
Abstract
During the development of the vertebrate nervous system, neural progenitors divide, generate progeny that exit mitosis, and then migrate to sites where they elaborate specific morphologies and synaptic connections. Mitotic exit in neurons is accompanied by an essential switch in ATP-dependent chromatin regulatory complexes from the neural progenitor Brg/Brm-associated factor (npBAF) to neuron-specific nBAF complexes that is in part driven by miR-9/9* and miR-124. Recapitulating this microRNA/chromatin switch in fibroblasts leads to their direct conversion to neurons. We have defined the kinetics of neuron-specific BAF complex assembly in the formation of induced neurons from mouse embryonic stem cells, human fibroblasts, and normal mouse neural differentiation and, using proteomic analysis, found that this switch also includes the removal of SS18 and its replacement by CREST at mitotic exit. We found that switching of chromatin remodeling mechanisms is highly correlated with a broad switch in the use of neurogenic transcription factors. Knock-down of SS18 in neural stem cells causes cell-cycle exit and failure to self-renew, whereas continued expression of SS18 in neurons blocks dendritic outgrowth, underlining the importance of subunit switching. Because dominant mutations in BAF subunits underlie widely different human neurologic diseases arising in different neuronal types, our studies suggest that the characteristics of these diseases must be interpreted in the context of the different BAF assemblies in neurons rather than a singular mammalian SWItch/sucrose nonfermentable (mSWI/SNF) complex.
Introduction
Combinatorics frequently underlie biologic specificity. Examples include the production of proteins from the combinatorial assembly of 20 aa and perhaps most remarkably the connectivity of the nervous system. Specific patterns of chromatin regulation can also be produced by combinatorial assembly of subunits of chromatin regulatory complexes. The first examples of this were the family of mammalian SWItch/sucrose nonfermentable (mSWI/SNF) or Brg/Brm-associated factor (BAF) complexes (Wang et al., 1996a; Wang et al., 1996b; Lemon et al., 2001; Lessard et al., 2007), which are made up of ∼15 subunits now known to be encoded by 28 genes (Lessard et al., 2007; Wu et al., 2009) and this study). These complexes are assembled from homologous subunits encoded by gene families in much the same way that letters are assembled into words to produce specific meanings. For specific complexes to have specific biological meanings, the subunits must not be readily exchanged, because this would produce a reduction in meaning in the same way that indiscriminately switching the “W” in the word “NOW” for “T” would produce confusion. Accordingly, BAF complexes appear to be assembled with nonexchangeable, dedicated subunits (Zhao et al., 1998). Perhaps the most distinctive and specific of these chromatin regulatory complexes is found in the mammalian nervous system, where neuron-specific BAF (nBAF) complexes are distinguished by nonexchangeable subunits apparently dedicated to these complexes: BAF53b, BAF45b, and BAF45c (Lessard et al., 2007).
Recent evidence suggests that the subunit composition of BAF complexes might have an instructive developmental role. In the nervous system, the transition from the neural progenitor BAF (npBAF) complexes in neural progenitors to nBAF complexes in neurons is controlled by two microRNAs, miR-9/9* and miR-124. These microRNAs bind to the 3′ untranslated region of BAF53a, repressing its expression and making way for substitution with the neuron-specific BAF53b (Yoo et al., 2009). Recapitulation of this microRNA/chromatin switch in human fibroblasts results in their conversion to functional neurons with as few as just one neurogenic factor, NeuroD2 (Yoo et al., 2011).
The possible instructive role of nBAF complexes is further supported by studies of human neurologic diseases. Recently, de novo mutations in BAF subunits have been identified in a number of neurological diseases. Five BAF subunits, BRG, BAF250a (ARID1a), BAF250b (ARID1b), BAF57, and BAF47, were linked to Coffin-Siris Syndrome (CSS), a sporadic intellectual disability syndrome (Santen et al., 2012; Tsurusaki et al., 2012). Eighty-seven percent of CSS patients in these studies had a mutation in a BAF subunit, underscoring how mutations in genes from the same protein complex can have similar clinical presentations. In addition, BRM, one of the two ATPases of BAF complexes, was found to be mutated in 36 of 44 (82%) cases of Nicolaides-Baraitser syndrome (NBS), which is characterized by mild to severe mental retardation, specific language difficulties, and microcephaly (Van Houdt et al., 2012). In addition, mutations in BAF250b are common in sporadic mental retardation (Halgren et al., 2012; Hoyer et al., 2012). Population genetic studies have also implicated BRM and BAF250b in schizophrenia (Koga et al., 2009; Loe-Mie et al., 2010) and exome sequencing of autistic individuals revealed mutations in BAF155, BAF170, BAF180, BAF250b, and REST (Neale et al., 2012; O'Roak et al., 2012), the latter of which controls miR-9/9* and miR-124 (Conaco et al., 2006). All of these mutations are dominant, perhaps paralleling the dominant role of Brg and BAF155 in neural development (Bultman et al., 2000; Kim et al., 2001).
nBAF chromatin-remodeling complexes have an evolutionary conserved role in dendrite morphogenesis. Drosophila BAP55, BAP60, Snr1, and Brm, the fly homologs of BAF53b, BAF60c, BAF47, and BRM, respectively, are required for routing of dendrites in the peripheral nervous system (Parrish et al., 2006). Furthermore, mutation of BAP55 causes perfect retargeting of CNS olfactory projection neuron dendrites from one glomerulus to another (Tea and Luo, 2011), a phenotype that was rescued by human BAF53b. BAF53b-null mice have impaired activity-dependent dendritic outgrowth (Wu et al., 2007). These studies suggest that failures of dendritic morphogenesis or targeting could underlie human neurologic disorders characterized by mutation of nBAF subunits.
To understand the mechanistic details of BAF subunit switching during neuronal differentiation, we undertook a proteomic and kinetic analysis of BAF complexes as neural progenitors become neurons. We show that SS18 is replaced by its paralog, CREST, near mitotic exit. We have related the switching of subunits to the expression patterns of classic neurogenic and neuronal genes to establish the characteristics of the genetic circuitry used during both differentiation of embryonic stem (ES) cells to neurons and direct reprogramming of human fibroblasts into neurons.
Materials and Methods
Affinity purification and mass spectrometry.
Affinity purification and mass spectrometry of endogenous BAF complexes were performed as described previously (Lessard et al., 2007; Ho et al., 2009b).
Preparation of ES cell and brain nuclear extracts.
