes. Freshly isolated mouse hippocampal astrocytes had a “bushy” appearance and were positive for GFAP staining (Fig. 2A) (Zhou and Kimelberg, 2001; Zhang et al., 2004). We stained astrocytes with antibodies against Syb2 and LAMP1. Freshly isolated astrocytes displayed punctate staining with Syb2 and LAMP1 similar to cultured cells, and there was almost no colocalization (n = 27 cells; Fig. 2B), indicating that both small vesicles and lysosomes probably exist in vivo.

Small vesicles and lysosomes in freshly isolated astrocytes. A, Representative images showing a freshly isolated astrocyte. Left, DIC image and DAPI (blue) staining. Right, GFAP staining (n = 17 cells). Scale bars, 10 μm. B, Double immunostaining of Syb2 (green; left) and LAMP1 (red; middle) in another freshly isolated astrocyte. Merged image at right. Syb2 did not colocalize with LAMP1 in freshly isolated astrocytes. Scale bars, 10 μm. The boxed areas are magnified in the insets.
FM labeled no small vesicles in astrocytes
FM dyes are used to monitor vesicle fusion from a wide range of cells from neuroendocrine cells to synaptic terminals (Cochilla et al., 1999; Aravanis et al., 2003; Rizzoli and Betz, 2004). In astrocytes, FM dye preferentially labels lysosomes (Zhang et al., 2007; Li et al., 2008). We determined whether FM labeled small vesicles as well. Cultured astrocytes were transfected with EGFP-LAMP1 plasmid and were transiently loaded with LysoTracker (Li et al., 2008). Almost all LysoTracker puncta (97 ± 1%; n = 20 cells) were positive for EGFP-LAMP1 (Fig. 3A), demonstrating that LysoTracker is a good lysosome marker in astrocytes. There were large lysosomes around the nucleus in astrocytes expressing EGFP-LAMP1 (Fálcon-Pérez et al., 2005). To prevent saturation of LysoTracker signals within these large lysosomes, we reduced the laser intensity for excitation, which led to very weak LysoTracker signals in smaller lysosomes. Thus, there appeared to be more EGFP-LAMP1 puncta than LysoTracker puncta.

FM1-43 labeled lysosomes, but not small vesicles. A, Colocalization of EGFP-LAMP1 with LysoTracker. Cultured astrocytes were transiently transfected with EGFP-LAMP1. Cells were loaded with LysoTracker, and EGFP-LAMP1-positive astrocytes were chosen for imaging. Left, EGFP-LAMP1 signals (green); middle, LysoTracker signals (red); right, merged image. B, Colocalization of FM1-43 and LysoTracker signals in astrocytes. Cells were loaded with FM1-43 and LysoTracker together. C, Distinct localizations of EGFP-Syb2 and LysoTracker in astrocytes. Cells were transiently transfected with EGFP-Syb2 and loaded with LysoTracker. Note that there was almost no colocalization of EGFP-Syb2 (left; green) and LysoTracker (middle; red) puncta. The boxed areas are magnified in the insets. Scale bars, 20 μm.
Next, we loaded FM1-43 and LysoTracker together and observed that most FM1-43 puncta (93 ± 1%; n = 23 cells) colocalized with LysoTracker (Fig. 3B). Finally, astrocytes were transfected with EGFP-Syb2 and transiently loaded with LysoTracker. We found almost no colocalization between the LysoTracker signal and the EGFP-Syb2 signal (2 ± 0.5%; n = 18 cells; Fig. 3C). These results indicated that FM1-43 colocalized with LysoTracker and selectively labeled lysosomes, but not small vesicles, in cultured astrocytes.
