
Abstract
Regulation of the microtubule cytoskeleton is of pivotal importance for neuronal development and function. One such regulatory mechanism centers on microtubule plus-end tracking proteins (+TIPs): structurally and functionally diverse regulatory factors, which can form complex macromolecular assemblies at the growing microtubule plus-ends. +TIPs modulate important properties of microtubules including their dynamics and their ability to control cell polarity, membrane transport and signaling. Several neurodevelopmental and neurodegenerative diseases are associated with mutations in +TIPs or with misregulation of these proteins. In this review, we focus on the role and regulation of +TIPs in neuronal development and associated disorders.
Similar content being viewed by others


Introduction
Microtubules (MTs) are one of the major types of filaments that constitute the eukaryotic cytoskeleton. Over the years, MTs have emerged as key players in cellular processes such as vesicle and organelle transport, DNA segregation during mitosis, cell migration and maintenance of cell polarity. Neurons are among the most complex and polarized cells, whose distinct morphology allows them to establish intercellular connections and propagate chemical and electrical signals across the nervous system. Mature neurons typically extend multiple processes, one of which (the axon) serves as a transmitter whereas others (the dendrites) act as receivers of input from other neurons. MTs are important for numerous functions in nerve cells (reviewed in [1–3]), such as long-range transport of cargo and neuron-specific processes like growth cone guidance. Indeed, MTs are indispensable for neurodevelopment, and many neurological diseases stem from defects in the MT cytoskeleton or its regulation.
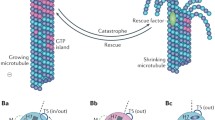
Evidence for the existence of MTs was first obtained from electron microscopy (EM) data in the 1950s (reviewed in [4]). Prior to the development of EM, fibrillar structures had already been described as part of the mitotic spindle and cytoplasm. However, interpretative differences and technical limitations of early microscopy made it challenging to identify unity among observations [4]. It therefore was not until 1963 that MTs were acknowledged as distinct structures and named by Slautterback, Ledbetter and Porter [5, 6]. Today, we know that MTs are hollow tubes with a diameter of approximately 25 nm. MTs are typically assembled from 13 laterally associating protofilaments, which in turn consist of α, β-tubulin dimers aligned in a head-to-tail fashion. As a consequence of tubulin dimer polarity, MTs possess polarity throughout, which results in distinct ends of the polymer: the minus- and the plus-end, exposing α- and β-tubulin, respectively. MTs alternate between rapid phases of growth and shrinkage, a behavior termed ‘dynamic instability’ [7]. A transition from shrinkage to growth is called a rescue, whereas the opposite transition is referred to as a catastrophe.
Dynamic instability allows MTs to be swiftly remodeled in response to environmental cues. The MT cytoskeleton is suitable for rapidly sensing and responding to changes in the intracellular environment. To this end, the MT cytoskeleton acts in concert with a large number of proteins (MAPs for MT-associated proteins) that either influence MTs themselves or relay signals from the MT cytoskeleton to other parts of the cell. MAPs are known to regulate MT behavior such as stability, assembly, bundling and targeting by associating with specific parts of the MT lattice or by interacting with the soluble tubulin pool. Well-characterized neuronal MAPs include MAP2 and tau, which maintain a polarized, mutually exclusive distribution and decorate MT bundles in dendrites and axons, respectively. Both proteins stabilize MTs and are able to induce MT bundling (reviewed in [8]). Abnormal phosphorylation of tau triggers its dissociation from MTs and causes tau to aggregate, resulting in the formation of potentially toxic tau deposits (neurofibrillary tangles) found in the brains of patients suffering from Alzheimer’s disease (AD) and other tauopathies. This process is accompanied by degradation of the axonal MT cytoskeleton, suggesting a model in which dissociation of tau results in MT instability. It should be noted, however, that the precise hierarchy of events during the onset of AD remains unclear. Additional roles for tau are still emerging and may shed new light on the biology of tauopathies (reviewed in [9]). Among these is the regulation of the subcellular distribution of MAPs that specifically bind to the growing MT plus-end [10], the subclass of MAPs that this review will focus on.
MT dynamics are most pronounced at the plus-end. Although growth events have been observed at the MT minus-end [11], in cells minus-ends are often anchored or stabilized, restricting their dynamic behavior [12]. At the growing plus-end, freshly polymerized MT stretches contain GTP-loaded β-tubulin as opposed to the GDP-bound subunits present in the MT lattice, resulting in a so-called GTP cap. Moreover, the structures of polymerizing and depolymerizing MT plus-ends are different [13]. The unique chemical environment of the polymerizing MT plus-end grants it its own interactome within the realm of MAPs, consisting of MT plus-end tracking proteins (+TIPs; reviewed in [14–16]). +TIPs display a large structural and functional variation between individual proteins. However, a common theme sets them apart from other MAPs: +TIPs associate with the polymerizing MT plus-end, where they act as powerful regulators of MT dynamics and MT interactions with other structures.
In this review, we use the neuronal MT cytoskeleton to illustrate the role of +TIPs in the development of one of the most polarized and complex cell types. Before discussing plus-end tracking mechanisms and highlighting the roles and regulation of various +TIPs in neurons, we touch upon the function of the MT cytoskeleton in the developing and mature nervous system. Moreover, we highlight the role of +TIPs in neurodegenerative and neurodevelopmental diseases. We conclude this review with an outlook on the future of neuronal +TIP research and briefly discuss the drug target potential of these pivotal proteins.
Microtubules in neurons
Neurons are derived from progenitor cells located in the ventricular zone deep inside the brain, necessitating young neurons to migrate large distances into remote regions. During their journey, neurons undergo dramatic changes in morphology and establish complex polarity. Even mature neurons must remain plastic as connections between neurons, synapses, are continuously rewired in response to stimuli. This intricate development relies heavily on both the MT and the actin cytoskeleton, on their crosstalk and on their accessory proteins. For a detailed analysis of the role of the cytoskeleton and in particular MTs during neuronal development, we refer the reader to a number of excellent reviews [1–3, 17]. Here, we briefly highlight some of the main events involving MTs during the maturation of multipolar neurons. It should be noted that the development of neurons extending a single process, unipolar neurons, is considerably different [18].
Microtubules during neurite formation and axon outgrowth
Neurons start out as spherical, unpolarized cells with a MT organization similar to that commonly found in mammalian cells (Fig. 1a). In young neurons, MTs mainly nucleate from the centrosome, with MT minus-ends pointing inward and plus-ends oriented towards the cell periphery [19]. Upon differentiation, neurons undergo symmetry breaking. During this event, the neuron extends multiple processes that start as small buds on the membrane and elongate to form thin protrusions [20]. These early protrusions, termed neurites, mature into axons and dendrites when the neuron polarizes. It has been proposed that neurite formation is powered, at least in part, by MT sliding. One model suggests that MAP2c (microtubule-associated protein 2c) induces stable MT bundles, which translocate to the membrane where they exert a dynein-dependent force to trigger protrusion formation [21]. Another model proposes that the motor protein kinesin-1 powers the displacement of MTs along other MTs, exerting a mechanical force on the membrane which results in neurite extension [22] (Fig. 1b).

Microtubule organization and function during neurodevelopment. Cultured dissociated neurons start out as spherical, unpolarized cells with MTs oriented with their plus-ends towards the plasma membrane (a). Upon symmetry breaking, neurite extension is thought to be facilitated by motor proteins, which were proposed to push MTs and thus exert a force on the membrane to form protrusions (b). Young neurons possess multiple neurites and maintain a mainly plus-end out MT orientation (c). While remaining neurites cycle between phases of growth and shrinkage, one neurite rapidly extends to form the axon. In this neurite, MTs become stabilized and MT bundles are decorated with the axon-specific MAP tau, while MTs remain oriented plus-end out. The rate of advance and the directionality of axon outgrowth is controlled by the growth cone, a specialized structure at the tip of the axon that contains a dynamic array of MTs. Local stabilization of a MT in one of the filopodia of the growth cone prompts the growth cone to turn in that direction (d). Later in development, the remaining neurites differentiate into dendrites. Dendrites acquire unique antiparallel MT bundles decorated by MAP2, presumably contributing to selective cargo trafficking (e). The post-synapse is present at the tips of dendritic spines. Targeting of dynamic MTs to spines triggers morphological changes and alters synaptic strength, possibly by allowing the delivery of specific cargo to the spine or activating signaling processes (f)
Shortly after neurite extension, the axon is formed (Fig. 1c). This event is preceded by local stabilization of MTs in the pre-axonal neurite [23]. As the newly formed axon starts to elongate, it relies on stable MT tracks for the transport of proteins and organelles necessary for the formation of new axonal segments [24]. The direction of axon outgrowth is determined by the growth cone, which probes the extracellular environment to allow non-random establishment of synaptic connections. Amidst the actin, which drives invasion of the extracellular matrix (reviewed in [25]), an array of MTs controls the direction in which the growth cone advances (Fig. 1d; [26]). The MT array assumes a looped conformation in pausing growth cones [27], while active growth cones maintain a dynamic MT array. These MTs probe the growth cone cortex and respond to guidance signals by being stabilized or destabilized, prompting the growth cone to turn towards or away from the guidance cue, respectively [24, 28]. As the axon matures further, it branches to allow higher interconnectivity. Branch formation is accomplished by splaying of tau-decorated MT bundles at branching sites. Here, dynamic MTs invade actin-rich areas close to the membrane to form a collateral branch [29]. Thus, while dynamic MTs play only a minor role in neurite outgrowth, they are crucial for axon polarization, pathfinding and branching.
Microtubules during dendrite maturation
Dendritic differentiation occurs later in development than axogenesis and prompts dramatic changes to the soon-to-be dendritic MT infrastructure. Prior to dendritogenesis, mammalian precursor neurites cycle between phases of growth and shrinkage and maintain ~80 % plus-end outFootnote 1 MT directionality [30]. Perhaps the most striking feature of the dendritic MT cytoskeleton is the appearance of MTs with their minus-ends oriented towards the dendritic tips [31, 32] (Fig. 1e). However, it should be noted that the ratio between minus- and plus-end out MT orientations changes per neuron type and even differs between regions of the same dendrite. In invertebrates, as much as ninety percent of dendritic MTs are oriented minus-end out [33, 34]. Differences in MT orientations between axon and dendrites likely contribute to proper targeting of specific cargo by enabling transport by specific motor proteins. While dendrites branch more than axons, the behavior of MTs during this process has been studied less extensively. Specialized Golgi compartments called Golgi outposts were identified as potential sites of MT nucleation in centrosome-free Drosophila neurons [35], and were shown to localize to dendritic branch points [36]. Since axon branching depends on dynamic MTs arising from splayed bundles, it seems likely that dynamic MTs nucleated at Golgi outposts fulfill a similar role in dendritic branching [35].
Microtubules and synapses
The formation of synapses, connections between the axon of one neuron and the dendrite of another, is the final and ongoing step in neuronal maturation. Excitatory synapses are formed on dendritic spines, which are actin-rich protrusions on the dendrite that scaffold the post-synaptic density. The absence of MAP2-positive MTs in dendritic spines raised the belief that the MT cytoskeleton remained confined to the dendritic shaft [37]. However, visualization of the behavior of MTs and MT plus-ends revealed that dynamic MTs transiently invade dendritic spines by polymerizing from proximal sites [38–41]. MT-depolymerizing drugs such as nocodazole markedly reduce the number of spines, while the number of dendrites remains unaffected [38]. Nocodazole also blocks the spine-inducing effect of the growth factor BDNF (brain-derived neurotrophic factor), while the MT stabilizing drug taxol enhances BDNF-induced spine formation [40]. Finally, depletion of end-binding protein 3 (EB3), a key scaffolding factor at the MT plus-end and regulator of MT dynamics discussed below, reduces the amount of spines [40]. These studies imply dynamic MTs as modulators of neuronal plasticity. +TIPs involved in synaptic remodeling may rely on dynamic MT plus-ends as a means of reaching spines targeted for remodeling [38]. Other candidate regulatory mechanisms involve the facilitation of cargo delivery to the postsynaptic terminal [1] (Fig. 1f), though it remains to be elucidated exactly which events are at play.
Microtubule plus-end tracking proteins in neurons
MT behavior during neurodevelopment has been catalogued extensively. However, research has only just begun to uncover which factors control MT dynamics and how MTs are able to relay intricate signals in neurons. Many of the cellular processes and the molecular mechanisms that underlie them remain unknown to date.
Since +TIPs have emerged as potent MT regulators, they are excellent candidates to control MTs and relay their signals during neuronal development and homeostasis. Indeed, numerous +TIPs have already been linked to neurodevelopmental functions (Table 1). New +TIPs are still discovered on a regular basis, and it is likely that additional roles for +TIPs in neurons will be uncovered in the future. Current knowledge suggests that, based on their mode of association with MTs, +TIPs can be divided into three categories: end-binding proteins (EBs), EB-dependent +TIPs and EB-independent +TIPs. It should be noted that even in the case of EB-independent +TIPs, there is crosstalk between these +TIPs and EBs albeit indirectly. Therefore, none of these categories can be regarded as fully independent.