Mouse CD1 embryonic brains of either sex were dissected in cold HBSS, transferred to cold hypotonic lysis buffer containing the following: 25 mm HEPES, pH 7.6, 5 mm MgCl2, 25 mm KCl, 0.05 mm EDTA, 10% glycerol, 0.1% NP40, 1 mm DTT, and protease inhibitors (complete mini tablets; Roche) supplemented with 1 mm PMSF, and homogenized with 10–15 strokes of a glass Dounce homogenizer. Nuclei were centrifuged at 800 × g for 3 min. Chromatin was precipitated in 10 mm HEPES, pH 7.6, 3 mm MgCl2, 100 mm KCl, 0.1 mm EDTA, 10% glycerol with 1:10 vol of 3 m ammonium sulfate, pH 7.9, for 30 min and centrifuged in an Ultracentrifuge at 100,000 rpm for 15 min. Proteins were then precipitated with 0.3 g/ml high-grade ammonium sulfate for 20 min and spun at 100,000 rpm for 15 min. Protein extracts were resuspended in immunoprecipitation buffer containing the following: 150 mm NaCl, 50 mm Tris-HCl, pH 8.0, 1 mm EDTA, 1% Triton X-100, and 1 mm DTT with protease inhibitors unless stated otherwise.
Glycerol gradient cosedimentation experiments.
For glycerol gradient sedimentation, embryonic day 10.5 (E10.5) or E18.5 brain nuclear extracts were diluted in 37.5 mm HEPES-KOH, pH 7.9, 90 mm KCl, 6.25 mm MgCl2, 0.05 mm EDTA, and 0.75 mm DTT. Approximately 500 μg (in 500 μl) of nuclear extract was applied to 10 ml 10–30% column gradients of glycerol in HEMG/100 buffer containing 100 mm KCl. Samples were centrifuged in a Beckman SW40Ti rotor at 32,000 rpm for 20 h at 4°C. Nineteen 500 μl fractions were collected from the top (no. 1) to the bottom (no. 19) of the gradients by hand. Fractions were separated by SDS-PAGE and analyzed by immunoblotting with anti-Brg, BAF250a, BAF170, BAF155, CREST, SS18, BAF53a, 53b, and Ezh2 specific antibodies.
Immunocytochemistry.
Cells were grown on glass coverslips coated with poly-ornithine, laminin, and fibronectin. Cells were fixed with 4% PFA, blocked, and permeabilized with 3% normal goat serum,5% BSA, 0.1% Triton X-100, and incubated with primary antibodies overnight, followed by goat anti-rabbit or mouse secondary antibodies (Invitrogen) and DAPI, washed with PBS, and mounted with 4% N-propyl gallate (Sigma) in 90% glycerol, 10% PBS and imaged on a Leica DM5000 fluorescent microscope.
Co-IP experiments.
Nuclear extracts were made as described above. Co-IP experiments were performed with 100–200 μg nuclear extracts precleared with either protein A or G beads (Sepharose or Dynabeads), incubated with primary antibodies at 4°C overnight, bound to protein A or G beads, washed 3 times, and interacting proteins eluted with 2× sample buffer and boiling (Fig. 2, Fig. 8) or 0.1 m glycine, pH 2.5 (Fig. 3), before SDS-PAGE and Western analysis.
Antibodies.
mouse anti-b-III tubulin (Tuj) (Covance, Western [W],1:30k; immunofluorescence [IF],1:20k), rabbit anti-BAF170 (39A Bethyl Laboratories, rabbit anti-BAF155 (-5 or -3-glycerol gradients; Crabtree Laboratory; W, 1:5k), mouse anti-Brg1-G7 (Santa Cruz Biotechnology; W, 1:2k), rabbit anti-SS18 (H80; Santa Cruz Biotechnology; W, 1:750; IF, 1:300), goat anti-CREST (M15; Santa Cruz Biotechnology; W, 1:1000; IF, 1:3k), CREST (Proteintech; IF, 1:2k), rabbit anti-BAF250a (H-90x SC98441; Santa Cruz Biotechnology; W, 1:3k), mouse anti-BAF250a (Santa Cruz Biotechnology; W, 1:1k), mouse anti-BAF250b (SC32762; Santa Cruz Biotechnology; W, 1:6k), mouse anti-BRM (Santa Cruz Biotechnology; W, 1:2k), rabbit anti-BAF53a (Novus; W, 1:5k; IF, 1:2k), rabbit anti-53b (fraction 2–3, BAF53a depleted; Crabtree laboratory; W, 1:250; IF, 1:250), GFAP (MAB3402; Millipore; IF, 1:1k), nestin (BD Biosciences; IF, 1:2k), mouse anti-V5 (Invitrogen), DAPI, and Alexa Fluor 488, Alexa Fluor 568 (Invitrogen) secondary antibodies (IF, 1:1k). IRDye secondary antibodies included: goat anti-Mouse 800CW (926–32210; LiCOR), goat anti-rabbit 800CW (926–32211; LiCOR, ), goat anti-mouse 680 (926–32220; LiCOR), and goat ani-rabbit 680 (926–32221; LiCOR, ; W, 1:30k). Western blots were imaged using the LiCOR Odyssey infrared imager.
Cell conversion.
The conversion of mouse ES cells into glutamatergic pyramidal neurons followed essentially the protocol in Bibel et al. (2007). Conversion of human fibroblasts into neurons followed the protocol in Yoo et al. (2011).
Quantitative RT-PCR.
For quantitative RT-PCR (qRT-PCR), total RNA was isolated using TRIzol reagent (Invitrogen). One microgram of total RNA was treated with DNase I to remove potential contamination of genomic DNA. One microgram of DNase I-treated RNA was reverse transcribed using a First Strand Synthesis kit (Invitrogen) and ultimately resuspended in 900 μl of water. qRT-PCR analysis was performed in duplicate using 1/100 of the reverse transcription reaction in an ABI Prism 7000 (Applied Biosystems) with FastStart Universal SYBR green Master Mix with ROX (Roche). Data were extracted from the linear range of amplification. All graphs of qRT-PCR data shown represent samples of RNA that were DNase treated, reverse transcribed, and amplified in parallel to avoid variation inherent in these procedures. The housekeeping gene Ppia was used for sample cDNA normalization because it changed the least across all data points of the time course. Error bars represent SEM of three biological replicates.
Primary neuron cell culture.
mouse E18.5 cortical neurons of either sex were isolated using the MACSTM neuron isolation kit. Dissociated cells were transfected with GFP or other constructs using Nucleofector (Amaxa shuttle) before plating on poly-l-ornithine, laminin-, and fibronectin-coated coverslips. Culture media contained DMEM/F12 with putrescine, 2-mercaptoethanol, transferrin, insulin, selenium, progesterone, MEM vitamin additive, and 5% FBS.
Dendrite outgrowth experiments.
Mus musculus SS18, Ascension number BC096742 and Mus musculus CREST ascension number BC053087 cDNA were ordered from OpenBioSystems and subcloned into the pCIG-IRES-eGFP plasmid. Then, 25 ng of the respective plasmids mixed with 475 ng pCIG-IRES-eGFP plasmid was transfected into 250,000 dissociated E18.5 mouse cortical neurons of either sex using Nucleofector (AMAXA shuttle) and plated onto coated coverslips. The neural cultures were grown for 6 d with or without 30 mm KCl for the last day. To define dendrite structure, cultures were fixed with 4% PFA and stained with anti-GFP (Invitrogen), anti-MAP2 (Sigma), and Alexa Fluor 488 and Alexa Fluor 568 (Invitrogen) secondary antibodies (1:1000). Dendrite analysis was done on pictures of GFP+/MAP2+ neurons using ImageJ and NeuronJ software.