Live imaging of small vesicle and lysosome fusion
FM1-43 selectively labeled lysosomes but not small vesicles in astrocytes (Fig. 3). This explained why previous studies using FM1-43 found only exocytosis signals of large vesicles or lysosomes in astrocytes (Chen et al., 2005; Zhang et al., 2007; Li et al., 2008). To investigate exocytosis of both small vesicles and lysosomes, we imaged synapto-pHluorin (Syb2-pHluorin) and EGFP-LAMP1 by TIRFM in cultured astrocytes (Fig. 4A). Almost no spontaneous fusion events were observed without any stimulation (data not shown). When mechanical stimulation using a patch-pipette was applied to astrocytes (Chen et al., 2005; Haydon and Carmignoto, 2006), there were numerous fusion events from both small vesicles and lysosomes (Fig. 4A,B). Interestingly, the kinetics of evoked exocytosis was faster for small vesicles than for lysosomes (latency of 50% total exocytosis after stimulation was 2.2 ± 0.2 s for small vesicles, n = 9 cells, and 2.9 ± 0.1 s for lysosomes, n = 9 cells, p < 0.01; Fig. 4C). No fusion events were observed in Ca2+-free solution (Fig. 4D), demonstrating that both types of exocytosis were Ca2+ dependent. To quantify fusion events in different astrocytes, we analyzed the fusion events per cell, since the average of the imaged area per transfected astrocyte was comparable between synapto-pHluorin-expressing cells (2579 ± 334 μm2) and EGFP-LAMP1-expressing cells (2666 ± 286 μm2) (small vesicles, n = 9 cells; lysosomes, n = 9 cells; p = 0.9). The total number of events for small vesicle fusion was 121 ± 18 per cell and for lysosome fusion was 42 ± 9 per cell (p < 0.01).

Small vesicles and lysosomes both underwent Ca2+-dependent exocytosis, as revealed by TIRFM. A, Representative events of small vesicle and lysosome fusion observed with synapto-pHluorin and EGFP-LAMP1. Each frame is 220 ms and the image size is 2.2 μm × 2.2 μm. Scale bar, 2 μm. B, Histograms of small vesicle and lysosome fusion events following mechanical simulation observed by TIRFM. Cultured astrocytes were transiently transfected with synapto-pHluorin or EGFP-LAMP1 to label small vesicles or lysosomes. Each column indicates the number of fusion events in each imaging frame. The red arrow marks the time when mechanical stimulation was applied. The bars depict mean ± SEM. C, Cumulative fusion plots (normalized) of small vesicles and lysosomes in single-color TIRFM. Red arrowhead marks the mechanical stimulation. The latencies between stimulation and 50% of total exocytosis were significantly different for small vesicles and lysosomes. **p < 0.01; Student's t test. D, Summary of evoked exocytosis of small vesicles and lysosomes in the presence or absence of 2.5 mm Ca2+. Total events within 30 frames (7.8 s) after mechanical stimulation were analyzed. ***p < 0.001; Student's t test. Data in B–D were from 9 cells for small vesicles and 10 cells for lysosomes. E, Representative image of dual-color TIRFM of small vesicle and lysosome fusion in a cultured astrocyte. The astrocyte was transiently transfected with Syb2-mOrange2 and EGFP-LAMP1. Ionomycin (2.5 μm) was applied in the bath to trigger vesicle fusion. Two representative fusion events are shown. In box 1, a lysosomal vesicle was exocytosed in the absence of small vesicle fusion. In box 2, only a small vesicle fusion was observed. Scale bars, 5 μm and 2 μm. F, Top graph shows summary plots of small vesicle (red) and lysosome (green) fusion in dual-color TIRFM. Bottom graph shows normalized cumulative events of small vesicle and lysosome fusion. Each point represents the number of fusion events of normalized results in 2 s averaged from 10 cells. The bars depict mean ± SEM. The latencies between stimulation and 50% of total exocytosis were significantly different for small vesicles and lysosomes. **p < 0.01; Student's t test.
To make direct comparison of the exocytosis kinetics between small vesicles and lysosomes, we visualized Syb2 and LAMP1 simultaneously in the same astrocytes by dual-color TIRFM. These astrocytes were cotransfected with plasmids of Syb2-mOrange2 and EGFP-LAMP1. We raised the intracellular calcium level with a calcium ionophore (2.5 μm ionomycin; added to the recording chamber using a pipette), and recorded the evoked exocytosis mediated by both Syb2-mOrange2 (small vesicles) and EGFP-LAMP1 (lysosomes) in the same cell (Fig. 4E). Distinct small vesicle and lysosome fusions were readily observed. For example, a lysosome fusion was revealed by a flash of EGFP-LAMP1 signal in the absence of Syb2-mOrange2; in contrast, a small vesicle fusion appeared with Syb2-mOrange2 only (Fig. 4E). Consistent with the experiments using single-color TIRFM images (Fig. 4C), small vesicles fused ∼1.5 times faster than lysosomes after ionomycin stimulation of the same cell (latency to 50% total exocytosis after stimulation: 46 ± 2 s for small vesicles vs 73 ± 6 s for lysosomes; n = 8 cells; p < 0.01; Fig. 4F, bottom graph). In addition, the total number of fusion events for small vesicles was 59 ± 7 and for lysosomes was 25 ± 3 in the same cell (p < 0.001; Fig. 4F, top graph). Note that the latency to 50% total exocytosis after stimulation was greater in these experiments (Fig. 4F, bottom graph) because bath application of ionomycin into the recording chamber was a much slower stimulus than direct mechanical stimulation of cells (Fig. 4C). Together, these results established that both small vesicles and lysosomes are Ca2+-regulated releasable vesicles and they possess distinct exocytosis kinetics.