End-binding proteins
EBs are at the core of the MT plus-end interactome. They are known to regulate MT behavior both autonomously and by providing a structural scaffold for other +TIPs [42–44]. EB plus-end tracking depends on an N-terminal calponin homology (CH) domain that grants MT affinity [45]. It has been shown that EB proteins associate with the MT plus-end by a CH-dependent nucleotide sensing mechanism [46, 47]. MT binding is regulated by a negatively charged C-terminal domain, which repels the negatively charged MT lattice and thereby contributes to specificity for the MT plus-end [48]. In addition, C-terminal coiled-coil and EB-homology domains mediate homo- and heterodimerization as well as interaction with other proteins including +TIPs [49, 50].
EBs mainly function as scaffolding proteins at the MT plus-end, where they form a hub for other +TIPs to associate with and thereby regulate local protein composition and MT dynamics. This is illustrated by the fact that EBs promote catastrophes when reconstituted with tubulin in biochemical preparations, while they reduce the number of catastrophes and promote continuous MT growth in cells. This suggests that EBs primarily act on other MT regulators in cells rather than autonomously [44]. Plus-end bound EBs rapidly exchange with the cytosolic pool, providing a rapidly remodeling platform for protein binding [43, 51]. Quantitative proteomic studies of non-neuronal cell lines have revealed that EBs are the most abundant plus-end binding proteins: the EB family outnumbers the second most abundant +TIPs by factors of approximately 7–30 in cultured fibroblasts [52, 53]. While technical limitations arising from sample heterogeneity have hampered large-scale quantitative proteomics studies in nerve cells [54], it is expected that EBs dominate neuronal MT plus-ends in a similar fashion as in cultured fibroblasts. The relatively high concentration of EBs compared to other +TIPs offers a simple explanation for how EB-decorated plus-ends are efficiently formed and maintained. Such a hub provides cells with an extra layer of control to regulate large numbers of +TIPs with minimal changes to the MT cytoskeleton itself, making it easier to retain MT integrity and reliant functions alongside.
In mammalian cells, the EB family is represented by three members (EB1, EB2 and EB3), which all bind to MT plus-ends but differ in their affinity for MT tips, phosphorylation and affinity for binding partners [44, 55–59]. EB1 and EB2 appear to be expressed ubiquitously, while EB3 is strongly expressed in muscle and brain tissue [60, 61]. During neurodevelopment, EB1 expression decreases while EB3 expression is upregulated [38]. Axon extension coincides with EB1 expression in neuroblastoma cells [62]. In Drosophila¸ depletion of EB1 impairs axon outgrowth and leads to the disorganization, but not loss, of MTs [63]. Other studies also point in the direction of a role for EB1 in axogenesis [64], and suggest a differential role of EB proteins in neurite formation. EB1 and EB3 have a positive role in neurite outgrowth, while EB2 has a negative effect [65], possibly because EB1 and EB3 have a higher affinity than EB2 for MT-stabilizing partners [56, 66]. EB3 has been specifically implicated in neuritogenesis in the context of actin-MT interactions [67], suggesting that the mechanisms underlying the importance of EBs for controlling neuronal morphogenesis can be quite complex.
Apart from scaffolding other +TIPs, EBs may also aid the capture of MT plus-ends for regulatory purposes. For example, MTs are proposed to rely on EB3 and Drebrin to enter actin-rich spines. Drebrin interacts with the growing MT plus-end via an unconventional interaction with EB3 [67], and is enriched in spines by binding to F-actin [68]. Drebrin localization becomes enhanced in spines upon NMDA (N-methyl-d-aspartate) receptor activity, whereupon Drebrin is believed to capture EB-decorated MT plus-ends near or in the spine neck and thereby guide dynamic MT entry into spines. Accordingly, Drebrin overexpression and increased amounts of F-actin upregulate the number of MT entries into spines [41].
Interestingly, another neuron-specific role for EB1/3 was reported that does not depend on plus-end tracking. EBs are enriched in the axon initial segment (AIS) of hippocampal neurons, where they contribute to AIS integrity and maintenance [69]. This possibly contributes to enhanced MT stability in the AIS [69], although the precise mechanism remains unclear.
EB-dependent +TIPs: CAP-Gly proteins
Most known EB-dependent +TIPs can be divided in two categories, depending on their mode of association with EB proteins. The first category consists of a minority of +TIPs that contain an evolutionarily conserved cytoskeletal-associated protein glycine-rich (CAP-Gly) domain, which associates with EEY/F motifs in the C-terminus of EB proteins and tubulin [70–73].
An example of one such CAP-Gly domain-containing +TIP is p150glued: the largest out of eleven subunits of the dynactin complex. Dynactin is essential to nearly all functions of dynein, the most prominent minus-end directed motor protein [74]. Dynein plays a particularly important role in axons, wherein transport into the soma relies on minus-end directed transport due to the uniform plus-end out orientation of MTs. It is likely that multiple mechanisms contribute to dynein activation at different locations along the neuron [74]. At the MT plus-end specifically, one such model explains how retrograde transport is initiated when the dynamic MT plus-end loaded with p150glued and other dynein regulators encounters minus-end directed cargos ([75] and reviewed in [74]). While the role of dynactin in dynein plus-end targeting appears to differ between organisms [74], research suggests that dynactin accumulation at the MT plus-ends in axons of murine dorsal root ganglia (DRG) neurons contributes to long-range retrograde transport by recruiting dynein to vesicles [76]. It has been postulated that this function is exerted by a neuron-specific p150glued isoform, which reduces the frequency of catastrophes and thus increases MT stability [77].
Although p150glued binds to EBs directly, its affinity for MT plus-ends appears not to be very high. In cells, p150glued is assisted in targeting the plus-ends by another CAP-Gly domain containing protein, cytoplasmic linker protein of 170 kDa or CLIP-170 [76, 78]. Mammals also express a protein closely related to CLIP-170: the neuronally enriched CLIP-115 [79]. Both CLIPs are +TIPs, but differ by the structure of their C-termini. Only CLIP-170 contains zinc-binding domains and an EEY/F motif, which mediate the interactions with the CAP-Gly domain of p150glued and with the dynein regulator LIS1, as well as autoinhibition [78, 79]. Both CLIPs also promote MT rescue [80], although the underlying mechanism is still obscure, as it should involve depolymerizing MT ends or the MT lattice, where CLIPs are not enriched. Of note, CLIP plus-end tracking behavior is less prominent in neuronal compared to non-neuronal cells [81], which would be compatible with a function that is not directly related to growing MT tips. In addition to regulating dynactin recruitment, CLIPs are enriched in axonal growth cones, where they stabilize MTs protruding into the actin-rich leading edge [82]. CLIPs are therefore necessary for axon formation and outgrowth as MT stabilization in the growth cone precedes engorgement and consolidation. CLIP-170 is involved in dendrite morphogenesis by regulating crosstalk between the actin cytoskeleton and dynamic MTs [83]. Given the importance of CLIPs for different processes in cultured neurons, the phenotypes of CLIP-115 and CLIP-170 knock-out mice are rather mild, though CLIP-115 knock-out animals do display behavioral phenotypes [84, 85]. The loss of CLIP-190, the Drosophila homologue of CLIP-170, causes no strong phenotype either [81]. This suggests that the neuronal function of CLIPs might be redundant with that of other MT regulators.
EB-dependent +TIPs: SxIP proteins
The largest subclass of EB-dependent +TIPs comprises proteins which utilize a short linear motif known as the SxIP motif (serine/threonine-any amino acid-isoleucine/leucine–proline) to bind the EB homology domain ([42] and reviewed in [14]). SxIP motifs are generally embedded in unstructured amino acid stretches enriched in proline, serine and basic residues, resulting in a positive charge [42]. Further computational analysis revealed that the nine amino acids surrounding the SxIP motif cannot contain acidic amino acids, and that at least one basic amino acid is present in the four amino acids preceding the motif [56]. Since discussing all currently identified SxIP +TIPs is beyond the scope of this review, we here focus on a selection of prominent examples to illustrate the broad range of neurodevelopmental functions of these +TIPs. All currently known neurodevelopmental functions are listed per +TIP in Table 1, along with their mode of association with the MT plus-end and the known neurological disease associations.
Among the most conserved SxIP proteins are CLASPs (cytoplasmic linker protein-associated proteins), the mammalian versions of which were discovered through their association with CLIPs [86]. Similar to CLIPs, there are two CLASP-encoding genes in mammals: CLASP1, which is expressed ubiquitously, and CLASP2, the products of which appear enriched in nervous tissue [86]. CLASPs utilize their SxIP motifs to bind EB1 and contain several additional TOG (tumor overexpressed gene) domains which can serve as tubulin-binding modules [87–89]. Different cell lines have revealed a function for CLASPs at the membrane, where they capture dynamic MT ends to promote MT rescue and pausing, and thus stabilize MTs [89, 90]. CLASP-mediated cortical MT stabilization is crucial to axon outgrowth and directionality and as such, CLASP was implicated in axon development in various organisms [90, 91]. Interestingly, CLASPs have affinity for both the MT plus-end and lattice, and differences in CLASP distribution and CLASP-MT associations inside the growth cone are able to direct axon growth status. CLASPs can be localized to the tips of growth cone filopodia, where they capture plus-ends of MTs to facilitate axon outgrowth. Conversely, in pausing growth cones, lattice-binding CLASP is present close to the end of the axon shaft to prevent MTs from protruding into the peripheral growth cone, thereby preventing outgrowth. The localization of CLASPs inside growth cones is regulated by kinases such as GSK3β and Abelson kinase [90, 92–94]. In addition to axon growth status, the direction of axon outgrowth is regulated by CLASP localization. The kinase-controlled, asymmetric distribution of CLASPs to the filopodia of a growth cone determines the sites of MT capture and thereby dictates the direction in which the axon advances [94, 95]. Additional functions for CLASPs include a role in synaptic functioning likely via global control of neuronal morphology [95] and maintenance of the Xenopus growth cone lamellipodium [91]. Furthermore, CLASP2 mediates MT capture at the postsynaptic membrane to promote transport of acetylcholine receptors to neuromuscular junctions [96].
Morphological changes during neurodevelopment are the result of complex interplay between different components of the cytoskeleton. This is in part facilitated by the Microtubule-Actin Crosslinking Factor proteins MACF1 (ACF7) and MACF2 (dystonin), known as spectraplakins. As spectraplakin nomenclature is complicated, we will here refer to MACF1/2. The reader is directed to Table 1 for a comprehensive list of alternative names for these proteins. Spectraplakins gain their name from membership of the spectrin family and their plakin repeats, which grant affinity for intermediate filaments [97]. Spectraplakins also contain CH domains to bind to actin, GAR (growth arrest-specific 2 protein-related region) domains to bind and stabilize MTs [98], and SxIP motifs to bind the MT plus-end via EBs. This places them at the heart of cytoskeletal crosstalk and renders them a popular subject for neurodevelopmental research. Indeed, homozygous MACF1 knockout mice are not viable, and mutant mice die from neuronal migration defects when MACF1 is depleted during development [99]. MACF1 is known to guide MTs along actin filaments and to mediate MT capture at actin-rich sites near the membrane [100], consistent with a role for MACF1 and its orthologs in growth cone MT organization and axon extension [101]. In case of the Drosophila MACF1 ortholog Shot, this function was shown to depend on its SxIP motifs and interaction with EB1 [63]. MACF1 has also been implicated in formation of growth cone filopodia, although for Shot this function does not rely on its actin- or MT-binding domains [101].
The second mammalian spectraplakin, MACF2 or dystonin, is best known for its role in the neurological disorder dystonia. MACF2 knockout mice develop dystonia and show repetitive muscle spasms [102], and mutations in MACF2 have been identified in patients with Hereditary Sensory Autonomic Neuropathy [103]. These pathologies are associated with the degeneration of sensory and autonomic nerves [102, 103]. Axons of MACF2 null mice degenerate as a result of MT fragmentation, which is believed to contribute to the dystonia phenotype independent of the neurofilament-binding functions of MACF2 [104]. MACF2 has also been implicated in retrograde axonal transport by interacting with p150glued [105]. This function may depend on MACF2’s EB-dependent association with the MT plus-end, as overexpression of peptides which competitively block EB–SxIP interactions inhibit retrograde transport of endosomes [106]. Another important factor in dystonia may be the neuron’s inability to regulate ER stress levels, Golgi integrity, MT acetylation and autophagy due to loss of a neuron-specific MACF2 isoform (BPAG1-a2) [107–109]. This particular isoform has an N-terminal trans-membrane domain and does not localize to MTs [110], although it does affect MTs near the centrosome via an association with MAP1B [108] and retains the C-terminal SxIP motifs. The same isoform was shown to partially rescue phenotypes in a dystonia musculorum mouse model [111]. On the other hand, another neuron-specific MACF2 splice variant (BPAG1n3) exclusively binds MTs and may be involved in sustaining axonal MT integrity [104], suggesting distinct functions for different MACF2 isoforms. The same holds true for the many splice variants of MACF1, and more research is needed to elucidate the contribution of spectraplakin plus-end tracking to their functions in both nerve cells and in other cell types.