Results
SS18 and CREST are mutually exclusive npBAF and nBAF subunits
An unbiased proteomic comparison was performed with murine ES cells, mouse embryonic fibroblasts (MEFs) and newborn (P0) brain extracts. BAF complexes were affinity purified using a high-affinity, highly specific Brg/Brm antibody (J1) that recognizes an epitope without complex disruption (Khavari et al., 1993). Brg/Brm-associated proteins were subjected to proteolytic digestion, analyzed by multidimensional liquid chromatography electrospray ionization-tandem mass spectrometry, and identified using SEQUEST (Yates et al., 1995) to determine the identity and relative abundance of each protein in the BAF complexes (Ranish et al., 2003).
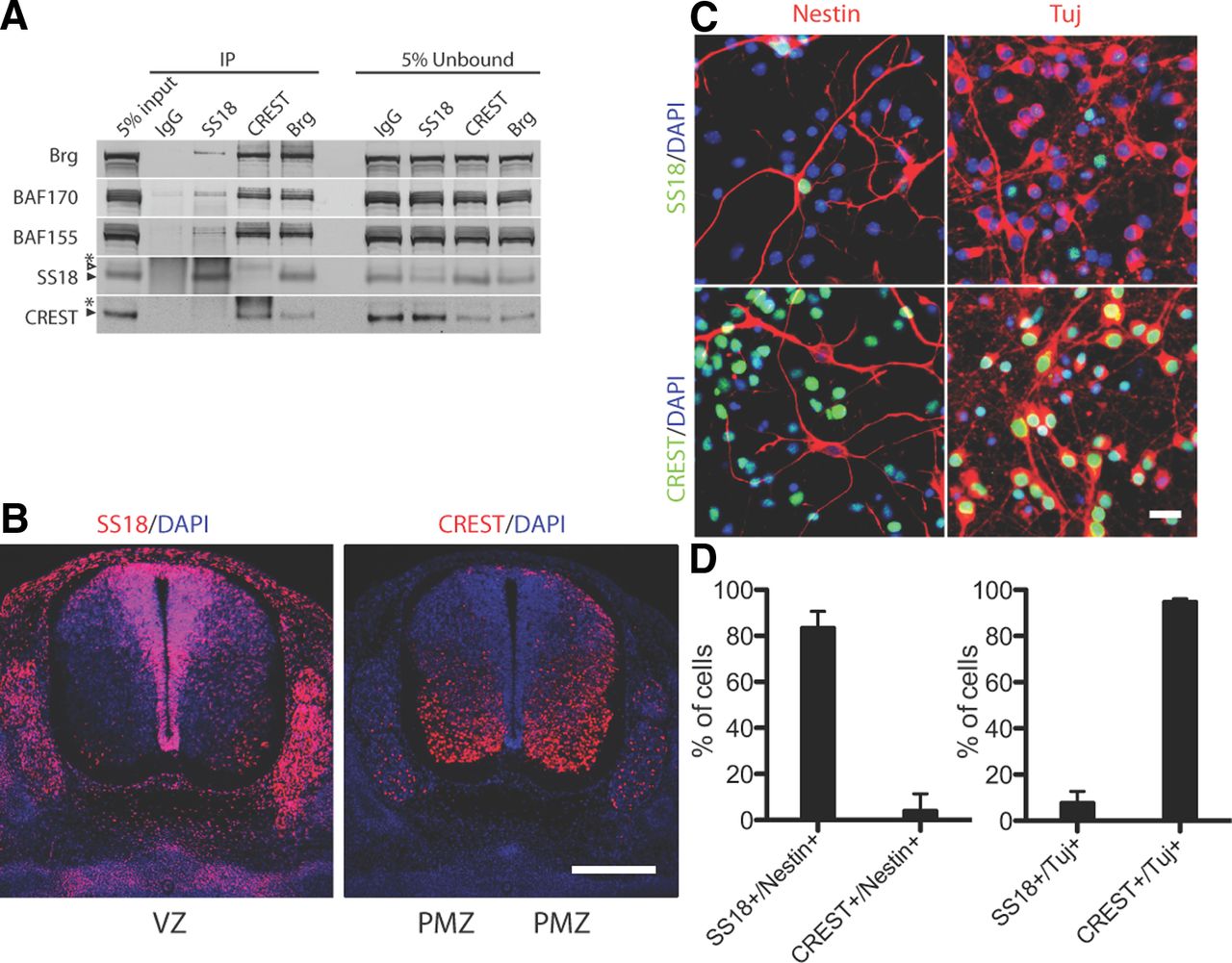
This analysis identified the paralogs SS18 (aka SYT) and CREST (SS18L1) (62% identical) as Brg/Brm-interacting proteins, which is in agreement with previous reports (Wu et al., 2007; Qiu and Ghosh, 2008; Middeljans et al., 2012). We identified unique SS18 peptides for ES cells (28 peptides, 39.3% coverage), MEFs (7 peptides, 13.7% coverage), but not for P0 brain nuclear extracts. In contrast, CREST peptides were most abundant in P0 brain (Fig. 1A), consistent with the expression pattern of CREST described previously (Aizawa et al., 2004). The reciprocal pattern of the recovered peptide abundance also suggested a switch in SS18/CREST expression during neural differentiation, in which SS18 is expressed in ES cells and MEFs whereas CREST is expressed in neurons.

Identification of novel BAF subunits SS18 and CREST. A, Table of mass-spectrometric analysis of proteins co-purified with Brg/Brm from mouse ES cells, MEFs, and newborn P0 whole brain. Shown is the percent coverage based on unique peptides. B, Glycerol gradient (10–30%) analysis of E10.5 and E18.5 mouse brain cortical nuclear extracts. SS18 but not CREST cosediments with npBAF complexes at 2MDa in E10.5 mouse brain nuclear extracts, whereas CREST cosediments with nBAF complexes in E18.5 mouse brain nuclear extracts. The Polycomb PRC2 complex protein Ezh2 was blotted as a control and peaks in ∼1 MDa fractions. *From other studies, these proteins are not found in pure undifferentiated ES cells nor MEFs. The presence of these proteins in ES cells could be from spontaneously differentiating cells in the ES cultures and neural cells in the MEFs. Glycerol gradients are representative of two separate protein purifications from each developmental time point.