Blockade of VGLUT inhibited glutamate release
Glutamate is the most important excitatory neurotransmitter in the brain. Glutamate released from astrocytes also plays an important role in the regulation of neural activity (Fiacco and McCarthy, 2004; Jourdain et al., 2007). We therefore set out to study the vesicular glutamate release in cultured astrocytes. It is known that small vesicles express VGLUT1 (Bezzi et al., 2004; Bowser and Khakh, 2007), and we determined whether this was also present in lysosomes. We transfected astrocytes with EGFP-VGLUT1 plasmid and stained the cells with antibodies against cellubrevin and LAMP1. EGFP-VGLUT1 colocalized with endogenous cellubrevin signals but did not colocalize with endogenous LAMP1 signals (Fig. 5A). These data implied that VGLUT was only expressed in small vesicles, and glutamate was pumped into small vesicles in astrocytes.

Blockade of VGLUT inhibited evoked glutamate release. A, Colocalization of EGFP-VGLUT1 with small vesicles but not lysosomes. Cultured astrocytes were transiently transfected with EGFP-VGLUT1 and stained with antibodies against cellubrevin (red; left, n = 15 cells) and LAMP1 (red; right, n = 17 cells). Merged images are shown. DAPI staining (for nuclei) is shown in blue to indicate cell location. The boxed areas are magnified in the insets. Scale bars, 20 μm. B, C, Representative recordings of glutamate release following mechanical stimulation in a control (left) and a trypan blue- or rose bengal-treated (right) cultured astrocyte. The glutamate release was recorded by patch-clamped sniffer HEK293 cells. The top traces show mechanical stimulation-induced glutamate currents in HEK293 cells; the bottom traces show integrated charge of the upper currents. Red arrows mark mechanical stimuli. D, E Representative traces of mechanical stimulation-induced intracellular Ca2+ rise in control and trypan blue- or rose bengal-treated astrocytes. In trypan blue experiments, intracellular Ca2+ was measured using Fluo-4 AM. In rose bengal experiments, intracellular Ca2+ was measured using Fura-2 AM. a.u., arbitrary units. F, G, Statistics of mechanical stimulation-induced intracellular Ca2+ rise. H, I, Statistics of peak of sniffer current (Ipeak) and total charge transfer (Q, integrated signal). **p < 0.01, ***p < 0.001; Student's t test. The bars depict mean ± SEM.
To investigate whether VGLUT has essential functions in glutamate release in astrocytes, we directly detected glutamate release using GluR-L497Y-expressing HEK293 cells. The mutant GluR has a much higher affinity for glutamate and can hardly be desensitized, making it a sensitive glutamate sensor (Stern-Bach et al., 1998; Lee et al., 2007). GluR-L497Y was cotransfected with AsRed2 into HEK293 cells for glutamate sniffer recording. Red HEK293 cells were first perfused with 200 μm glutamate (10 s) to screen for GluR-L497Y expression. When astrocytes were mechanically stimulated, reliable glutamate currents (∼3 nA) were recorded from the sniffer cells (Fig. 5B, control).