+TIPs also play important roles in mature neurons, as neurons remain plastic throughout their lifespan and remodel synapses in response to both intra- and extracellular cues. One such +TIP with a function at the synapse is p140Cap, whose name is a combination of its molecular weight and ‘Cas-associated protein’ (Cap; Cas for Crk-associated substrate). p140Cap is regarded as a tumor suppressor protein due to its function as an inhibitor of Src kinase, which is involved in cell migration and growth [112]. In addition to its potential to associate with MT plus-ends via an SxIP/EB3-mediated interaction, p140Cap binds actin fibers and localizes to actin-rich dendritic spines of hippocampal neurons [38, 112]. p140Cap knockout mice display impaired learning and memory functions, and spine defects have been observed in the absence of p140Cap both in cultured primary neurons and in knockout mice [38, 113]. Synaptosomes prepared from p140Cap−/−- mice reveal hyperactivation and hyperphosphorylation of Src kinase and its substrate cortactin, respectively, as well as reduced RhoA activity [113]. Research suggests that p140Cap forms a synaptic complex with and increases the interaction between Src kinase and Citron-N, a protein known to scaffold the actin remodeling machinery, and as such controls spine morphology [113]. EB3 appears to function upstream of the spine remodeling process as synaptic phenotypes of EB3 knockdown mimic those of p140Cap knockdown, and can be rescued by simultaneous overexpression of p140Cap or Citron-N [38, 113]. Notably, while overexpressed p140Cap tracks MT plus-ends in neurons, this is rarely the case for endogenous p140Cap. p140Cap’s affinity for local binding partners in spines is likely sufficiently high to prevent cytoplasmic diffusion necessary for MT plus-end tracking behavior [38, 114]. In addition to its function at the postsynapse, p140Cap likely plays a role at the presynapse where it interacts with several proteins implicated in synaptic vesicle secretion [115, 116].
Another interesting SxIP-containing partner of EB1 is the Stromal interaction molecule 1 or STIM1 [117], a transmembrane ER protein which contains one SxIP motif and can thus link EB-decorated MT plus-ends to the ER membrane. STIM1 regulates store operated calcium entry (SOCE) in neurons and is necessary to resupply the ER with calcium by opening plasma membrane channels after calcium release during synaptic signaling [118, 119]. The function of the interaction of STIM1 with MT tips is not yet entirely clear. STIM1 participates in ER tubule extension by coupling growing MT plus-ends to the ER [117], and it is possible that such ER remodeling contributes to SOCE in certain cell types by bringing STIM1 in the vicinity of the plasma membrane. Interestingly, in HEK293T cells changes in MT dynamics affect STIM1’s association with calcium channels [120], suggesting that in some cell types MT dynamics are important for calcium signaling, possibly via the action of +TIPs. It remains to be verified, however, what the exact contribution of STIM1’s plus-end tracking behavior is to SOCE in neurons. Part of the answer may come from the function of STIM2, a STIM1 homolog with 63 % sequence identity but without conservation of the SxIP motif. STIM2 regulates SOCE instead of STIM1 in pyramidal neurons of the neocortex [121], suggesting that plus-end tracking is not required for STIM functioning during SOCE in neurons. However, remodeling of the ER was observed along MTs during SOCE [122]. As STIM1 was found to remodel the ER via its interaction with EB1 in HeLa cells [117], there is a possibility STIM1 may exert additional, potentially plus-end tracking-dependent functions in neurons. This is underlined by the finding that STIM1 participates in growth cone steering, and that this effect is only coupled to effects on SOCE for certain guidance cues [118].
To summarize, SxIP motif-containing proteins represent a large and heterogeneous group of EB-dependent +TIPs, which exert different functions at different stages of neurodevelopment. Although many roles have been identified (Table 1), it is not always clear what the contribution of plus-end tracking behavior is to each of these functions.
Other +TIPs
While EB proteins are responsible for targeting a large variety of proteins to MT tips, several major classes of MT plus-end interacting factors target the growing MT plus-end via other mechanisms. These include the MT polymerase ch-TOG/XMAP215 and certain kinesin motor proteins, such as kinesin-4, -8 and -13 family members (see below). In addition, for some MT- or tubulin-binding proteins, such as doublecortin and stathmin, specific interaction with MT plus-ends was established or proposed based on in vitro reconstitution experiments [123, 124]. However, this was not demonstrated in cells, and is therefore not further discussed here.
ch-TOG/XMAP215 can track the growing plus-ends of MTs directly by recognizing the outmost MT tips [125, 126], or indirectly, via EB proteins. In mammalian cells, MT plus-end tracking of ch-TOG is facilitated by binding to SLAIN proteins, which themselves are EB-dependent, SxIP-containing +TIPs that have the ability to bind a number of other +TIPs [127]. Both SLAIN1/2 and ch-TOG are enriched in mammalian brain tissue, and promote MT growth by positioning the MT polymerase at the tip of the growing MT [128]. Like the majority of +TIPs implicated in maintaining MT cytoskeleton integrity, ch-TOG plays a role in axon outgrowth. Depletion of ch-TOG increases catastrophe rates in all subcellular compartments of rat hippocampal neurons, while reducing MT growth rates [128]. Axon outgrowth defects are apparent in young neurons depleted of ch-TOG, as well as in those overexpressing a dominant negative SLAIN construct that prevents ch-TOG from accumulating at the MT plus-end [128]. In Xenopus, the ch-TOG homolog XMAP215 is necessary for MTs to resist axon retraction induced by contractile actin forces and thereby promotes persistent axon outgrowth [129]. Interestingly, depletion of XMAP215 results in an increased rate of MT plus-end displacement specifically in the growth cone but not in axons. This effect does not seem to depend on XMAP215’s MT-polymerizing function, which relies on plus-end localization, but rather seems to be a result of an additional role for Xenopus XMAP215 in MT sliding [129].
The kinesin-4 family member KIF21A is also involved in axon development. In HeLa cells, KIF21A is part of a cortical MT-anchoring complex that includes CLASP, where KIF21A acts as a growth inhibitor to prevent further polymerization of MTs that reach the cell cortex [130]. Missense mutations in KIF21A cause Congenital Fibrosis of the Extraocular Muscles type 1 (CFEOM1), a disease characterized by the patients’ inability to control eye movements due to defects in oculomotor nerve development [131]. These mutations were found to prevent KIF21A autoinhibition, promoting increased cortical MT growth inhibition via an EB-independent interaction of KIF21A with the MT plus-end [130, 132]. Mutant KIF21A results in growth cone and axon pathfinding defects in cultured neurons and knockin mice, suggesting that improper innervation of extraocular muscles in CFEOM1 is a result of misregulation of MT dynamics by KIF21A [130, 132].
Regulation of interactions between microtubules and +TIPs
It is clear that elaborate control of the MT cytoskeleton, which involves tight regulation of interactions between +TIPs and MTs, is of pivotal importance to neurons.
Since many +TIPs use the same mode of association with EBs, competitive binding between +TIPs from the same subclass is a major factor in the regulation of interactions. This is illustrated by the use of SxIP-motif containing peptides to disrupt +TIP complexes and MT dynamics in the literature (e.g. [133, 134]), and such competition was proposed between CLIP-115 and CLIP-170, for example [85]. However, competition between different subclasses has also been reported. For instance, the SxIP-motif containing Neuron Navigator (NAV) +TIPs have been shown to displace p150glued from MT plus-ends upon overexpression [135], although p150glued relies on CAP-Gly domains to associate with EBs. Small SxIP peptides can also abolish p150glued and CLIP binding to MT plus-ends in in vitro reconstitution assays, confirming that binding sites on EBs for different +TIPs of different subclasses overlap at least partly [133]. Additionally, it will be interesting to see whether +TIPs can provide indirect feedback to other +TIPs by impacting cytoskeleton dynamics or by affecting the conformation of EB proteins.
Not all dominant +TIP-EB interactions negatively impact the recruitment of other +TIPs. For instance, EB1 is sufficient to independently recruit both p150glued and CLIP-170 to the MT plus-end in reconstitution assays using purified proteins, but p150glued binds the plus-end tighter in the presence of CLIP-170 because of an additional interaction between the two +TIPs [133]. A similar mechanism is employed by SLAIN2, which likely uses interactions with multiple +TIPs to overcome the issue of competition [127]. Intuitively, multiplying the number of binding modules should also increase the affinity of +TIPs for the MT plus-end. This has been shown for both the repetition of SxIP motifs within individual proteins and for increases in the number of SxIP motifs via oligomerization [42].
Apart from effects arising from the presence of other +TIPs at the MT plus-end, modifications of MTs or +TIPs provide additional layers of control. At the MT level, post-translational tubulin modifications favor binding of certain +TIPs over others. For example, CAP-Gly +TIPs only associate with MT plus-ends containing tyrosinated α-tubulin [71, 136]. Presence of the C-terminal tyrosine on α-tubulin also promotes MT interaction with another +TIP, the SxIP-containing kinesin-13 KIF2C/MCAK (mitotic centromere-associated kinesin), which has a MT-destabilizing function [137]. MAPs present on the MT lattice may also contribute to regulation of +TIP binding: MAP1B is able to capture cytosolic EBs and immobilize them along MTs, effectively lowering the concentration of EBs at the plus-end of the MT and thereby fine-tuning axon outgrowth [138]. Likewise, MAP2 recruits EBs to the MT lattice in dendrites upon synaptic stimulation [139] and tau was recently reported to bind to EB1. Tau expression levels may also regulate EB localization, as high levels of tau result in EB immobilization along the MT lattice [10].
At the +TIP level, phosphorylation is considered the classic mechanism to regulate binding. Phosphorylation of +TIPs results in unfavorable electrostatic interactions with negatively charged MTs and may promote +TIP accumulation at the MT plus-end rather than along the MT lattice, or abrogate binding altogether [15, 114]. Phosphorylation of +TIPs in the vicinity of SxIP motifs can also suppress binding to the negatively charged C-terminal part of EBs and thus the plus-end tracking [42]. Many +TIPs involved in axon outgrowth are substrates of GSK3β [92, 140–142], a kinase involved in prominent signaling pathways such as the phosphoinositide 3-kinase (PI3K)/Akt pathway and Wnt signaling. In addition to the previously discussed role of CLASP as a GSK3β substrate during axon outgrowth, GSK3β exerts control on the +TIP APC during growth cone advance [140, 143]. Binding of the spectraplakin +TIP MACF1 to MTs is also under control of GSK3β, and the GSK3β-MACF1 interaction plays a role in pyramidal neuron migration [142, 144]. Interestingly, both APC and MACF1 were suggested to regulate GSK3β activity during Wnt signaling [145, 146], hinting at the existence of complex feedback loops between +TIPs and signaling pathways during neurodevelopment.
A second kinase with strong connections to +TIP regulation is the Abelson kinase (Abl). Together with its substrate, Abelson interacting protein (Abi), Abl orchestrates actin dynamics important for Drosophila axon guidance and synaptogenesis [90] and appears to link +TIPs to the actin remodeling machinery. The +TIP NAV2 can promote actin polymerization by interacting with Abi at sites targeted by pioneer MTs [147]. Abl also interacts with p140Cap and is required for p140Cap-mediated actin remodeling [148], although this interaction has not yet been explored in neurons. Interestingly, Abl controls axon guidance via CLASP, which is also under control of GSK3β during the same process [90, 93]. Possibly, multiple signaling pathways and kinases act in parallel on the same +TIPs to allow additional levels of control.
Although phosphorylation remains the best-studied mechanism for regulation of MT-+TIP associations, other types of regulation have started to gain attention. For example, EB1 acetylation has been postulated to regulate binding of the SxIP motif-containing +TIP DDA3 during directional cell migration [149]. Intracellular calcium levels determine whether the MACF2 isoform BPAG1n4 localizes to the lattice or to the plus-end of MTs [150]. Finally, since individual EB proteins display different affinities for their binding partners [56, 151], their expression levels can also affect the composition of +TIP networks in manner dependent on the cell type or developmental stage.
Microtubule plus-end tracking proteins in brain diseases
For certain neurological disorders, MT dynamics have been examined by live imaging of +TIPs. In the case of multiple sclerosis, a neuroinflammatory condition associated with axonal transport defects and motor neuron degeneration, the number of EB3-positive MT plus-ends was found to increase and their directionality was altered in swollen axons of mouse models [152]. MT plus-end dynamics were also investigated in C. elegans after axon damage, which can occur after spinal cord injury or stroke in humans. As expected, axon severing generated a large amount of dynamic MT plus-ends at the newly formed tip of the axon, but interestingly axon regrowth depended on the C. elegans EB homolog EBP-1 and could be inhibited by overexpression the MT catastrophe-promoting protein EFA-6 (Exchange Factor for Arf6) [153]. Consistently, axon injury or stress induced by the expression of expanded polyglutamine proteins led to increased MT dynamics, which had a neuroprotective role by delaying or counteracting neuron degeneration in Drosophila [154].