Because specific assemblies of subunits provide the biochemical underpinnings for biologic specificity, we determined whether SS18 and CREST were dedicated BAF subunits by examining their sedimentation on glycerol gradients (Fig. 1B). This analysis is useful for differentiating between interacting proteins that might exchange quickly and subunits that are structurally part of a protein complex. This is an important functional point because it defines the potential for new biologic meaning to emerge from different complex assemblies. In E10.5 cortical preparations (composed of >95% Pax-6+, BAF53a+ neural progenitors; data not shown), SS18 sedimentation is in fractions 15 and 16 with 2 MegaDalton markers. Brg/Brm, the core ATPases of these BAF complexes, and the subunits BAF250a (Arid1a), BAF250b (Arid1b), BAF170, BAF155, and BAF53a also cosediment in these fractions. There was no SS18 in the earlier fractions in which monomeric proteins would sediment. Polybromo-1 (PBrm/BAF180) sediments primarily in a distinct higher molecular weight complex termed PBAF. CREST was not present in E10.5 cortical preparations nor was BAF53b. In contrast, E18.5 cortical preparations composed of 75% postmitotic, BAF53b+ neurons (data not shown), CREST, and BAF53b cosedimented exclusively with Brg. The appearance of low levels of SS18 in the E18.5 gradients was due to neural progenitors and glia present at E18.5. Therefore, during the course of neuronal development, CREST is activated and incorporated into nBAF complexes. These studies indicate that SS18 and CREST are dedicated to the 2 MDa Brg/Brm-based BAF complexes.
One could imagine that SS18 and CREST can heterodimerize and are both present within a single complex or that there is a sterically specific concavity within BAF complexes that is occupied by either SS18 or CREST—but never both. To test these possibilities, we conducted co-IP experiments with antibodies against SS18, CREST, or Brg in mouse E15.5 cortical nuclear extracts. Because the E15.5 mouse cortex contains a mixture of neural progenitors and neurons, this experiment tests both the mutual exclusivity of the subunits and the exchangeability of subunits. Antibodies to SS18, CREST, and Brg co-IP the core BAF subunits Brg, BAF155, and BAF170. Only antibodies to Brg co-IP both SS18 and CREST, whereas antibodies to SS18 co-IP SS18 but not CREST and antibodies to CREST co-IP CREST but not SS18 (Fig. 2A). This failure to co-IP homologous subunits indicates that SS18 and CREST are mutually exclusive BAF subunits, with few if any complexes having both SS18 and CREST. These data also suggest the subunits are not easily exchangeable between complexes in vitro.

SS18 and CREST are mutually exclusive, nonexchangeable subunits of npBAF and nBAF complexes, respectively. A, Co-IP from E15.5 brain cortical extracts; Brg co-IPs SS18 and CREST whereas CREST does not co-IP SS18 and SS18 does not co-IP CREST. Black arrowheads mark the SS18 and CREST proteins. Empty arrowhead marks SS18 Ab cross-reacting with CREST (CREST runs above SS18 on these Western blots). *Immunoglobin heavy chain. B, Immunostaining on cross sections of E12.5 spinal cords. SS18 is expressed specifically in neural progenitors in ventricular zone (VZ) and is replaced by the homologous subunit CREST in postmitotic neurons in the postmitotic zone (PMZ). Scale bar, 250 μm. C, Mutual exclusive expression pattern of SS18 and CREST as determined by costaining primary mouse E15.5 cortical cells cultured for 4 d in vitro with neural markers. SS18 is expressed in Nestin+ NS/progenitors, whereas CREST is specifically expressed in Tuj+ neurons. Scale bar, 20 μm. D, Quantification of staining in C. Data are ± SD of three fields of view each, with ∼100 cells each field.
To determine whether the mutually exclusive assembly was due to distinct patterns of expression, we determined the cellular localization and the cell-type specificity of SS18 and CREST. In the developing spinal cord between E10.5 and E16.5, SS18 was specifically expressed in the proliferating neural progenitors of the ventricular zone. In differentiated neurons of the postmitotic zone, SS18 was repressed and CREST was expressed (Fig. 2B). In primary isolated cortical neural cultures, SS18 was present in >75% of Nestin+ neural progenitors and <5% of β-tubulin III+ (Tuj+) neurons. Conversely, CREST was exclusively dedicated to Tuj+ neurons (Fig. 2C,D). Therefore, we conclude that SS18 is an es/npBAF subunit and CREST is an nBAF subunit, and different cell types are characterized by specific BAF complexes that have the potential for unique biologic functions.
SS18 and CREST are subunits of the npBAF and nBAF complexes, respectively
To define the stability of SS18 and CREST incorporation into npBAF and nBAF complexes, respectively, we determined their resistance to increasing concentrations of the denaturant urea. Urea will denature most proteins at ∼5 m and will partially denature many proteins at lower concentrations. Therefore, the sensitivity of a protein–protein interaction to urea is a measure of the degree to which the interaction depends on the native state of the protein. Interactions that survive high concentrations of urea are often found to reflect cofolding within complexes such as the ribosome (Petermann and Pavlovec, 1971) and are useful in differentiating a subunit from an associated, exchangeable protein. BAF complex core subunits remain associated with Brg >1 m urea (core subunits also remain associated under highly stringent IP conditions of 0.3 m NaCl, 1% NP40, 0.5% deoxycholate, and 0.1% SDS; Wang et al., 1996a; Zhao et al., 1998; Lessard et al., 2007). We treated ES cells (Fig. 3A), E10.5 neural progenitors, E18.5 neurons (Fig. 3B), and P1.5 cortical cell (Fig. 3C) nuclear extracts with increasing amounts of urea for 10 min at room temperature before IP with an antibody to Brg. The bona fide core BAF subunits BAF155 and BAF170 remained associated with Brg up to 2.5 m urea, whereas SS18 and CREST interactions survived up to 5 m urea, indicating extensive and stable protein–protein interactions that are unlikely to turn over quickly supporting the SS18 and CREST co-IP results.

SS18 and CREST remain associated with Brg up to 5 m urea. A, Denaturation analysis of BAF complexes in ES cells. SS18 and BAF53a remain associated with Brg at 5 m urea, whereas BAF155 comes off in 2.5 m urea. Nuclear extracts were prepared from E14 ES cells as described in the text. Nuclear extracts were subjected to different concentrations of urea for 10 min at 25°C before IP, diluted 10× with IP buffer, incubated with anti-Brg (G7) antibodies overnight at 4C. Brg co-IP proteins were isolated, electrophoresed, and probed with antibodies to the labeled proteins. B, Denaturation analysis of npBAF complexes in E10.5 neural progenitors or E18.5 neurons. npBAF containing SS18 in E10.5 nuclear extracts switches to nBAF containing CREST in E18.5 neurons. SS18 remains associated with Brg to 5 m urea in neural progenitors, whereas CREST remains associated with Brg to 5 m urea in neurons. No CREST is detected at E10.5 and very little SS18 is detected at E18.5. TBP is blotted as a loading control. C, Denaturation analysis of nBAF complexes in P1.5 brain extracts. CREST remains associated with Brg up to 5 m urea, whereas BAF170 comes off at 2.5 m urea. Western blots are representative of three separate protein purifications from the respective cell types.
These studies indicate that the two subunits are likely to have interlocking interactions dependent upon their native folded state within BAF complexes and are not dependent upon BAF155 or BAF170 to be bound to the complexes. They also indicate that the SS18 and CREST subunits within BAF complexes have approximately the same stability as the subunits of ribosomes.