We assessed glutamate release when blocking VGLUT with trypan blue or rose bengal (Roseth et al., 1998; Ogita et al., 2001). Consistent with a previous report (Montana et al., 2004), blockade of VGLUT with trypan blue or rose bengal greatly inhibited evoked glutamate currents (Fig. 5B–I). In trypan blue experiments, the peak of mechanical stimulation-induced sniffer current was inhibited from 3.3 ± 0.5 nA to 1.3 ± 0.5 nA (control, n = 6 cells; trypan blue, n = 7 cells; p < 0.01). Integrated total charge was reduced from 86 ± 10 nC to 14 ± 7 nC (p < 0.001), while the mechanical stimulation-induced intracellular Ca2+ rise was unaffected (control, 4.8 ± 0.5 n = 9 cells; trypan blue, 5.5 ± 0.6 n = 10 cells; p > 0.05; Fig. 5B,D,F,H). Glutamate-induced currents in GluR-L497Y-expressing HEK293 cells used for sniffer recording in control and trypan blue-treated astrocytes were without significant differences (peak current: control, 3.0 ± 0.4 nA, n = 6; trypan blue, 2.8 ± 0.5 nA, n = 7; p = 0.87; integrated total charge: control, 39.4 ± 5.8 nA, n = 6; trypan blue, 43.8 ± 2.2 nA, n = 7; p = 0.36). In rose bengal experiments, the peak sniffer current was inhibited from 2.9 ± 0.1 nA to 0.6 ± 0.2 nA (control, n = 10 cells; rose bengal, n = 6 cells; p < 0.001). Integrated total charge was reduced from 78 ± 11 nC to 11 ± 3 nC (p < 0.001), while the mechanical stimulation-induced intracellular Ca2+ rise was unaffected (control, 2.2 ± 0.2 a.u., n = 8 cells; rose bengal-treated cells, 2.0 ± 0.1 a.u., n = 15 cells; p > 0.05; Fig. 5C,E,G,I). Glutamate-induced currents in GluR-L497Y-expressing HEK293 cells used for sniffer recording in control and rose bengal-treated astrocytes were without significant differences (peak current: control, 3.0 ± 0.7 nA, n = 10; rose bengal: 2.7 ± 0.5 nA, n = 6; p = 0.7; integrated total charge: control, 43.3 ± 6.4 nA, n = 10; rose bengal, 54.7 ± 8.5 nA, n = 6; p = 0.5). These experiments suggested that VGLUTs in small vesicles are essential for Ca2+-dependent glutamate release.
Small vesicle exocytosis was required for glutamate release
TeNT, a Clostridium toxin, is widely used to selectively cleave Syb2 and cellubrevin and to block small vesicle exocytosis (Schiavo et al., 2000). Syb2 in synaptic vesicles is essential for vesicle fusion and neurotransmitter release (Schoch et al., 2001). Glutamate release in astrocytes in situ and in culture is sensitive to TeNT treatment as well (Montana et al., 2004, 2006; Haydon and Carmignoto, 2006; Xu et al., 2007; Zhao et al., 2009). We assessed the effect of TeNT on small vesicle and lysosome fusion, as well as Ca2+-dependent glutamate release in our experimental conditions. Plasmid of TeNT was cotransfected with AsRed2 into cultured astrocytes. As a negative control, AsRed2 alone was transfected into astrocytes.
First, we determined the TeNT effects on both types of vesicle exocytosis by TIRFM. Cultured astrocytes were transfected with plasmid of synapto-pHluorin or EGFP-LMAP1 alone, or cotransfected with synapto-pHluorin and TeNT, EGFP-LAMP1, and TeNT. To compare the signals of synapto-pHluorin or EGFP-LAMP1 between control astrocytes and TeNT-cotransfected astrocytes, we used the same exposure time and excitation strength in this TIRFM experiment. Coexpression of TeNT with synapto-pHluorin greatly attenuated the synapto-pHluorin signal, while the EGFP-LAMP1 signal was not altered (Fig. 6A,B). This is consistent with the specific cleavage of VAMP2 and cellubrevin by TeNT (Schiavo et al., 2000). After cleavage, VAMP2 and cellubrevin are degraded (Zhao et al., 2009), and in our study, the synapto-pHluorin signal was almost abolished under TIRFM (Fig. 6A). We next tested whether lysosome fusion was affected by TeNT. The averaged numbers of induced fusion events marked by EGFP-LAMP1 with or without TeNT were 23 ± 2 (n = 8 cells) and 24 ± 3 (n = 7 cells) (Fig. 6C; p > 0.05). The average of the imaged area per transfected astrocyte was similar (control, 2923 ± 328 μm2; TeNT, 3085 ± 275 μm2; p > 0.05; Fig. 6D).