While such studies strongly imply that regulation of MT dynamics is important in the response to neurological damage and disease, the underlying mechanisms are still poorly understood. Due to their diverse range of neurodevelopmental functions, many +TIPs are involved in human neurological disorders (Table 1), with mutations in certain +TIPs found to directly cause neurological disease. In this section, we highlight two +TIPs and associated diseases for which the role of plus-end tracking has been investigated at least to some extent: p150glued and tau-tubulin kinase 2 (TTBK2). In addition we consider TTBK1, the closest homolog of TTBK2.
p150glued in Perry syndrome and hereditary motor neuropathy 7B
As described earlier in this review, p150glued is an essential component of the retrograde axonal transport machinery by regulating and positioning dynein at MT plus-ends. Mutations in the CAP-Gly domain of p150glued, which is required for p150glued binding to EBs and MTs [155, 156], cause two distinct neurological disorders: Perry syndrome and hereditary motor neuropathy 7B (HMN7B). HMN7B affects motor neurons. Symptoms ensue in early adulthood and include muscle atrophy, vocal fold paralysis and breathing difficulties [157]. By contrast, Perry syndrome (reviewed in [158]) mainly affects neurons in the substantia nigra but not motor neurons. This rare syndrome manifests itself around 46 years of age and is associated with parkinsonism, depression, hypoventilation and weight loss.
Interestingly, the p150glued mutations that give rise to these different conditions are in close proximity: G59S mutations cause HMN7B [157], and G71(R/E/A), T72P and Q74P mutations were identified in Perry syndrome patients [159]. Differences between these two diseases may in part be explained by the effect of these mutations on the stability of p150glued. In case of the HMN7B mutation, mutant p150glued aggregates and is incorporated into inclusion bodies [160, 161]. However, p150glued’s global folding and stability is largely unaffected by Perry syndrome mutations [161]. This observation is reflected by the presence of dynactin aggregates in motor neurons of HMN7B patients [162], which are believed to contribute to cell death [160]. Such inclusions are less common in Perry syndrome patients [159]. All mutations disturb the interactions between p150glued and EB proteins or MTs [157, 159, 163], but functional differences between mutated forms of p150glued were also reported. The HMN7B mutation globally perturbs axonal transport by disturbing dynactin binding to dynein when mutant p150glued is incorporated in the dynactin complex. Conversely, Perry syndrome mutations do not affect global axonal transport, but G71R p150glued has a dominant negative effect on the initiation of retrograde trafficking from distal axon tips [163]. This function directly depends on p150glued’s interaction with EB proteins at the MT plus-end [76]. Another potentially disease-related mechanism is the MT catastrophe-suppressing function of p150glued, which is disturbed by the Perry syndrome mutation Q74P [77]. p150glued’s ability to suppress catastrophes relies on its binding to MTs and free tubulin dimers, and is independent of EB proteins. p150glued isoforms harboring all domains necessary for these interactions are primarily expressed in neurons, rendering the effect neuron-specific.
p150glued’s involvement in Perry syndrome and HMN7B is an excellent example of how deficiencies in plus-end tracking can result in neurological disease. It underlines the importance of investigating the role of plus-end tracking for +TIPs implicated in disorders of the brain, yet surprisingly, p150glued remains the single best-studied case.
TTBK1/2 in spinocerebellar ataxia and Alzheimer’s disease
Two other, closely related +TIPs firmly implicated in brain disease are tau-tubulin kinase 1 and 2 (TTBK1 and TTBK2). TTBK1/2 belong to the casein kinase 1 group and share 60 % sequence identity, primarily between their N-terminal kinase domains (reviewed in [164]). Both TTBK proteins contain two SxIP motives in their C-terminal tail [56], which is only present in vertebrates [164].
TTBK2 was the first tau-tubulin kinase to be identified, phosphorylating MAP2 and α-casein in addition to tau and tubulin [165]. TTBK2 is expressed ubiquitously [166] and can phosphorylate tau at sites identified in paired helical filament tau, a hyperphosphorylated tau variant found in the brains of Alzheimer’s disease (AD) patients [166]. Mutations in TTBK2, which yield a mutant protein truncated after the N-terminal kinase domain, cause spinocerebellar ataxia type 11 (SCA11) [167]: a rare neurodegenerative disease of which symptoms include pronunciation difficulties, involuntary eye movement and ataxia [168]. So far, the brain of one SCA11 patient has been examined and revealed the presence of tau deposits among other signs of pathological aging and cerebellar degeneration, raising the possibility that aberrant tau phosphorylation by mutant TTBK2 contributes to SCA11 pathology [167]. In addition, TTBK2 was more recently shown to be required for the formation of cilia by promoting the removal of the centriolar capping protein CEP110 to allow axoneme extension [169]. Diseases resulting from cilia defects, termed ciliopathies, often affect the brain (reviewed in [170]), and mice harboring a mutation that prematurely truncates TTBK2 lack certain neural cell types known to be lost in cilia-depleted animal models [169]. Since the SCA11-mutated form of TTBK2 is unable to initiate ciliogenesis and can interfere with the ciliary function of full-length TTBK2, TTBK2’s function in cilia formation provides a second explanation for the symptoms of SCA11 [169]. However, the EB1-TTBK2 interaction plays no role in the recruitment of TTBK2 to the basal body or in the subsequent initiation of ciliogenesis [171]. Recently, TTBK2 was found to phosphorylate the kinesin-13 family member KIF2A at the plus-end of MTs in an EB-dependent manner. This interaction regulates the binding and thereby depolymerization of MTs by KIF2A in HeLa cells [172]. The same study suggested that TTBK2 can exist in an auto-inhibited conformation, wherein the kinase domain is folded back onto the tail domain containing the SxIP motifs. Binding of EB would liberate the kinase domain and promote kinase activity, presenting an attractive mechanism for how TTBK kinase activity can be regulated at the plus-end of MTs [172]. Interestingly, overexpressed TTBK2 is also able to displace EB1 from the MT plus-end, and TTBK2 affinity to MTs itself appears to be regulated by its autophosphorylation, suggesting that TTBK2 has the potential to regulate +TIP-MT associations [56]. These studies clearly signal the importance of TTBK2’s interaction with EBs and MT plus-ends. The direct relevance of plus-end tracking for neuronal functions of TTBK2 other than ciliogenesis has not been investigated. It is thus currently unclear whether the functions attributed to TTBK2, such as the earlier discussed pathological tau phosphorylation, a role in neuronal migration [172], regulation of the retrieval of synaptotagmin-1 during synaptic vesicle endocytosis [173] or the control of the activity of transport channels including BGT1, a betaine/γ-amino-butyric acid (GABA) transporter [174], require TTBK2 plus-end tracking.
Full-length TTBK1 was only characterized in 2006 as a neuron-specific kinase with the ability to phosphorylate tau on sites associated with AD [175]. During AD, tau becomes hyperphosphorylated, causing it to detach from MTs and form non-soluble aggregates in the cytosol. Certain genetic variations of TTBK1 have been linked to lower risk of developing AD [176, 177], suggesting that TTBK1 and variations in its expression levels may play a role in Alzheimer pathology. Full-length TTBK1 co-localizes with EB1 and bundles MTs at high expression levels independent of its kinase activity [56], but the contribution of TTBK1 plus-end tracking and MT modulation has never been investigated with respect to its function or involvement in disease.
More recently, TTBK1 and TTBK2 were also shown to phosphorylate TDP-43 (transactive response DNA binding protein of 43 kDa) [178], the phosphorylation-driven aggregation of which is a hallmark of amyotrophic lateral sclerosis and frontotemporal lobar degeneration as well as numerous other neurodegenerative diseases [179]. Understanding the cell biology of these kinases both in health and disease, as well as mapping the full range of substrates of TTBKs will be a crucial step towards the identification of targets for therapeutic intervention.
Future outlook
As versatile regulators of the MT cytoskeleton, +TIPs play important roles in all aspects of neurodevelopment. It is therefore not surprising that +TIPs are involved in many neurological diseases (Table 1). Although more is known about how individual +TIPs affect MT behavior and integrity, in many cases this knowledge is yet to be extrapolated to neuronal MT networks and in some cases insight is lacking altogether.
It is crucial to develop such understanding as pharmacological intervention to inhibit or promote +TIP activity, and thereby affect specific subsets or properties of MTs, likely provides a more elegant and refined approach to therapy development than the use of MT targeting agents that globally impact MT dynamics. The influence of low doses of MT-targeting drugs on neurons has been studied extensively and was found to impact nearly all processes in developing and mature neurons. For example, treatment with 15–75 nM of the MT destabilizer nocodazole impairs the formation (but not outgrowth) of neurites in young dissociated neurons [23]. At 200 nM, nocodazole was also reported to inhibit primary dendrite formation [83] and has the ability to transform mature, mushroom-headed spines of dendrites into filopodia [38]. These morphological spine changes are paired with decreased synaptic response [38]. The MT stabilizing drug taxol, on the other hand, is known to induce the formation of multiple axons in young dissociated neurons at concentrations as low as 3 nM [23], and nanomolar concentrations of both MT stabilizing and destabilizing drugs affect axon pathfinding [28]. The above effects on neurons were mostly found to be a result of altered MT dynamics, rather than drug-specific effects on other cellular processes. It is therefore reasonable to assume that treatment with other MT-targeting agents would result in similar effects on neuronal development and functioning.
MT-targeting agents do provide evidence that drugs altering MT dynamics may be successful in treating brain diseases. For example, the MT stabilizing, blood–brain barrier-penetrant drug epothilone D shows promise as a therapeutic agent in neurological disorders, as epothilone D treatment partially relieves pathology in AD mouse models [180]. Recently, the closely related MT-stabilizing agent epothilone B also proved to stimulate regeneration of axons after spinal cord injury in rats [181]. However, in contrast to the global effect of MT-targeting drugs which can yield unintended side effects, targeting individual +TIPs that are restricted to certain subcellular localizations or tissues, expressed at certain times or influence only limited aspects of MT behavior may be preferable. Even when +TIPs are not involved in establishing pathogenesis, they can potentially be stimulated or inhibited to counteract MT-related disease mechanisms. Of note, it has recently been proposed that the neuroprotective peptide NAP possibly exerts its effects via EBs. NAP affects the brain-specific tubulin pool as well as MT stability and dynamics [182, 183], and has been described to have beneficial effects in tauopathy animal models [184–186]. NAP is believed to bind EB3 via an alternative SxIP motif, Ser-Ile-Pro, and EB silencing in PC12 cells obliterates the protective effect of NAP to zinc toxicity. Although the precise mechanism remains unclear, these data suggest that NAP’s binding to EBs may contribute to its neuroprotective properties [187].
In summary, +TIPs may offer a more precise approach to interfere with pathological MT-related processes, rather than conventional MT-targeting agents currently used in the clinic. In order to truly establish their potential as therapeutic targets for treatment against neurological disorders, more research is needed into the functions and mechanisms of action of an ever-growing pool of +TIPs. New +TIPs are still being recognized on a large scale (e.g. [56]), rendering the field of +TIP research an exciting and promising one for years to come.
Notes
With the terminology ‘plus-end out’, we designate MTs with their plus-end oriented towards the cell periphery, or towards dendritic or axonal tips. ‘Minus-end out’ indicates MTs of which the minus-end is oriented towards the cell periphery or process tips.