Ex vivo production of neurons also involves substitution of SS18-containing npBAF complexes for CREST-containing nBAF complexes
To further define the coregulation of SS18/CREST with other npBAF and nBAF subunits during the course of neural differentiation, we set up an ex vivo neural differentiation system that converts ES cells into glutamatergic neurons (Bibel et al., 2007). In this ES-to-neuron system, cells differentiate into Nestin+/Pax6+, radial glial cell-like neural progenitor cells, when treated with retinoic acid (RA; Fig. 4A). As they did in vivo, these neural progenitors differentiated into neurons that form functional synaptic contacts. We used this system to visualize the kinetics of npBAF repression and nBAF activation with antibodies to BAF subunits, the neural progenitor marker Nestin, or the neuron marker Tuj (Fig. 4B). We determined the timing of mitotic exit by 5-ethynyl-2′-deoxyuridine (EdU) pulsing to be between day 9 and day 10 (Fig. 4C). Interestingly, this time point corresponds to the switch from npBAF subunits to nBAF subunits. At this time, expression of npBAF subunits (SS18, BAF45a, BAF53a) was repressed as the nBAF subunits (CREST, BAF45b/c, BAF53b) were activated. However, we noted a low level of expression of BAF45c in neural progenitors, the appearance of which was earlier than that of CREST, BAF45b, or BAF53b.

Kinetics of induction and loss of nBAF and npBAF subunits after neural induction of ES cells. A, Schematic of neural induction of ex vivo ES cell-to-neuron differentiation time course. Open arrowhead denotes cell aggregate dissociation and transfer of neural progenitors to neural differentiation media and poly-ornithine-, laminin-, and fibronectin-coated plates. B, SS18 protein intensity decreases when cells differentiate into Tuj+ neurons. CREST is first detected on day 9 in Tuj+ neurons with rapidly increasing intensity as the cells become more mature neurons. BAF53a/45a decrease in neurons and BAF53b/45b are activated only in Tuj+ neurons. BAF53a colocalizes with Nestin+ NS/progenitor cells. BAF45c is activated before BAF45b and before cells becoming Tuj+. C, Proliferation measured by EdU incorporation during the switch from Nestin+ NS/progenitors to Tuj+ neurons. Cell cycle exit corresponds with the timing of the npBAF to nBAF switch. Cells were pulsed with EdU for 2 h before being fixed. Data are shown ± SEM. Images and EdU staining are representative of three separate ex vivo differentiations.
To further examine the nature of the switching mechanism, we measured mRNA and protein levels during the switch. The npBAF to nBAF switch is rapid as nBAF subunits are robustly activated and npBAF subunits are repressed. In these induced neurons, SS18 transcription was reduced to 25% of maximum at day 10 and protein levels decreased significantly on day 11 (Fig. 5A,B). In this neuronal differentiation system, on day 12 among viable cells, 80 ± 3% were Tuj+ neurons, <1% were Nestin+ neural progenitors, and 19 ± 3% were non-neural lineage cells. The non-neural cells were SS18+ (data not shown). Therefore, the residual SS18 protein detected by the Western blots was from non-neural cells in the culture. In a reciprocal fashion to SS18, CREST was activated to 80% of maximum by day 10, with robust protein accumulation observed as the cells differentiated into neurons on day 10 (Fig. 5A,B). Because the SS18/CREST switch is similar to the BAF53a/53b switch, we wondered how this ES-to-neuron differentiation system recapitulated the essential genetic circuitry we defined previously for the miRNA/chromatin switch during neural development. At the top of this circuit, REST, which with its co-repressors restricts the activation of neuronal genes to neurons by silencing its target genes in non-neurons, is downregulated as neural progenitors differentiate into neurons (Chong et al., 1995; Schoenherr and Anderson, 1995; Chen et al., 1998; Lunyak et al., 2002; Kuwabara et al., 2004; Ballas et al., 2005). The REST target genes miR-9/9* and miR-124 are derepressed, which leads to repression of BAF53a and substitution with BAF53b during neuronal differentiation (Conaco et al., 2006; Yoo et al., 2009).

Kinetics of transcriptional induction and repression of nBAF and npBAF genes, miRNAs, cell-type marker genes, and neurogenic transcription factors after neural induction of ES cells. A, qRT-PCR analysis of mRNA and microRNAs during neural induction time course. Green panel denotes neural progenitor stage, red panel denotes neuron stage, and vertical line delineates time of cell cycle exit. B, Protein analysis by Western blot. BAF45c is the first nBAF subunit to be activated and BAF53b is the last to be activated. Data are representative of three separate ex vivo differentiations. C, BAF45c is activated independent of RA induction. BAF45b and BAF53b are not activated without RA induction. D, BAF250a, 250b, BAF60a, BAF60b, and BAF60c expression kinetics. qRT-PCR is presented as a percentage of the maximum transcript level ± SEM of three separate ex vivo differentiations.
In the ex vivo differentiation system, the neural progenitor genes Nestin and Pax6 were activated as cells passed through the neural progenitor stage. The proneural genes Neurogenin2 (Ngn2) and Ascl1 preceded the activation of the neural differentiation genes NeuroD2 and Myt1L, recapitulating their in vivo order of activation (Bertrand et al., 2002). REST levels were reduced at the mRNA and protein level 4 d after LIF removal. On day 6, as cells entered the Nestin+/Pax6+ neural progenitor phase 2 d after the addition of RA, REST mRNA was reduced to 25% of ES cell levels with marked reduction in REST protein. On day 7, the REST target genes miR-9/9* were derepressed and activated and, on day 8, miR-124 was activated. Therefore miR-124 activation lagged approximately 1 d behind miR-9/9*. The additional REST target genes BAF53b and BAF60c (Johnson et al., 2008) were activated on day 8 and 9, respectively, when REST transcript levels dropped <10% of maximum (Fig. 5A,B).
BAF53a repression was observed on day 10 as the miRNAs rapidly approached their maximal levels, reflecting the essential role of miR-9/9* and miR-124 for BAF53a repression. The miRNAs were robustly activated: miR-9/9* was upregulated ∼1000-fold and miR-124 was upregulated ∼100-fold. The reduction of BAF53a protein lagged behind its transcript kinetics, possibly because of the stability of this protein and the presence in non-neural cells in the culture. Interestingly, BAF45c was induced before miR-9/9* and miR-124 and was activated in the absence of RA, albeit not as robustly (Fig. 5C), indicating that the activation of BAF45c must be independent of RA induction and the expression of miR-9/9* and miR-124. miR-9/9*, miR-124, CREST, BAF45b, and BAF53b were not activated in the absence of RA induction (Fig. 5C and data not shown). Therefore, there are at least two upstream operators that bring about the npBAF to nBAF switch, one RA dependent and one RA independent. The switch to nBAF subunits occurs on day 10, as measured by transcript and protein (Fig. 5) corresponding to immunostaining and cell cycle exit data (Fig. 4). We conclude the ex vivo ES-to-neuron differentiation system recapitulates the sequence and timing of the in vivo microRNA/chromatin switch.