TeNT did not affect lysosome fusion. A, Synapto-pHluorin signal in control and TeNT-treated cultured astrocytes (control, n = 14 cells; TeNT, n = 15 cells). White outline marks an astrocyte according to epifluorescence microscopy. Scale bars, 10 μm. B, EGFP-LAMP1 signals in control and TeNT-treated cultured astrocytes (control, n = 14 cells; TeNT, n = 12 cells). Scale bars, 10 μm. C, Statistics of number of mechanical simulation-induced fusion events marked by EGFP-LAMP1 in control and TeNT-treated astrocytes. All events occurring within 10 s after the stimulus were counted. D, Statistics of average imaged area in control and TeNT-treated astrocytes. n.s., p > 0.05; Student's t test. Bars depict mean ± SEM.
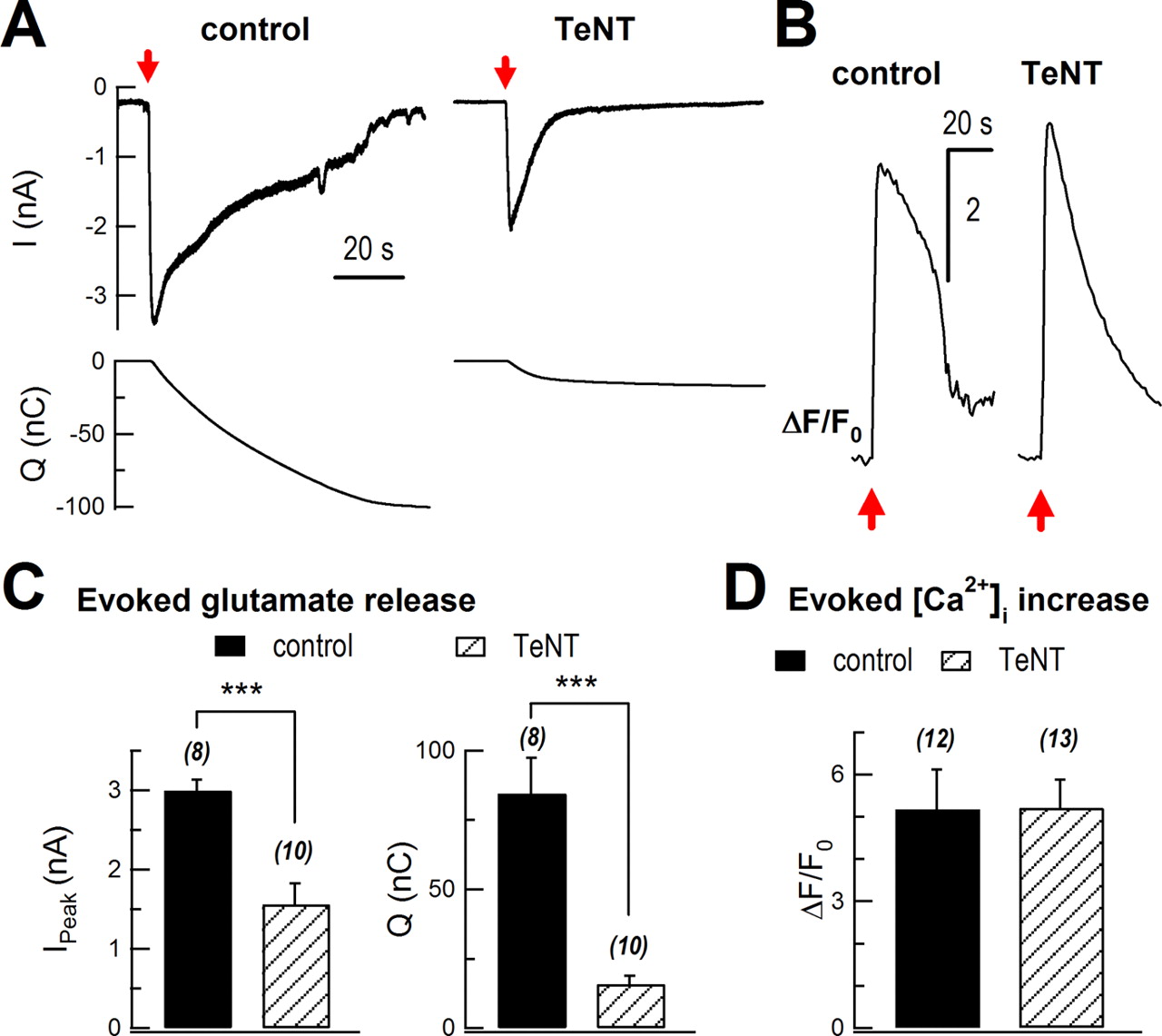
Next, we determined the effects of TeNT on glutamate release. Following a mechanical stimulation, glutamate release was greatly attenuated in TeNT-expressing cells (Fig. 7A). The peak of sniffer current was inhibited from 3.0 ± 0.1 nA to 1.6 ± 0.3 nA (control, n = 8 cells; TeNT, n = 10 cells; p < 0.001). Integrated charge of glutamate sniffer current was markedly inhibited by 81% (84 ± 13 nC vs 16 ± 3 nC; p < 0.001; Fig. 7C), while the evoked [Ca2+]i rise remained similar (5.2 ± 0.9 vs 5.2 ± 0.7; control, n = 12 cells; TeNT, n = 13 cells; p > 0.05; Fig. 7B,D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TeNT inhibited evoked glutamate release. A, Sniffer current recordings of evoked glutamate release in control (AsRed2-transfected) and TeNT-treated (cotransfection of AsRed and TeNT) cultured astrocytes. Sniffer recordings were performed 72 h after transfection. The top traces show mechanical stimulation-induced glutamate currents in HEK293 cells, while the bottom traces show integration of the upper currents. Red arrows mark mechanical stimulation. B, Representative traces of mechanical stimulation-induced intracellular Ca2+ rise in control and TeNT-treated astrocytes. Intracellular Ca2+ was measured with Fluo-4 AM. C, Statistics of peak of sniffer current (Ipeak) and total charge transfer (Q, integrated signal). D, Statistics of mechanical stimulation-induced intracellular Ca2+ rise. **p < 0.01, ***p < 0.001; Student's t test. Bars depict mean ± SEM.