Abbreviations
- Abi:
-
Abelson interacting protein
- Abl:
-
Abelson kinase
- AD:
-
Alzheimer’s disease
- AIS:
-
Axon initial segment
- BDNF:
-
Brain-derived neurotrophic factor
- CAMSAP:
-
Calmodulin-regulated spectrin-associated protein
- CAP-Gly:
-
Cytoskeletal-associated protein glycine-rich
- CEP:
-
Centrosomal protein
- CFEOM1:
-
Congenital fibrosis of the extraocular muscles type 1
- CH:
-
Calponin homology
- CLASP:
-
Cytoplasmic linker protein-associated protein
- CLIP:
-
Cytoplasmic linker protein
- DRG:
-
Dorsal root ganglia
- EB:
-
End-binding protein
- EFA-6:
-
Exchange factor for Arf6
- EM:
-
Electron microscopy
- ER:
-
Endoplasmic reticulum
- GAR:
-
Growth arrest-specific 2 protein-related region
- GSK3β:
-
Glycogen synthase kinase 3 beta
- HMN7B:
-
Hereditary motor neuropathy 7B
- MACF:
-
Microtubule-actin crosslinking factor
- MAP:
-
Microtubule-associated protein
- MCAK:
-
Mitotic centromere-associated kinesin
- MT:
-
Microtubule
- NAV:
-
Neuron navigator
- NMDA:
-
N-Methyl-d-aspartate
- PI3K:
-
Phosphoinositide 3-kinase
- SCA11:
-
Spinocerebellar ataxia type 11
- SOCE:
-
Store operated calcium entry
- STIM1:
-
Stromal interaction molecule 1
- TDP-43:
-
Transactive response DNA binding protein of 43 kDa
- +TIP:
-
Microtubule plus-end tracking protein
- TOG:
-
Tumor overexpressed gene
- TTBK:
-
Tau-tubulin kinase
References
Hoogenraad CC, Bradke F (2009) Control of neuronal polarity and plasticity–a renaissance for microtubules? Trends Cell Biol 19(12):669–676. doi:10.1016/j.tcb.2009.08.006
Conde C, Caceres A (2009) Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci 10(5):319–332. http://www.nature.com/nrn/journal/v10/n5/suppinfo/nrn2631_S1.html
Kapitein LC, Hoogenraad CC (2015) Building the neuronal microtubule cytoskeleton. Neuron 87(3):492–506. doi:10.1016/j.neuron.2015.05.046
Wells WA (2005) Microtubules get a name. J Cell Biol 168(6):852–853. doi:10.1083/jcb1686fta1
Slautterback DB (1963) Cytoplasmic microtubules. I. Hydra. J Cell Biol 18:367–388
Ledbetter MC, Porter KR (1963) A “microtubule” in plant cell fine structure. J Cell Biol 19(1):239–250
Desai A, Mitchison TJ (1997) Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13:83–117. doi:10.1146/annurev.cellbio.13.1.83
Dehmelt L, Halpain S (2005) The MAP2/Tau family of microtubule-associated proteins. Genome Biol 6(1):204. doi:10.1186/gb-2004-6-1-204
Lee G, Leugers CJ (2012) Tau and tauopathies, chapter 8. In: David BT (ed) Progress in molecular biology and translational science, vol 107. Academic Press, New York, pp 263–293. doi:10.1016/B978-0-12-385883-2.00004-7
Sayas CL, Tortosa E, Bollati F, Ramirez-Rios S, Arnal I, Avila J (2015) Tau regulates the localization and function of End-binding proteins 1 and 3 in developing neuronal cells. J Neurochem 133(5):653–667. doi:10.1111/jnc.13091
Jiang K, Hua S, Mohan R, Grigoriev I, Yau KW, Liu Q, Katrukha EA, Altelaar AF, Heck AJ, Hoogenraad CC, Akhmanova A (2014) Microtubule minus-end stabilization by polymerization-driven CAMSAP deposition. Dev Cell 28(3):295–309. doi:10.1016/j.devcel.2014.01.001
Akhmanova A, Hoogenraad CC (2015) Microtubule minus-end-targeting proteins. Curr Biol 25(4):R162–R171. doi:10.1016/j.cub.2014.12.027
Nogales E (2000) Structural insights into microtubule function. Annu Rev Biochem 69:277–302. doi:10.1146/annurev.biochem.69.1.277
Kumar P, Wittmann T (2012) +TIPs: SxIPping along microtubule ends. Trends Cell Biol 22(8):418–428. doi:10.1016/j.tcb.2012.05.005
Akhmanova A, Steinmetz MO (2008) Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol 9(4):309–322. doi:10.1038/nrm2369
Akhmanova A, Steinmetz MO (2015) Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 16(12):711–726. doi:10.1038/nrm4084
Poulain FE, Sobel A (2010) The microtubule network and neuronal morphogenesis: dynamic and coordinated orchestration through multiple players. Mol Cell Neurosci 43(1):15–32. doi:10.1016/j.mcn.2009.07.012
Sanchez-Soriano N, Bottenberg W, Fiala A, Haessler U, Kerassoviti A, Knust E, Lohr R, Prokop A (2005) Are dendrites in Drosophila homologous to vertebrate dendrites? Dev Biol 288(1):126–138. doi:10.1016/j.ydbio.2005.09.026
Kuijpers M, Hoogenraad CC (2011) Centrosomes, microtubules and neuronal development. Mol Cell Neurosci 48(4):349–358. doi:10.1016/j.mcn.2011.05.004
Gotz M, Huttner WB (2005) The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6(10):777–788. doi:10.1038/nrm1739
Dehmelt L, Nalbant P, Steffen W, Halpain S (2006) A microtubule-based, dynein-dependent force induces local cell protrusions: implications for neurite initiation. Brain Cell Biol 35(1):39–56. doi:10.1007/s11068-006-9001-0
Lu W, Fox P, Lakonishok M, Davidson MW, Gelfand VI (2013) Initial neurite outgrowth in Drosophila neurons is driven by kinesin-powered microtubule sliding. Curr Biol 23(11):1018–1023. doi:10.1016/j.cub.2013.04.050
Witte H, Neukirchen D, Bradke F (2008) Microtubule stabilization specifies initial neuronal polarization. J Cell Biol 180(3):619–632. doi:10.1083/jcb.200707042
Geraldo S, Gordon-Weeks PR (2009) Cytoskeletal dynamics in growth-cone steering. J Cell Sci 122(Pt 20):3595–3604. doi:10.1242/jcs.042309
Dent EW, Gertler FB (2003) Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 40(2):209–227
Tanaka E, Ho T, Kirschner MW (1995) The role of microtubule dynamics in growth cone motility and axonal growth. J Cell Biol 128(1–2):139–155
Tanaka EM, Kirschner MW (1991) Microtubule behavior in the growth cones of living neurons during axon elongation. J Cell Biol 115(2):345–363
Buck KB, Zheng JQ (2002) Growth cone turning induced by direct local modification of microtubule dynamics. J Neurosci 22(21):9358–9367
Dent EW, Kalil K (2001) Axon branching requires interactions between dynamic microtubules and actin filaments. J Neurosci 21(24):9757–9769
Yau KW, Schatzle P, Tortosa E, Pages S, Holtmaat A, Kapitein LC, Hoogenraad CC (2016) Dendrites in vitro and in vivo contain microtubules of opposite polarity and axon formation correlates with uniform plus-end-out microtubule orientation. J Neurosci 36(4):1071–1085. doi:10.1523/jneurosci.2430-15.2016
Baas PW, Deitch JS, Black MM, Banker GA (1988) Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci USA 85(21):8335–8339
Baas PW, Black MM, Banker GA (1989) Changes in microtubule polarity orientation during the development of hippocampal neurons in culture. J Cell Biol 109(6 Pt 1):3085–3094
Stone MC, Roegiers F, Rolls MM (2008) Microtubules have opposite orientation in axons and dendrites of Drosophila neurons. Mol Biol Cell 19(10):4122–4129. doi:10.1091/mbc.E07-10-1079
Maniar TA, Kaplan M, Wang GJ, Shen K, Wei L, Shaw JE, Koushika SP, Bargmann CI (2012) UNC-33 (CRMP) and ankyrin organize microtubules and localize kinesin to polarize axon-dendrite sorting. Nat Neurosci 15(1):48–56. http://www.nature.com/neuro/journal/v15/n1/abs/nn.2970.html#supplementary-information
Ori-McKenney KM, Jan LY, Jan YN (2012) Golgi outposts shape dendrite morphology by functioning as sites of acentrosomal microtubule nucleation in neurons. Neuron 76(5):921–930. doi:10.1016/j.neuron.2012.10.008
Ye B, Zhang Y, Song W, Younger SH, Jan LY, Jan YN (2007) Growing dendrites and axons differ in their reliance on the secretory pathway. Cell 130(4):717–729. doi:10.1016/j.cell.2007.06.032
Matus A (2000) Actin-based plasticity in dendritic spines. Science (New York, NY) 290(5492):754–758
Jaworski J, Kapitein LC, Gouveia SM, Dortland BR, Wulf PS, Grigoriev I, Camera P, Spangler SA, Di Stefano P, Demmers J, Krugers H, Defilippi P, Akhmanova A, Hoogenraad CC (2009) Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 61(1):85–100. doi:10.1016/j.neuron.2008.11.013
Hu X, Viesselmann C, Nam S, Merriam E, Dent EW (2008) Activity-dependent dynamic microtubule invasion of dendritic spines. J Neurosci 28(49):13094–13105. doi:10.1523/jneurosci.3074-08.2008
Gu J, Firestein BL, Zheng JQ (2008) Microtubules in dendritic spine development. J Neurosci 28(46):12120–12124. doi:10.1523/jneurosci.2509-08.2008
Merriam EB, Millette M, Lumbard DC, Saengsawang W, Fothergill T, Hu X, Ferhat L, Dent EW (2013) Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J Neurosci 33(42):16471–16482. doi:10.1523/jneurosci.0661-13.2013
Honnappa S, Gouveia SM, Weisbrich A, Damberger FF, Bhavesh NS, Jawhari H, Grigoriev I, van Rijssel FJ, Buey RM, Lawera A, Jelesarov I, Winkler FK, Wuthrich K, Akhmanova A, Steinmetz MO (2009) An EB1-binding motif acts as a microtubule tip localization signal. Cell 138(2):366–376. doi:10.1016/j.cell.2009.04.065
Bieling P, Laan L, Schek H, Munteanu EL, Sandblad L, Dogterom M, Brunner D, Surrey T (2007) Reconstitution of a microtubule plus-end tracking system in vitro. Nature 450(7172):1100–1105. doi:10.1038/nature06386
Komarova Y, De Groot CO, Grigoriev I, Gouveia SM, Munteanu EL, Schober JM, Honnappa S, Buey RM, Hoogenraad CC, Dogterom M, Borisy GG, Steinmetz MO, Akhmanova A (2009) Mammalian end binding proteins control persistent microtubule growth. J Cell Biol 184(5):691–706. doi:10.1083/jcb.200807179
Slep KC, Vale RD (2007) Structural basis of microtubule plus end tracking by XMAP215, CLIP-170, and EB1. Mol Cell 27(6):976–991. doi:10.1016/j.molcel.2007.07.023
Maurer SP, Fourniol FJ, Bohner G, Moores CA, Surrey T (2012) EBs recognize a nucleotide-dependent structural cap at growing microtubule ends. Cell 149(2):371–382. doi:10.1016/j.cell.2012.02.049
Zhang R, Alushin GM, Brown A, Nogales E (2015) Mechanistic origin of microtubule dynamic instability and its modulation by EB proteins. Cell 162(4):849–859. doi:10.1016/j.cell.2015.07.012
Buey RM, Mohan R, Leslie K, Walzthoeni T, Missimer JH, Menzel A, Bjelic S, Bargsten K, Grigoriev I, Smal I, Meijering E, Aebersold R, Akhmanova A, Steinmetz MO (2011) Insights into EB1 structure and the role of its C-terminal domain for discriminating microtubule tips from the lattice. Mol Biol Cell 22(16):2912–2923. doi:10.1091/mbc.E11-01-0017
De Groot CO, Jelesarov I, Damberger FF, Bjelic S, Scharer MA, Bhavesh NS, Grigoriev I, Buey RM, Wuthrich K, Capitani G, Akhmanova A, Steinmetz MO (2010) Molecular insights into mammalian end-binding protein heterodimerization. J Biol Chem 285(8):5802–5814. doi:10.1074/jbc.M109.068130
Slep KC, Rogers SL, Elliott SL, Ohkura H, Kolodziej PA, Vale RD (2005) Structural determinants for EB1-mediated recruitment of APC and spectraplakins to the microtubule plus end. J Cell Biol 168(4):587–598. doi:10.1083/jcb.200410114
Dragestein KA, van Cappellen WA, van Haren J, Tsibidis GD, Akhmanova A, Knoch TA, Grosveld F, Galjart N (2008) Dynamic behavior of GFP-CLIP-170 reveals fast protein turnover on microtubule plus ends. J Cell Biol 180(4):729–737. doi:10.1083/jcb.200707203
Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol 7:548. doi:10.1038/msb.2011.81
Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R (2011) The quantitative proteome of a human cell line. Mol Syst Biol 7:549. doi:10.1038/msb.2011.82
Craft GE, Chen A, Nairn AC (2013) Recent advances in quantitative neuroproteomics. Methods (San Diego, Calif) 61(3):186–218. doi:10.1016/j.ymeth.2013.04.008
Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N (2003) Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein). J Neurosci 23(7):2655–2664
Jiang K, Toedt G, Montenegro Gouveia S, Davey NE, Hua S, van der Vaart B, Grigoriev I, Larsen J, Pedersen LB, Bezstarosti K, Lince-Faria M, Demmers J, Steinmetz MO, Gibson TJ, Akhmanova A (2012) A Proteome-wide screen for mammalian SxIP motif-containing microtubule plus-end tracking proteins. Curr Biol 22(19):1800–1807. doi:10.1016/j.cub.2012.07.047
Yue J, Xie M, Gou X, Lee P, Schneider MD, Wu X (2014) Microtubules regulate focal adhesion dynamics through MAP4K4. Dev Cell 31(5):572–585. doi:10.1016/j.devcel.2014.10.025
Komarova YA, Huang F, Geyer M, Daneshjou N, Garcia A, Idalino L, Kreutz B, Mehta D, Malik AB (2012) VE-cadherin signaling induces EB3 phosphorylation to suppress microtubule growth and assemble adherens junctions. Mol Cell 48(6):914–925. doi:10.1016/j.molcel.2012.10.011
Ban R, Matsuzaki H, Akashi T, Sakashita G, Taniguchi H, Park SY, Tanaka H, Furukawa K, Urano T (2009) Mitotic regulation of the stability of microtubule plus-end tracking protein EB3 by ubiquitin ligase SIAH-1 and Aurora mitotic kinases. J Biol Chem 284(41):28367–28381. doi:10.1074/jbc.M109.000273
Nakagawa H, Koyama K, Murata Y, Morito M, Akiyama T, Nakamura Y (2000) EB3, a novel member of the EB1 family preferentially expressed in the central nervous system, binds to a CNS-specific APC homologue. Oncogene 19(2):210–216. doi:10.1038/sj.onc.1203308
Su LK, Qi Y (2001) Characterization of human MAPRE genes and their proteins. Genomics 71(2):142–149. doi:10.1006/geno.2000.6428
Morrison EE, Moncur PM, Askham JM (2002) EB1 identifies sites of microtubule polymerisation during neurite development. Brain Res Mol Brain Res 98(1–2):145–152
Alves-Silva J, Sanchez-Soriano N, Beaven R, Klein M, Parkin J, Millard TH, Bellen HJ, Venken KJ, Ballestrem C, Kammerer RA, Prokop A (2012) Spectraplakins promote microtubule-mediated axonal growth by functioning as structural microtubule-associated proteins and EB1-dependent +TIPs (tip interacting proteins). J Neurosci 32(27):9143–9158. doi:10.1523/jneurosci.0416-12.2012
Jimenez-Mateos EM, Paglini G, Gonzalez-Billault C, Caceres A, Avila J (2005) End binding protein-1 (EB1) complements microtubule-associated protein-1B during axonogenesis. J Neurosci Res 80(3):350–359. doi:10.1002/jnr.20453
Arens J, Duong T-T, Dehmelt L (2013) A morphometric screen identifies specific roles for microtubule-regulating genes in neuronal development of P19 stem cells. PLoS One 8(11):e79796. doi:10.1371/journal.pone.0079796
Laketa V, Simpson JC, Bechtel S, Wiemann S, Pepperkok R (2007) High-content microscopy identifies new neurite outgrowth regulators. Mol Biol Cell 18(1):242–252. doi:10.1091/mbc.E06-08-0666
Geraldo S, Khanzada UK, Parsons M, Chilton JK, Gordon-Weeks PR (2008) Targeting of the F-actin-binding protein drebrin by the microtubule plus-tip protein EB3 is required for neuritogenesis. Nat Cell Biol 10(10):1181–1189. doi:10.1038/ncb1778
Kojima N, Shirao T (2007) Synaptic dysfunction and disruption of postsynaptic drebrin–actin complex: a study of neurological disorders accompanied by cognitive deficits. Neurosci Res 58(1):1–5. doi:10.1016/j.neures.2007.02.003
Leterrier C, Vacher H, Fache MP, d’Ortoli SA, Castets F, Autillo-Touati A, Dargent B (2011) End-binding proteins EB3 and EB1 link microtubules to ankyrin G in the axon initial segment. Proc Natl Acad Sci USA 108(21):8826–8831. doi:10.1073/pnas.1018671108
Bieling P, Kandels-Lewis S, Telley IA, van Dijk J, Janke C, Surrey T (2008) CLIP-170 tracks growing microtubule ends by dynamically recognizing composite EB1/tubulin-binding sites. J Cell Biol 183(7):1223–1233. doi:10.1083/jcb.200809190
Weisbrich A, Honnappa S, Jaussi R, Okhrimenko O, Frey D, Jelesarov I, Akhmanova A, Steinmetz MO (2007) Structure-function relationship of CAP-Gly domains. Nat Struct Mol Biol 14(10):959–967. doi:10.1038/nsmb1291
Mishima M, Maesaki R, Kasa M, Watanabe T, Fukata M, Kaibuchi K, Hakoshima T (2007) Structural basis for tubulin recognition by cytoplasmic linker protein 170 and its autoinhibition. Proc Natl Acad Sci USA 104(25):10346–10351. doi:10.1073/pnas.0703876104
Honnappa S, Okhrimenko O, Jaussi R, Jawhari H, Jelesarov I, Winkler FK, Steinmetz MO (2006) Key interaction modes of dynamic +TIP networks. Mol Cell 23(5):663–671. doi:10.1016/j.molcel.2006.07.013
Kardon JR, Vale RD (2009) Regulators of the cytoplasmic dynein motor. Nat Rev Mol Cell Biol 10(12):854–865. doi:10.1038/nrm2804
Vaughan PS, Miura P, Henderson M, Byrne B, Vaughan KT (2002) A role for regulated binding of p150(Glued) to microtubule plus ends in organelle transport. J Cell Biol 158(2):305–319. doi:10.1083/jcb.200201029
Moughamian AJ, Osborn GE, Lazarus JE, Maday S, Holzbaur EL (2013) Ordered recruitment of dynactin to the microtubule plus-end is required for efficient initiation of retrograde axonal transport. J Neurosci 33(32):13190–13203. doi:10.1523/jneurosci.0935-13.2013
Lazarus JE, Moughamian AJ, Tokito MK, Holzbaur EL (2013) Dynactin subunit p150(Glued) is a neuron-specific anti-catastrophe factor. PLoS Biol 11(7):e1001611. doi:10.1371/journal.pbio.1001611
Lansbergen G, Komarova Y, Modesti M, Wyman C, Hoogenraad CC, Goodson HV, Lemaitre RP, Drechsel DN, van Munster E, Gadella TW Jr, Grosveld F, Galjart N, Borisy GG, Akhmanova A (2004) Conformational changes in CLIP-170 regulate its binding to microtubules and dynactin localization. J Cell Biol 166(7):1003–1014. doi:10.1083/jcb.200402082
De Zeeuw CI, Hoogenraad CC, Goedknegt E, Hertzberg E, Neubauer A, Grosveld F, Galjart N (1997) CLIP-115, a novel brain-specific cytoplasmic linker protein, mediates the localization of dendritic lamellar bodies. Neuron 19(6):1187–1199. doi:10.1016/S0896-6273(00)80411-0
Komarova YA, Akhmanova AS, Kojima S, Galjart N, Borisy GG (2002) Cytoplasmic linker proteins promote microtubule rescue in vivo. J Cell Biol 159(4):589–599. doi:10.1083/jcb.200208058
Beaven R, Dzhindzhev NS, Qu Y, Hahn I, Dajas-Bailador F, Ohkura H, Prokop A (2015) Drosophila CLIP-190 and mammalian CLIP-170 display reduced microtubule plus end association in the nervous system. Mol Biol Cell 26(8):1491–1508. doi:10.1091/mbc.E14-06-1083
Neukirchen D, Bradke F (2011) Cytoplasmic linker proteins regulate neuronal polarization through microtubule and growth cone dynamics. J Neurosci 31(4):1528–1538. doi:10.1523/jneurosci.3983-10.2011
Swiech L, Blazejczyk M, Urbanska M, Pietruszka P, Dortland BR, Malik AR, Wulf PS, Hoogenraad CC, Jaworski J (2011) CLIP-170 and IQGAP1 cooperatively regulate dendrite morphology. J Neurosci 31(12):4555–4568. doi:10.1523/jneurosci.6582-10.2011
Akhmanova A, Mausset-Bonnefont AL, van Cappellen W, Keijzer N, Hoogenraad CC, Stepanova T, Drabek K, van der Wees J, Mommaas M, Onderwater J, van der Meulen H, Tanenbaum ME, Medema RH, Hoogerbrugge J, Vreeburg J, Uringa EJ, Grootegoed JA, Grosveld F, Galjart N (2005) The microtubule plus-end-tracking protein CLIP-170 associates with the spermatid manchette and is essential for spermatogenesis. Genes Dev 19(20):2501–2515. doi:10.1101/gad.344505
Hoogenraad CC, Koekkoek B, Akhmanova A, Krugers H, Dortland B, Miedema M, van Alphen A, Kistler WM, Jaegle M, Koutsourakis M, Van Camp N, Verhoye M, van der Linden A, Kaverina I, Grosveld F, De Zeeuw CI, Galjart N (2002) Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nat Genet 32(1):116–127. doi:10.1038/ng954
Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, Vermeulen W, Burgering BM, De Zeeuw CI, Grosveld F, Galjart N (2001) Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell 104(6):923–935
Leano JB, Rogers SL, Slep KC (2013) A cryptic TOG domain with a distinct architecture underlies CLASP-dependent bipolar spindle formation. Structure (London, England: 1993) 21(6):939–950. doi:10.1016/j.str.2013.04.018
Al-Bassam J, Chang F (2011) Regulation of microtubule dynamics by TOG-domain proteins XMAP215/Dis1 and CLASP. Trends Cell Biol 21(10):604–614. doi:10.1016/j.tcb.2011.06.007
Mimori-Kiyosue Y, Grigoriev I, Lansbergen G, Sasaki H, Matsui C, Severin F, Galjart N, Grosveld F, Vorobjev I, Tsukita S, Akhmanova A (2005) CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J Cell Biol 168(1):141–153. doi:10.1083/jcb.200405094
Lee H, Engel U, Rusch J, Scherrer S, Sheard K, Van Vactor D (2004) The microtubule plus end tracking protein Orbit/MAST/CLASP acts downstream of the tyrosine kinase Abl in mediating axon guidance. Neuron 42(6):913–926. doi:10.1016/j.neuron.2004.05.020
Marx A, Godinez WJ, Tsimashchuk V, Bankhead P, Rohr K, Engel U (2013) Xenopus cytoplasmic linker-associated protein 1 (XCLASP1) promotes axon elongation and advance of pioneer microtubules. Mol Biol Cell 24(10):1544–1558. doi:10.1091/mbc.E12-08-0573
Watanabe T, Noritake J, Kakeno M, Matsui T, Harada T, Wang S, Itoh N, Sato K, Matsuzawa K, Iwamatsu A, Galjart N, Kaibuchi K (2009) Phosphorylation of CLASP2 by GSK-3beta regulates its interaction with IQGAP1, EB1 and microtubules. J Cell Sci 122(Pt 16):2969–2979. doi:10.1242/jcs.046649
Hur EM, Saijilafu Lee BD, Kim SJ, Xu WL, Zhou FQ (2011) GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev 25(18):1968–1981. doi:10.1101/gad.17015911
Engel U, Zhan Y, Long JB, Boyle SN, Ballif BA, Dorey K, Gygi SP, Koleske AJ, Vanvactor D (2014) Abelson phosphorylation of CLASP2 modulates its association with microtubules and actin. Cytoskeleton (Hoboken, NJ) 71(3):195–209. doi:10.1002/cm.21164
Beffert U, Dillon GM, Sullivan JM, Stuart CE, Gilbert JP, Kambouris JA, Ho A (2012) Microtubule plus-end tracking protein CLASP2 regulates neuronal polarity and synaptic function. J Neurosci 32(40):13906–13916. doi:10.1523/jneurosci.2108-12.2012
Schmidt N, Basu S, Sladecek S, Gatti S, van Haren J, Treves S, Pielage J, Galjart N, Brenner HR (2012) Agrin regulates CLASP2-mediated capture of microtubules at the neuromuscular junction synaptic membrane. J Cell Biol 198(3):421–437. doi:10.1083/jcb.201111130
Suozzi KC, Wu X, Fuchs E (2012) Spectraplakins: master orchestrators of cytoskeletal dynamics. J Cell Biol 197(4):465–475. doi:10.1083/jcb.201112034
Sun D, Leung CL, Liem RK (2001) Characterization of the microtubule binding domain of microtubule actin crosslinking factor (MACF): identification of a novel group of microtubule associated proteins. J Cell Sci 114(Pt 1):161–172
Goryunov D, He CZ, Lin CS, Leung CL, Liem RK (2010) Nervous-tissue-specific elimination of microtubule-actin crosslinking factor 1a results in multiple developmental defects in the mouse brain. Mol Cell Neurosci 44(1):1–14. doi:10.1016/j.mcn.2010.01.010
Kodama A, Karakesisoglou I, Wong E, Vaezi A, Fuchs E (2003) ACF7: an essential integrator of microtubule dynamics. Cell 115(3):343–354
Sanchez-Soriano N, Travis M, Dajas-Bailador F, Goncalves-Pimentel C, Whitmarsh AJ, Prokop A (2009) Mouse ACF7 and drosophila short stop modulate filopodia formation and microtubule organisation during neuronal growth. J Cell Sci 122(Pt 14):2534–2542. doi:10.1242/jcs.046268