The dynamic regulation during ex vivo differentiation was determined for the BAF subunits BAF250a (Arid1a) and BAF250b (Arid1b), which have been genetically linked to mental retardation and autism (see Discussion). BAF250a was more abundant than BAF250b in ES cells. However, BAF250b mRNA was induced 25-fold in neurons relative to ES cells (Fig. 5D). BAF250b protein levels were 2× higher in E18.5 neurons relative to E10.5 neural progenitors (Fig. 3B, Western blot densitometry normalized to TBP levels). Therefore, BAF250b was transcriptionally activated during neuronal differentiation. In addition, BAF250b appeared to be regulated at the level of pre-mRNA splice variation as the relative intensities of the BAF250b bands changed from E10.5 neural progenitors to E18.5 neurons (Fig. 1B, Fig. 3B). BAF250b has 4 predicted splice variants (http://www.ensembl.org). Interestingly, the lowest-molecular-weight band was most tightly associated with BAF complexes because it remained associated with Brg in 5 m urea (Fig. 3B).
The ex vivo differentiation system also recapitulates previously observed expression patterns for BAF60a, BAF60b, and BAF60c. BAF60a and BAF60b were expressed in ES cells whereas BAF60c was not (Ho et al., 2009b). BAF60c was upregulated ∼300-fold in day 10 neurons relative to ES cells (Fig. 5B,D) in agreement with previously reported expression in brain and muscle (Wang et al., 1996b; Lickert et al., 2004). Conversely, BAF60b decreased with neural differentiation, consistent with previous results that BAF60b is reduced in cells of the neural lineage (Lessard et al., 2007).
Recently, we found that human fibroblasts can be induced to neurons with the addition of miR-9/9*and miR-124 and neurogenic factors (Yoo et al., 2011). During the conversion to induced neurons (iNs), BAF45a was replaced by BAF45b and BAF45c and BAF53a by BAF53b. We wondered whether SS18 and CREST also switched with the addition of miR-9/9*-124 and production of iN. Indeed, CREST was transcriptionally activated >10× in induced neurons, whereas SS18 was transcriptionally reduced 3× (Fig. 6A). The robustness of this switch is such that the level of CREST expression in the iN is similar to that found in an adult human brain. Therefore, CREST replaces SS18 during in vivo neural development, during ex vivo ES-to-neuron differentiation, and also during the conversion of human fibroblasts to neurons.

SS18 and CREST switch during artificial neuron development. Human fibroblasts were infected with lentivirus containing miR-9/9* and miR-124. Eighteen days later, mRNA was isolated. qRT-PCR analysis shows the switch from SS18-containing BAF complexes in fibroblasts to predominantly CREST-containing complexes after miR-9/9* and miR-124 addition to human fibroblasts. After the switch, the transcript profile is similar to that of an adult human brain sample. Data are shown ± SEM of three separate neuronal inductions. *p < 0.05, Student's t test.
SS18 is required for neural stem cell self-renewal
SS18 is a dosage-sensitive gene, being haploinsufficient on the C57bl6 background with a phenotype of embryonic lethality between E8.5 and E9.5 (De Bruijn, 2006). We were interested in the role of SS18 in neural stem (NS) cells, but also wondered whether there was a difference in the requirement for SS18 in ES cells compared with NS cells because SS18-null mice develop well past the peri-implantation stage. Therefore, we hypothesized that NS cells are more sensitive to reduction in SS18 levels than ES cells. To test the role of SS18 in ES and NS cell self-renewal, we used shRNA knock-down delivered by lentiviral vectors to ES cells and primary neurospheres (NS cells).
Three shRNAs against SS18 were tested, reducing SS18 to 79 ± 4% to 20 ± 1% of the wild-type (WT) protein level. In ES cells, shSS18 knock-down compared with shScramble control resulted in a cell-cycle progression phenotype with reduced S-phase entry and reduced progression from the G2 to the M phase (Fig. 7A). These reductions in SS18 protein reduced the ES cell proliferation rates, but were compatible with ES cell culture viability, because the ES cell cultures continued to proliferate for at least two passages (Fig. 7B). In NS cells, knock-down of SS18 to 79 ± 4% of WT protein levels resulted in a cell-cycle progression phenotype with reduced S-phase entry (26.8% WT versus 14.2% SS18 knock-down) and reduced progression from the G2 to the M phase (2.4% in G2 for WT vs 8.5% in G2 for SS18 knock-down; Fig. 7C). A measure of NS cell self-renewal is the ability to form secondary neurospheres after dissociation to single cells. shScramble-treated NS cells formed robust secondary neurospheres, but shSS18-treated NS cells failed to form secondary neurospheres (Fig. 7D). Therefore, in ES cells, SS18 protein could be reduced 80% and still proliferate, albeit more slowly. However, NS cells were extremely SS18-dosage sensitive, with ∼25% reduction causing cell-cycle phenotypes and inhibiting NS cell self-renewal. We conclude that NS cells are more sensitive than ES cells to reduced SS18 protein levels, and that this selective sensitivity is consistent with the pathogenesis of several human neurologic diseases and syndromes, including microcephaly, resulting from dominant haploinsufficient mutations in npBAF complexes.

SS18 knock-down causes a G2-M-phase block and reduced rate of S-phase entry in ES and NS cells and results in failure of NS cells to self-renew. A, ES cell cycle analysis. Bottom left, G1; top, S-phase (DNA synthesis); bottom right, G2-mitosis. Lentivirus infection with shScramble shSS18#1. shSS18 knock-down causes G2-M-phase block and slower progression into the S-phase. SS18 protein quantification of ES cells. Data are representative of three separate experiments done in duplicate. B, Reduction of SS18 protein with shSS18 slows proliferation rate of ES cells. C, Neurosphere NS cell-cycle analysis. Infection with shSS18#1 caused a G2-M block and reduced S-phase entry. Shown is SS18 protein quantification of NS cells. The SS18 protein level was reduced ∼25%, indicating SS18 haploinsufficiency. Data are shown ± SEM. D, Secondary neurosphere formation is blocked by SS18 knock-down, indicating failure of NS cell self-renewal. Neurosphere NS cell analysis is representative of two separate experiments done in duplicate. Scale bar, 100 μm.
Proper combinatorial assembly of nBAF complexes with CREST is essential for dendritic outgrowth in neurons
Both BAF53b and CREST have been shown to be required for activity-dependent dendritic growth (Aizawa et al., 2004; Wu et al., 2007). We hypothesized that SS18 is transcriptionally repressed at mitotic exit to make way for CREST incorporation into nBAF complexes, where CREST performs a unique activity that cannot be replaced by SS18. To test this hypothesis, we forced SS18 expression in primary cortical neurons and examined whether dendritic growth was compromised. Depolarization with KCl induced a robust increase in total dendritic length in vector and CREST-transfected neurons, whereas SS18-transfected neurons had a significant reduction in activity-dependent dendritic growth and branch number increase (Fig. 8A–C). Exogenous SS18 and CREST protein incorporated into BAF complexes (Fig. 8D) and were exclusively nuclear in the primary cortical neurons (Fig. 8E). Forced expression of SS18 decreased endogenous CREST protein to 50% of control levels in depolarized cortical neurons, suggesting that exogenous SS18 competes with CREST for incorporation into nBAF complexes (Fig. 8D,E). These data indicate that extended expression of SS18 past its normal repression at mitotic exit leads to failure of dendritic outgrowth and that SS18 and CREST are not functionally redundant as subunits in cell-type specific BAF complexes. The data also indicate that producing confused nBAF complexes with SS18 reduces the biological specificity of the nBAF composite surface and compromises the neuron's ability to respond productively to depolarization.