Together, our results showed that VGLUT, Syb2, and cellubrevin, which selectively localized to small vesicles, were necessary for Ca2+-dependent glutamate release in cultured astrocytes.
Discussion
In astrocytes, the types of vesicles used in Ca2+-regulated exocytosis are under debate (Bezzi et al., 2004; Li et al., 2008; Marchaland et al., 2008; Parpura and Zorec, 2010). Combining TIRFM with glutamate sniffer recording, we found that astrocytes efficiently released two types of Ca2+-dependent vesicles, small vesicles and lysosomes, following a single stimulation. The small vesicles in astrocytes responded to [Ca2+]i elevation faster and more frequently than the lysosomes. They were also necessary for glutamate release in astrocytes.
Immunocytochemistry in cultured and freshly isolated astrocytes showed that both small vesicles, which were Syb2-, cellubrevin- and VGLUT1-positive, and lysosomes, which were LAMP1- and FM 1–43-positive, existed in the same astrocytes (Figs. 1⇑–3). In TIRFM experiments, we visualized small vesicles with either synapto-pHluorin (Syb2-pHluorin) or Syb2-mOrange2, and lysosomes with EGFP-LAMP1. They both underwent robust exocytosis upon a single stimulation that increased [Ca2+]i (Fig. 4). Separate TIRFM imaging of small vesicles and lysosomes, and dual-color imaging of both vesicles, gave consistent results in vesicle fusion kinetics. Altogether, there were 1–2 times more small vesicle fusion events than lysosomal events (Fig. 4B,F, top graph) and exocytosis of small vesicles was ∼1.5 times faster than that of lysosomes (Fig. 4C,F, bottom graph).
When FM1-43 staining was performed (Fig. 3), only lysosomes were labeled, consistent with previous reports (Zhang et al., 2007; Li et al., 2008). Although we observed a more frequent fusion of small vesicles with TIRFM, FM1-43 failed to label this type of vesicle in astrocytes. The apparent staining difference in astrocytes and neurons remains to be resolved.