Guo L, Degenstein L, Dowling J, Yu QC, Wollmann R, Perman B, Fuchs E (1995) Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell 81(2):233–243
Edvardson S, Cinnamon Y, Jalas C, Shaag A, Maayan C, Axelrod FB, Elpeleg O (2012) Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann Neurol 71(4):569–572. doi:10.1002/ana.23524
Yang Y, Bauer C, Strasser G, Wollman R, Julien JP, Fuchs E (1999) Integrators of the cytoskeleton that stabilize microtubules. Cell 98(2):229–238
Liu JJ, Ding J, Kowal AS, Nardine T, Allen E, Delcroix JD, Wu C, Mobley W, Fuchs E, Yang Y (2003) BPAG1n4 is essential for retrograde axonal transport in sensory neurons. J Cell Biol 163(2):223–229. doi:10.1083/jcb.200306075
Kapur M, Maloney MT, Wang W, Chen X, Millan I, Mooney T, Yang J, Yang Y (2014) A SxIP motif interaction at the microtubule plus end is important for processive retrograde axonal transport. Cell Mol Life Sci 71(20):4043–4054. doi:10.1007/s00018-014-1611-6
Ryan SD, Ferrier A, Sato T, O’Meara RW, De Repentigny Y, Jiang SX, Hou ST, Kothary R (2012) Neuronal dystonin isoform 2 is a mediator of endoplasmic reticulum structure and function. Mol Biol Cell 23(4):553–566. doi:10.1091/mbc.E11-06-0573
Ryan SD, Bhanot K, Ferrier A, De Repentigny Y, Chu A, Blais A, Kothary R (2012) Microtubule stability, Golgi organization, and transport flux require dystonin-a2-MAP1B interaction. J Cell Biol 196(6):727–742. doi:10.1083/jcb.201107096
Ferrier A, De Repentigny Y, Lynch-Godrei A, Gibeault S, Eid W, Kuo D, Zha X, Kothary R (2015) Disruption in the autophagic process underlies the sensory neuropathy in dystonia musculorum mice. Autophagy 11(7):1025–1036. doi:10.1080/15548627.2015.1052207
Young KG, Kothary R (2008) Dystonin/Bpag1 is a necessary endoplasmic reticulum/nuclear envelope protein in sensory neurons. Exp Cell Res 314(15):2750–2761. doi:10.1016/j.yexcr.2008.06.021
Ferrier A, Sato T, De Repentigny Y, Gibeault S, Bhanot K, O’Meara RW, Lynch-Godrei A, Kornfeld SF, Young KG, Kothary R (2014) Transgenic expression of neuronal dystonin isoform 2 partially rescues the disease phenotype of the Dystonia musculorum mouse model of hereditary sensory autonomic neuropathy VI. Hum Mol Genet 23(10):2694–2710. doi:10.1093/hmg/ddt663
Di Stefano P, Damiano L, Cabodi S, Aramu S, Tordella L, Praduroux A, Piva R, Cavallo F, Forni G, Silengo L, Tarone G, Turco E, Defilippi P (2007) p140Cap protein suppresses tumour cell properties, regulating Csk and Src kinase activity. EMBO J 26(12):2843–2855. doi:10.1038/sj.emboj.7601724
Repetto D, Camera P, Melani R, Morello N, Russo I, Calcagno E, Tomasoni R, Bianchi F, Berto G, Giustetto M, Berardi N, Pizzorusso T, Matteoli M, Di Stefano P, Missler M, Turco E, Di Cunto F, Defilippi P (2014) p140Cap regulates memory and synaptic plasticity through Src-mediated and citron-N-mediated actin reorganization. J Neurosci 34(4):1542–1553. doi:10.1523/jneurosci.2341-13.2014
Akhmanova A, Hoogenraad CC (2005) Microtubule plus-end-tracking proteins: mechanisms and functions. Curr Opin Cell Biol 17(1):47–54. doi:10.1016/j.ceb.2004.11.001
Ito H, Atsuzawa K, Sudo K, Di Stefano P, Iwamoto I, Morishita R, Takei S, Semba R, Defilippi P, Asano T, Usuda N, Nagata K (2008) Characterization of a multidomain adaptor protein, p140Cap, as part of a pre-synaptic complex. J Neurochem 107(1):61–72. doi:10.1111/j.1471-4159.2008.05585.x
Chin LS, Nugent RD, Raynor MC, Vavalle JP, Li L (2000) SNIP, a novel SNAP-25-interacting protein implicated in regulated exocytosis. J Biol Chem 275(2):1191–1200
Grigoriev I, Gouveia SM, van der Vaart B, Demmers J, Smyth JT, Honnappa S, Splinter D, Steinmetz MO, Putney JW Jr, Hoogenraad CC, Akhmanova A (2008) STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol 18(3):177–182. doi:10.1016/j.cub.2007.12.050
Mitchell CB, Gasperini RJ, Small DH, Foa L (2012) STIM1 is necessary for store-operated calcium entry in turning growth cones. J Neurochem 122(6):1155–1166. doi:10.1111/j.1471-4159.2012.07840.x
Hartmann J, Karl RM, Alexander RP, Adelsberger H, Brill MS, Ruhlmann C, Ansel A, Sakimura K, Baba Y, Kurosaki T, Misgeld T, Konnerth A (2014) STIM1 controls neuronal Ca(2)(+) signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 82(3):635–644. doi:10.1016/j.neuron.2014.03.027
Galan C, Dionisio N, Smani T, Salido GM, Rosado JA (2011) The cytoskeleton plays a modulatory role in the association between STIM1 and the Ca2+ channel subunits Orai1 and TRPC1. Biochem Pharmacol 82(4):400–410. doi:10.1016/j.bcp.2011.05.017
Berna-Erro A, Braun A, Kraft R, Kleinschnitz C, Schuhmann MK, Stegner D, Wultsch T, Eilers J, Meuth SG, Stoll G, Nieswandt B (2009) STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci Signal 2(93):ra67. doi:10.1126/scisignal.2000522
Shen WW, Frieden M, Demaurex N (2011) Remodelling of the endoplasmic reticulum during store-operated calcium entry. Biol Cell 103(8):365–380. doi:10.1042/bc20100152
Bechstedt S, Brouhard GJ (2012) Doublecortin recognizes the 13-protofilament microtubule cooperatively and tracks microtubule ends. Dev Cell 23(1):181–192. doi:10.1016/j.devcel.2012.05.006
Gupta KK, Li C, Duan A, Alberico EO, Kim OV, Alber MS, Goodson HV (2013) Mechanism for the catastrophe-promoting activity of the microtubule destabilizer Op18/stathmin. Proc Natl Acad Sci USA 110(51):20449–20454. doi:10.1073/pnas.1309958110
Brouhard GJ, Stear JH, Noetzel TL, Al-Bassam J, Kinoshita K, Harrison SC, Howard J, Hyman AA (2008) XMAP215 is a processive microtubule polymerase. Cell 132(1):79–88. doi:10.1016/j.cell.2007.11.043
Maurer Sebastian P, Cade Nicholas I, Bohner G, Gustafsson N, Boutant E, Surrey T (2014) EB1 accelerates two conformational transitions important for microtubule maturation and dynamics. Curr Biol 24(4):372–384. doi:10.1016/j.cub.2013.12.042
van der Vaart B, Manatschal C, Grigoriev I, Olieric V, Gouveia SM, Bjelic S, Demmers J, Vorobjev I, Hoogenraad CC, Steinmetz MO, Akhmanova A (2011) SLAIN2 links microtubule plus end-tracking proteins and controls microtubule growth in interphase. J Cell Biol 193(6):1083–1099. doi:10.1083/jcb.201012179
van der Vaart B, Franker MA, Kuijpers M, Hua S, Bouchet BP, Jiang K, Grigoriev I, Hoogenraad CC, Akhmanova A (2012) Microtubule plus-end tracking proteins SLAIN1/2 and ch-TOG promote axonal development. J Neurosci 32(42):14722–14728. doi:10.1523/jneurosci.1240-12.2012
Lowery LA, Stout A, Faris AE, Ding L, Baird MA, Davidson MW, Danuser G, Van Vactor D (2013) Growth cone-specific functions of XMAP215 in restricting microtubule dynamics and promoting axonal outgrowth. Neural Dev 8:22. doi:10.1186/1749-8104-8-22
van der Vaart B, van Riel WE, Doodhi H, Kevenaar JT, Katrukha EA, Gumy L, Bouchet BP, Grigoriev I, Spangler SA, Yu KL, Wulf PS, Wu J, Lansbergen G, van Battum EY, Pasterkamp RJ, Mimori-Kiyosue Y, Demmers J, Olieric N, Maly IV, Hoogenraad CC, Akhmanova A (2013) CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev Cell 27(2):145–160. doi:10.1016/j.devcel.2013.09.010
Yamada K, Andrews C, Chan WM, McKeown CA, Magli A, de Berardinis T, Loewenstein A, Lazar M, O’Keefe M, Letson R, London A, Ruttum M, Matsumoto N, Saito N, Morris L, Del Monte M, Johnson RH, Uyama E, Houtman WA, de Vries B, Carlow TJ, Hart BL, Krawiecki N, Shoffner J, Vogel MC, Katowitz J, Goldstein SM, Levin AV, Sener EC, Ozturk BT, Akarsu AN, Brodsky MC, Hanisch F, Cruse RP, Zubcov AA, Robb RM, Roggenkaemper P, Gottlob I, Kowal L, Battu R, Traboulsi EI, Franceschini P, Newlin A, Demer JL, Engle EC (2003) Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat Genet 35(4):318–321. doi:10.1038/ng1261
Cheng L, Desai J, Miranda CJ, Duncan JS, Qiu W, Nugent AA, Kolpak AL, Wu CC, Drokhlyansky E, Delisle MM, Chan WM, Wei Y, Propst F, Reck-Peterson SL, Fritzsch B, Engle EC (2014) Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron 82(2):334–349. doi:10.1016/j.neuron.2014.02.038
Duellberg C, Trokter M, Jha R, Sen I, Steinmetz MO, Surrey T (2014) Reconstitution of a hierarchical +TIP interaction network controlling microtubule end tracking of dynein. Nat Cell Biol 16(8):804–811. doi:10.1038/ncb2999
Lesniewska K, Warbrick E, Ohkura H (2014) Peptide aptamers define distinct EB1- and EB3-binding motifs and interfere with microtubule dynamics. Mol Biol Cell 25(7):1025–1036. doi:10.1091/mbc.E13-08-0504
van Haren J, Draegestein K, Keijzer N, Abrahams JP, Grosveld F, Peeters PJ, Moechars D, Galjart N (2009) Mammalian Navigators are microtubule plus-end tracking proteins that can reorganize the cytoskeleton to induce neurite-like extensions. Cell Motil Cytoskelet 66(10):824–838. doi:10.1002/cm.20370
Peris L, Thery M, Faure J, Saoudi Y, Lafanechere L, Chilton JK, Gordon-Weeks P, Galjart N, Bornens M, Wordeman L, Wehland J, Andrieux A, Job D (2006) Tubulin tyrosination is a major factor affecting the recruitment of CAP-Gly proteins at microtubule plus ends. J Cell Biol 174(6):839–849. doi:10.1083/jcb.200512058
Peris L, Wagenbach M, Lafanechere L, Brocard J, Moore AT, Kozielski F, Job D, Wordeman L, Andrieux A (2009) Motor-dependent microtubule disassembly driven by tubulin tyrosination. J Cell Biol 185(7):1159–1166. doi:10.1083/jcb.200902142
Tortosa E, Galjart N, Avila J, Sayas CL (2013) MAP1B regulates microtubule dynamics by sequestering EB1/3 in the cytosol of developing neuronal cells. EMBO J 32(9):1293–1306. doi:10.1038/emboj.2013.76
Kapitein LC, Yau KW, Gouveia SM, van der Zwan WA, Wulf PS, Keijzer N, Demmers J, Jaworski J, Akhmanova A, Hoogenraad CC (2011) NMDA receptor activation suppresses microtubule growth and spine entry. J Neurosci 31(22):8194–8209. doi:10.1523/jneurosci.6215-10.2011
Zumbrunn J, Kinoshita K, Hyman AA, Nathke IS (2001) Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr Biol 11(1):44–49
Kumar P, Lyle KS, Gierke S, Matov A, Danuser G, Wittmann T (2009) GSK3beta phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J Cell Biol 184(6):895–908. doi:10.1083/jcb.200901042
Wu X, Shen QT, Oristian DS, Lu CP, Zheng Q, Wang HW, Fuchs E (2011) Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3beta. Cell 144(3):341–352. doi:10.1016/j.cell.2010.12.033
Asada N, Sanada K (2010) LKB1-mediated spatial control of GSK3beta and adenomatous polyposis coli contributes to centrosomal forward movement and neuronal migration in the developing neocortex. J Neurosci 30(26):8852–8865. doi:10.1523/jneurosci.6140-09.2010
Ka M, Jung EM, Mueller U, Kim WY (2014) MACF1 regulates the migration of pyramidal neurons via microtubule dynamics and GSK-3 signaling. Dev Biol 395(1):4–18. doi:10.1016/j.ydbio.2014.09.009
Valvezan AJ, Zhang F, Diehl JA, Klein PS (2012) Adenomatous polyposis coli (APC) regulates multiple signaling pathways by enhancing glycogen synthase kinase-3 (GSK-3) activity. J Biol Chem 287(6):3823–3832. doi:10.1074/jbc.M111.323337
Chen HJ, Lin CM, Lin CS, Perez-Olle R, Leung CL, Liem RK (2006) The role of microtubule actin cross-linking factor 1 (MACF1) in the Wnt signaling pathway. Genes Dev 20(14):1933–1945. doi:10.1101/gad.1411206