SS18 expression prevents KCl-induced dendritic outgrowth. A, Total dendrite length. Primary E18.5 cortical neurons were isolated, transfected with iresGFP, SS18-iresGFP, or CREST-iresGFP where indicated, cultured for 6 d with 30 mm KCl for the last day where indicated. Control vector and CREST overexpression do not affect dendrite outgrowth in response to KCl depolarization. SS18 expression reduces activity-dependent dendritic outgrowth in response to KCl depolarization. B, Number of branch points per cell, a measure of dendritic arbor complexity, is also reduced. The average values are from three independent experiments, with 50–75 GFP+ neurons scored per condition per experiment. Data are shown ± SEM. *p < 0.02, **p < 0.002, ***p < 0.0005, Student's t test. C, Representative images of neurons with or without KCl. D, Nuclear extracts of ES cells infected with V5-tagged SS18 and CREST integrate into BAF complexes, as evidenced by co-IP with Brg and BAF155. E, Exogenous SS18 and CREST protein in cortical neurons is nuclear and SS18 expression in neurons decreases CREST protein levels after depolarization with KCl. Arrow points to neuron nuclei with decreased CREST. Images are of KCl-depolarized samples. F, Quantification CREST protein level expression (average fluorescence intensity in nuclei). Forced SS18 expression causes significant decrease in CREST protein with depolarization. The average values are from two independent experiments, with 20–30 GFP+ neurons scored per condition per experiment. Dara are shown ± SEM. *p < 0.02, **p < 0.002, ***p < 0.0005, Student's t test. Scale bar in C, 50 μm; in E, 20 μm.
Discussion
The following lines of evidence support our conclusion that SS18 and CREST are stable, dedicated, and functional subunits of the npBAF and nBAF complexes, respectively: (1) both proteins cosediment exclusively with Brm/Brg on glycerol gradients of whole-brain preparations, indicating that they are dedicated to these complexes; (2) they resist dissociation from BAF complexes even under partially denaturing conditions, indicating that they make stable, possibly cofolded interactions; (3) they do not co-IP each other, indicating that they are mutually exclusive subunits; and (4) SS18 cannot substitute for CREST in supporting dendritic growth. From these studies and their patterns of expression, we conclude that SS18 is a subunit of esBAF and npBAF complexes and that CREST is a subunit of nBAF complexes as illustrated in Fig. 9A. Although we and others have detected SS18 and CREST in studies of BAF complexes, it was not apparent that they were dedicated, functional, nonexchangable subunits rather than simply being interacting proteins. From our proteomic analysis, we conclude that NS/progenitors contain at least 36 different npBAF complexes and that neurons contain at least 36 different nBAF complexes. We do not know if these complexes are present in subtypes of neural progenitors and neurons or if each neuron has all possible assemblies. From a limited analysis of different brain regions and inspection of the Allen Brain Atlas (http://www.brain-map.org/), we favor multiple assemblies in each neural progenitor or neuron. These findings indicate that the spectrum of neurologic diseases due to mutations in npBAF and nBAF complexes must be interpreted in light of the emergent features of different combinatorial assemblies of these complexes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary of ordered events in npBAF to nBAF complex switching. A, The npBAF to nBAF switch involves three subunits regulated by miR9/9* and miR-124. Combinatorial assembly of npBAF and nBAF subunits could create up to 36 distinct npBAF or nBAF complexes. B, In a negative cascade network motif, downstream genes are sequentially repressed and activated when their regulator reaches the relevant threshold. The conversion of ES cells to neurons illuminates the sequential repression and activation of the genes of the triple-negative miRNA/chromatin switch, which is a negative cascade network. BAF53b is under independent control for transcription, but the presence of BAF53a will drive BAF53b from the BAF complex and destabilize 53b protein (B.T.S, G.R.C, unpublished results).
SS18 and CREST appear to be functionally exclusive, because expression of SS18 in postmitotic neurons inhibits activity-dependent dendritic outgrowth, whereas expression of CREST does not. In fact, SS18 expression in neurons phenocopied the CREST loss-of-function phenotype that is indicative of a dominant-negative function (Aizawa et al., 2004). We have recently identified two mutations in CREST in amyotrophic lateral sclerosis (ALS, also known as Lou Gehrig's disease) patients (Chesi et al., 2013). We find that these CREST mutants strongly block dendrite outgrowth even in the presence of endogenous WT CREST, suggesting a dominant-negative mode of action. One of the mutations terminates the final 9 aa of CREST, which has been shown to be necessary for interaction of CREST with CBP, a histone acetylase (Aizawa et al., 2004). The finding that CREST is entirely bound to nBAF complexes and resists dissociation even under denaturing conditions indicates that CBP is targeted to sites of action by nBAF complexes in a way comparable to the targeting of STAT3 to its DNA binding sites by the prior action of esBAF in ES cells (Ho et al., 2011).
Neither SS18 nor CREST have orthologs in yeast, flies, or worms. Although BAF complexes are frequently referred to as the mSWI/SNF complex, only six of the subunits have yeast homologs and other subunits have homologs in the RSC and SWR1 complexes. In most cases, the new BAF subunits appeared either in insects or fish. An SS18 homolog is present in the zebrafish and fugu genomes and is present in birds, Xenopus, and mammals (UCSC Genome Browser; http://www.genome.ucsc.edu/). However, CREST is present in neither the zebrafish nor fugu genomes but is present in the coelacanth genome. Coelacanths are lobe-finned fishes more closely related to tetrapods than to ray-finned fishes. Therefore, SS18 and CREST appear to have originated to deal with some aspect of chromatin regulation that has emerged relatively recently in tetrapod evolution. The exclusive expression of CREST in the nervous system and the extraordinary sensitivity of neural progenitors to reduction in SS18 levels indicates that this recently evolved chromatin function might be used in the development of the tetrapod motor nervous system. Neither SS18 nor CREST is required for nucleosome remodeling in vitro, indicating they may play a role in genome targeting or some as-yet-unrecognized functions of these complexes.