Glutamate release from astrocytes regulates neuronal activity in situ (Fiacco and McCarthy, 2004; Jourdain et al., 2007). Multiple pathways of vesicular and nonvesicular release of glutamate have been proposed in astrocytes (Anderson and Swanson, 2000; Duan et al., 2003; Ye et al., 2003; Bezzi et al., 2004; Chen et al., 2005; Kimelberg et al., 2006). To study the mechanism of glutamate release in astrocytes, we monitored it with the high-affinity glutamate receptor GluR1-L497Y expressed in HEK293 cells as a sensitive biosensor (Stern-Bach et al., 1998; Lee et al., 2007). Glutamate is pumped into vesicles by VGLUT. Both trypan blue and rose bengal, specific VGLUT blockers, inhibited mechanical stimulation-induced glutamate release (Fig. 5B–I) (Ogita et al., 2001; Montana et al., 2004). In astrocytes, VGLUTs are known to localize in small vesicles (Fig. 5A) (Bezzi et al., 2004). Furthermore, we detected no VGLUT1 on lysosomes (Fig. 5A), implying that glutamate was mainly stored in small vesicles. Similarly, selective cleavage of Syb2 and cellubrevin with TeNT greatly inhibited the evoked glutamate release (Fig. 7), while lysosome morphology and fusion remained intact under TIRFM (Fig. 6B–D). Note that a small evoked signal of rapid glutamate release remained even after trypan blue, rose bengal, or TeNT treatment (Figs. 5B,C, 7A). This residual glutamate release might come from nonvesicular pathway(s), such as anion channels (Kimelberg et al., 2006), hemichannels (Ye et al., 2003), P2X7 channels (Duan et al., 2003) or reversed glutamate transporters (Anderson and Swanson, 2000). Since the residual glutamate only accounted for ∼15–20% of total glutamate release, vesicular small vesicles play the major role in astrocyte glutamate release.
Neuronal terminals mainly use synaptic vesicles to release neurotransmitters and the molecular mechanism of synaptic vesicle fusion is complex (Zhai and Bellen, 2004; Groffen et al., 2010; Pang and Südhof, 2010; Yang et al., 2010; Vyleta and Smith, 2011). It is interesting that small vesicles in astrocytes are similar to synaptic vesicles: they both express Syb2 and VGLUT and are of the same size under the electron microscope (20–30 nm) (Bezzi et al., 2004; Sudhof, 2004; Marchaland et al., 2008). Future studies should identify the molecular mechanisms of small vesicle and lysosome exocytosis in astrocytes.
In this study, we directly visualized two types of Ca2+-dependent vesicles, small vesicles and lysosomes, which underwent regulated exocytosis after a single stimulation in the same astrocyte. While Ca2+ regulated the fusion of both types, small vesicles fused at higher frequency and shorter latency than lysosomes. This provides a novel insight into mechanisms of vesicular exocytosis in astrocytes. Previously, lysosomes were shown to store ATP and release it when they fuse (Zhang et al., 2007). On the other hand, the Syb2-labled small vesicles play an essential role in glutamate release (Bezzi et al., 2004; Marchaland et al., 2008) (Figs. 5⇑–7). Gliotransmitter release from astrocytes can be triggered by GPCR agonists (Bowser and Khakh, 2007; Xu et al., 2007; Marchaland et al., 2008) in addition to direct elevation of intracellular calcium levels by mechanical stimulation and the calcium ionophore. It is interesting that activation of different GPCRs in astrocytes favors different forms of small vesicle exocytosis (Bowser and Khakh, 2007). Future studies will aim to understand whether different types of stimulation preferentially induce fusion from one or both compartments, and to determine whether exocytosis of small vesicles and lysosomes play specific roles in brain function in vivo.
Footnotes
- Received December 8, 2010.
- Revision received May 5, 2011.
- Accepted June 1, 2011.
This work was supported by grants from the National Basic Research Program of China (2006CB500800, 2007CB512100) and the Natural Science Foundation of China (NSFC) (30728009, 30970660, 30770521, 30770788, and 30830043), PKU-THU Joint Center for Life Science, and A*STAR Biomedical Research Council. We thank Drs. Tao Xu (IBP, China), Lu Yang (IBP), Gero Miesenböck (Oxford University), Haoxing Xu (University of Michigan), Jeffrey D. Erickson (LSU Health Sciences Center), and Stephen F. Traynelis (Emory University) for gifts of valuable reagents; Liangyi Chen (IBP) and Lin Liu (IBP) for technical assistance in dual-color TIRFM; and Iain C. Bruce (Zhejiang University) for comments on this manuscript.
- Correspondence should be addressed to any one of the following: Dr. Claire Xi Zhang or Dr. Zhuan Zhou, Institute of Molecular Medicine, Peking University, 5 Yiheyuan Road, Beijing 100871, China, zzhou{at}pku.edu.cn or clairexz{at}pku.edu.cn; or Dr. Weiping Han, Laboratory of Metabolic Medicine, Singapore Bioimaging Consortium, Agency for Science, Technology, and Research, Singapore 138667, E-mail: weiping_han{at}sbic.a-star.edu.sg
- Copyright © 2011 the authors 0270-6474/11/3110593-09$15.00/0