The appearance of CREST in the lobe-finned coelacanth is interesting because it, like all tetrapods, has limb and finger bones covered with muscle and skin compared with ray-finned fishes, which have slender bony fin rays (lepidotrichs) covered with only skin. One hypothesis is that, during the tetrapod transition, CREST co-evolved with tetrapod limb development. Indeed, the majority of the putative regulatory elements located near CREST appear to originate in the tetrapod ancestor (Lowe et al., 2011). CREST may play a specific role in motor neuron chromatin regulation, where it pairs with motor neuron factors necessary for limb neuromuscular development in a way similar to the essential pairing of BAF60c with myoD to activate the skeletal muscle program (Albini et al., 2013) or esBAF with Sox2 and Oct4 in ES cells (Ho et al., 2009a). Perhaps this requirement for CREST in motor neurons is why mutant CREST causes ALS, a fatal motor neuron disease characterized by loss of coordination and atrophy of limbs. Interestingly, CREST-null mice also have coordination defects and smaller cerebellums (Aizawa et al., 2004). In contrast to the roles of BAF complexes in mental retardation and microcephaly, ALS is thought to result from deterioration in neural function and connectivity. Therefore, BAF complexes appear to function during normal human neural development and are also required to maintain circuitry and/or viability over a lifetime. However, additional studies will be required to confirm these possibilities.
SS18 and CREST subunits are 62% identical, yet SS18 cannot substitute for CREST in postmitotic neurons. SS18 has 65 proline residues and CREST has 27. Proline residues make proteins more rigid, so the secondary structure of SS18 will be more rigid than that of CREST and thus could give rise to different functions. In addition, the paralogs may have different posttranslational modifications, because SS18 has 31 serine and 8 threonine residues compared with 53 serines and 22 threonines in CREST. There are two splice variants of SS18, the short isoform (lacking exon 8) being the more abundant of the two (Fig. 5B). Both SS18 splice variants functioned as a dominant-negative and blocked activity-dependent dendritic outgrowth (Fig. 8 and data not shown). This indicates that exon 8, which CREST contains, does not confer CREST-like function to SS18. Therefore, the unique functionality of SS18 and CREST must come from other biochemical differences.
Recent exome sequencing studies have revealed frequent mutations in BAF complex subunits in CSS, NBS, and sporadic mental retardation. In addition, mutations have been reported in Brg and Brm in schizophrenia and in BAF155, BAF170, BAF180, and REST in human autism (Neale et al., 2012; O'Roak et al., 2012). These mutations were dominant and considered to be dominant-negatives. In the NBS study, the authors suggested the dominant mutations in BRM (one of two ATPases that in neurons are components of neural-specific nBAF complexes) to be dominant-negatives and also affect Brg-based complexes. Genetic dominance can have many different mechanistic underpinnings, including the production of dominant-negative proteins that can block WT protein function, abnormal polymerization processes in which the mutated protein blocks polymer extension, and haploinsufficiency due to a nonsense mutation in a protein involved in a rate-limiting process. To clarify this issue, we reexamined our proteomic analysis done with purified nBAF complexes. We found that peptides from Brg exceeded peptides recovered from Brm by more than threefold. Because both are large proteins, these differences are unlikely to be due to peptide recovery differences and likely reflect the endogenous levels of the proteins. Many of these BRM dominant mutations are surprisingly conservative amino acid changes and cluster to the ATP binding and Helicase domains (Ronan et al., 2013), suggesting that rather than being strong dominant-negatives, the mutations affect a rate-limiting catalytic process. ATP-dependent chromatin remodeling is well suited for a rate-limiting role in biological processes, because the rate of chromatin alteration may be limited by the intrinsic rate of energy utilization by these complexes rather than by ATP levels, which are generally not rate limiting. The dominant role of BAF subunits in human diseases fits nicely with the observation that, in mice, mutations in Brg, BAF155, and BAF53a all have dominant phenotypes and are apparently haploinsufficient. Rate-limiting functions are often instructive, so these considerations are consistent with the apparent instructive role of the microRNA/chromatin switch in converting fibroblasts to neurons (Yoo et al., 2011). Interestingly, the ALS mutations we have described also appear to be dominant (Chesi et al., 2013).
Our kinetic analysis of conversion of ES cells to neurons reveals the nature of the negative cascade network motif underlying the triple-negative genetic circuit we described as contributing to neural fate determination (Yoo et al., 2009; Yoo et al., 2011). The timing of repression of REST, activation of mir-9/9* and miR-124, subsequent repression of BAF53a, and stabilization of BAF53b protein nicely fit the negative cascade network motif model in which downstream genes are sequentially repressed and activated when their regulators reach the relevant threshold (Shoval and Alon, 2010; Fig. 9B). Our kinetic analysis also indicates that more than a single pathway controls the microRNA/chromatin switch. This follows from the observation that BAF45c is induced without RA and before miR-9/9* and miR-124. The requirement for two pathways to bring about this epigenetic switch indicates that the npBAF to nBAF switch could be a hub or integration site for neurogenesis, which is supported by recent genome sequencing efforts in human neurologic diseases.
The discovery that CREST is a dedicated, nonexchangeable subunit of nBAF complexes indicates that these complexes have three neuronal-specific “letters” endowing them with the ability to provide specific patterns of gene expression in neurons. These complexes in neurons are additionally polymorphic by virtue of alternative assembly of other subunits such as Brg versus Brm, BAF250a versus BAF250b versus BAF200, BAF60a versus BAF60b versus BAF60c, and BAF45b versus BAF45c. Therefore, they should be referred to as npBAF complexes and nBAF complexes, each with a complexity of 36 possible assemblies based on gene numbers (Fig. 9A). An even larger number of combinatorial possibilities might result from the extensive alternative splicing shown for BAF250b and SS18. The functional target(s) of the neuron-specific surfaces of the nBAF complexes is not known, but is likely to reflect the emergent features of complexes (i.e., those that emerge not from the subunit itself, but rather from the composite surface produced by each subunit and its adjacent subunits). Our findings indicate that the widely different neurologic diseases causally or possibly related to mutations in BAF subunits (mental retardation, schizophrenia, autism, ALS, and microcephaly) must be interpreted in the context of these different BAF assemblies rather than a monomorphic mSWI/SNF complex.
Footnotes
- Received March 21, 2013.
- Revision received May 8, 2013.
- Accepted May 15, 2013.
This work was supported by grants from the Howard Hughes Medical Institute, the National Institutes of Health (Grant #CA163915 and Grant #NS046789), and the California Institute of Regenerative Medicine (Grant #CIRM R34-05886 to G.R.C.). B.T.S. was supported by the Developmental and Neonatal Biology Training Program (Grant #T32 HD007249 from the National Institutes of Health). A.X.S. was supported by the Agency of Science, Technology and Research of Singapore (A*STAR). A.S.Y. was a fellow of Helen Hay Whitney Foundation. We thank all members of the Crabtree Laboratory for engaging discussions on the subject, especially Julie Lessard, Jiang Wu, and Ary Shalizi for helpful suggestions.
- Correspondence should be addressed to Gerald R. Crabtree, Investigator, Howard Hughes Medical Institute, Rm B211, Beckman Center HHMI, 279 Campus Drive, Stanford University School of Medicine, Stanford, CA 94305-5323. crabtree{at}stanford.edu
- Copyright © 2013 the authors 0270-6474/13/3310348-14$15.00/0