Stringham EG, Schmidt KL (2009) Navigating the cell: UNC-53 and the navigators, a family of cytoskeletal regulators with multiple roles in cell migration, outgrowth and trafficking. Cell Adhes Migr 3(4):342–346
Repetto D, Aramu S, Boeri Erba E, Sharma N, Grasso S, Russo I, Jensen ON, Cabodi S, Turco E, Di Stefano P, Defilippi P (2013) Mapping of p140Cap phosphorylation sites: the EPLYA and EGLYA motifs have a key role in tyrosine phosphorylation and Csk binding, and are substrates of the Abl kinase. PLoS One 8(1):e54931. doi:10.1371/journal.pone.0054931
Zhang L, Shao H, Zhu T, Xia P, Wang Z, Liu L, Yan M, Hill DL, Fang G, Chen Z, Wang D, Yao X (2013) DDA3 associates with microtubule plus ends and orchestrates microtubule dynamics and directional cell migration. Sci Rep 3:1681. doi:10.1038/srep01681
Kapur M, Wang W, Maloney MT, Millan I, Lundin VF, Tran TA, Yang Y (2012) Calcium tips the balance: a microtubule plus end to lattice binding switch operates in the carboxyl terminus of BPAG1n4. EMBO Rep 13(11):1021–1029. doi:10.1038/embor.2012.140
Bjelic S, De Groot CO, Scharer MA, Jaussi R, Bargsten K, Salzmann M, Frey D, Capitani G, Kammerer RA, Steinmetz MO (2012) Interaction of mammalian end binding proteins with CAP-Gly domains of CLIP-170 and p150(glued). J Struct Biol 177(1):160–167. doi:10.1016/j.jsb.2011.11.010
Sorbara CD, Wagner NE, Ladwig A, Nikic I, Merkler D, Kleele T, Marinkovic P, Naumann R, Godinho L, Bareyre FM, Bishop D, Misgeld T, Kerschensteiner M (2014) Pervasive axonal transport deficits in multiple sclerosis models. Neuron 84(6):1183–1190. doi:10.1016/j.neuron.2014.11.006
Chen L, Wang Z, Ghosh-Roy A, Hubert T, Yan D, O’Rourke S, Bowerman B, Wu Z, Jin Y, Chisholm AD (2011) Axon regeneration pathways identified by systematic genetic screening in C. elegans. Neuron 71(6):1043–1057. doi:10.1016/j.neuron.2011.07.009
Chen L, Stone MC, Tao J, Rolls MM (2012) Axon injury and stress trigger a microtubule-based neuroprotective pathway. Proc Natl Acad Sci USA 109(29):11842–11847. doi:10.1073/pnas.1121180109
Waterman-Storer CM, Karki S, Holzbaur EL (1995) The p150Glued component of the dynactin complex binds to both microtubules and the actin-related protein centractin (Arp-1). Proc Natl Acad Sci USA 92(5):1634–1638
Askham JM, Vaughan KT, Goodson HV, Morrison EE (2002) Evidence that an interaction between EB1 and p150(Glued) is required for the formation and maintenance of a radial microtubule array anchored at the centrosome. Mol Biol Cell 13(10):3627–3645. doi:10.1091/mbc.E02-01-0061
Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH Jr, Ludlow CL, Fischbeck KH (2003) Mutant dynactin in motor neuron disease. Nat Genet 33(4):455–456. doi:10.1038/ng1123
Wider C, Wszolek ZK (2008) Rapidly progressive familial parkinsonism with central hypoventilation, depression and weight loss (Perry syndrome)—a literature review. Parkinson Relat Disord 14(1):1–7. doi:10.1016/j.parkreldis.2007.07.014
Farrer MJ, Hulihan MM, Kachergus JM, Dachsel JC, Stoessl AJ, Grantier LL, Calne S, Calne DB, Lechevalier B, Chapon F, Tsuboi Y, Yamada T, Gutmann L, Elibol B, Bhatia KP, Wider C, Vilarino-Guell C, Ross OA, Brown LA, Castanedes-Casey M, Dickson DW, Wszolek ZK (2009) DCTN1 mutations in Perry syndrome. Nat Genet 41(2):163–165. doi:10.1038/ng.293
Levy JR, Sumner CJ, Caviston JP, Tokito MK, Ranganathan S, Ligon LA, Wallace KE, LaMonte BH, Harmison GG, Puls I, Fischbeck KH, Holzbaur ELF (2006) A motor neuron disease–associated mutation in p150(Glued) perturbs dynactin function and induces protein aggregation. J Cell Biol 172(5):733–745. doi:10.1083/jcb.200511068
Ahmed S, Sun S, Siglin AE, Polenova T, Williams JC (2010) Disease-associated mutations in the p150(Glued) subunit destabilize the CAP-gly domain. Biochemistry 49(25):5083–5085. doi:10.1021/bi100235z
Puls I, Oh SJ, Sumner CJ, Wallace KE, Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer A, Powers R, Finnegan K, Holzbaur EL, Fischbeck KH, Ludlow CL (2005) Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol 57(5):687–694. doi:10.1002/ana.20468
Moughamian AJ, Holzbaur EL (2012) Dynactin is required for transport initiation from the distal axon. Neuron 74(2):331–343. doi:10.1016/j.neuron.2012.02.025
Ikezu S, Ikezu T (2014) Tau-tubulin kinase. Front Mol Neurosci 7:33. doi:10.3389/fnmol.2014.00033
Takahashi M, Tomizawa K, Sato K, Ohtake A, Omori A (1995) A novel tau-tubulin kinase from bovine brain. FEBS Lett 372(1):59–64
Tomizawa K, Omori A, Ohtake A, Sato K, Takahashi M (2001) Tau-tubulin kinase phosphorylates tau at Ser-208 and Ser-210, sites found in paired helical filament-tau. FEBS Lett 492(3):221–227
Houlden H, Johnson J, Gardner-Thorpe C, Lashley T, Hernandez D, Worth P, Singleton AB, Hilton DA, Holton J, Revesz T, Davis MB, Giunti P, Wood NW (2007) Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat Genet 39(12):1434–1436. doi:10.1038/ng.2007.43
Schöls L, Bauer P, Schmidt T, Schulte T, Riess O (2004) Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 3(5):291–304. doi:10.1016/S1474-4422(04)00737-9
Goetz SC, Liem KF Jr, Anderson KV (2012) The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell 151(4):847–858. doi:10.1016/j.cell.2012.10.010
Waters AM, Beales PL (2011) Ciliopathies: an expanding disease spectrum. Pediatr Nephrol 26(7):1039–1056. doi:10.1007/s00467-010-1731-7
Oda T, Chiba S, Nagai T, Mizuno K (2014) Binding to Cep164, but not EB1, is essential for centriolar localization of TTBK2 and its function in ciliogenesis. Genes Cells 19(12):927–940. doi:10.1111/gtc.12191
Watanabe T, Kakeno M, Matsui T, Sugiyama I, Arimura N, Matsuzawa K, Shirahige A, Ishidate F, Nishioka T, Taya S, Hoshino M, Kaibuchi K (2015) TTBK2 with EB1/3 regulates microtubule dynamics in migrating cells through KIF2A phosphorylation. J Cell Biol 210(5):737–751. doi:10.1083/jcb.201412075
Zhang N, Gordon SL, Fritsch MJ, Esoof N, Campbell DG, Gourlay R, Velupillai S, Macartney T, Peggie M, van Aalten DMF, Cousin MA, Alessi DR (2015) Phosphorylation of synaptic vesicle protein 2A at Thr84 by casein kinase 1 family kinases controls the specific retrieval of synaptotagmin-1. J Neurosci 35(6):2492–2507. doi:10.1523/jneurosci.4248-14.2015
Almilaji A, Munoz C, Hosseinzadeh Z, Lang F (2013) Upregulation of Na+, Cl(−)-coupled betaine/gamma-amino-butyric acid transporter BGT1 by Tau tubulin kinase 2. Cell Physiol Biochem 32(2):334–343. doi:10.1159/000354441
Sato S, Cerny RL, Buescher JL, Ikezu T (2006) Tau-tubulin kinase 1 (TTBK1), a neuron-specific tau kinase candidate, is involved in tau phosphorylation and aggregation. J Neurochem 98(5):1573–1584. doi:10.1111/j.1471-4159.2006.04059.x
Yu NN, Yu JT, Xiao JT, Zhang HW, Lu RC, Jiang H, Xing ZH, Tan L (2011) Tau-tubulin kinase-1 gene variants are associated with Alzheimer’s disease in Han Chinese. Neurosci Lett 491(1):83–86. doi:10.1016/j.neulet.2011.01.011
Vazquez-Higuera JL, Martinez-Garcia A, Sanchez-Juan P, Rodriguez-Rodriguez E, Mateo I, Pozueta A, Frank A, Valdivieso F, Berciano J, Bullido MJ, Combarros O (2011) Genetic variations in tau-tubulin kinase-1 are linked to Alzheimer’s disease in a Spanish case-control cohort. Neurobiol Aging 32(3):550 e555–559. doi:10.1016/j.neurobiolaging.2009.12.021
Liachko NF, McMillan PJ, Strovas TJ, Loomis E, Greenup L, Murrell JR, Ghetti B, Raskind MA, Montine TJ, Bird TD, Leverenz JB, Kraemer BC (2014) The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP-43. PLoS Genet 10(12):e1004803. doi:10.1371/journal.pgen.1004803
Lee EB, Lee VMY, Trojanowski JQ (2012) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13(1):38–50. doi:10.1038/nrn3121
Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS, Hogan AM, Xie SX, Ballatore C, Smith AB 3rd, Lee VM, Brunden KR (2012) The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci 32(11):3601–3611. doi:10.1523/jneurosci.4922-11.2012
Ruschel J, Hellal F, Flynn KC, Dupraz S, Elliott DA, Tedeschi A, Bates M, Sliwinski C, Brook G, Dobrindt K, Peitz M, Brustle O, Norenberg MD, Blesch A, Weidner N, Bunge MB, Bixby JL, Bradke F (2015) Axonal regeneration. Systemic administration of epothilone B promotes axon regeneration after spinal cord injury. Science (New York, NY) 348(6232):347–352. doi:10.1126/science.aaa2958
Divinski I, Holtser-Cochav M, Vulih-Schultzman I, Steingart RA, Gozes I (2006) Peptide neuroprotection through specific interaction with brain tubulin. J Neurochem 98(3):973–984. doi:10.1111/j.1471-4159.2006.03936.x
Oz S, Ivashko-Pachima Y, Gozes I (2012) The ADNP derived peptide, NAP modulates the tubulin pool: implication for neurotrophic and neuroprotective activities. PLoS One 7(12):e51458. doi:10.1371/journal.pone.0051458
Jouroukhin Y, Ostritsky R, Assaf Y, Pelled G, Giladi E, Gozes I (2013) NAP (davunetide) modifies disease progression in a mouse model of severe neurodegeneration: protection against impairments in axonal transport. Neurobiol Dis 56:79–94. doi:10.1016/j.nbd.2013.04.012
Matsuoka Y, Gray AJ, Hirata-Fukae C, Minami SS, Waterhouse EG, Mattson MP, LaFerla FM, Gozes I, Aisen PS (2007) Intranasal NAP administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of Alzheimer’s disease at early pathological stage. J Mol Neurosci 31(2):165–170
Shiryaev N, Jouroukhin Y, Giladi E, Polyzoidou E, Grigoriadis NC, Rosenmann H, Gozes I (2009) NAP protects memory, increases soluble tau and reduces tau hyperphosphorylation in a tauopathy model. Neurobiol Dis 34(2):381–388. doi:10.1016/j.nbd.2009.02.011
Oz S, Kapitansky O, Ivashco-Pachima Y, Malishkevich A, Giladi E, Skalka N, Rosin-Arbesfeld R, Mittelman L, Segev O, Hirsch JA, Gozes I (2014) The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol Psychiatry 19(10):1115–1124. doi:10.1038/mp.2014.97
Pfister AS, Tanneberger K, Schambony A, Behrens J (2012) Amer2 protein is a novel negative regulator of Wnt/beta-catenin signaling involved in neuroectodermal patterning. J Biol Chem 287(3):1734–1741. doi:10.1074/jbc.M111.308650
Preitner N, Quan J, Nowakowski DW, Hancock ML, Shi J, Tcherkezian J, Young-Pearse TL, Flanagan JG (2014) APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell 158(2):368–382. doi:10.1016/j.cell.2014.05.042
Purro SA, Ciani L, Hoyos-Flight M, Stamatakou E, Siomou E, Salinas PC (2008) Wnt regulates axon behavior through changes in microtubule growth directionality: a new role for adenomatous polyposis coli. J Neurosci 28(34):8644–8654. doi:10.1523/jneurosci.2320-08.2008
Koester MP, Muller O, Pollerberg GE (2007) Adenomatous polyposis coli is differentially distributed in growth cones and modulates their steering. J Neurosci 27(46):12590–12600. doi:10.1523/jneurosci.2250-07.2007
Shi SH, Cheng T, Jan LY, Jan YN (2004) APC and GSK-3beta are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr Biol 14(22):2025–2032. doi:10.1016/j.cub.2004.11.009
Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD (2004) NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron 42(6):897–912. doi:10.1016/j.neuron.2004.05.011
Votin V, Nelson WJ, Barth AI (2005) Neurite outgrowth involves adenomatous polyposis coli protein and beta-catenin. J Cell Sci 118(Pt 24):5699–5708. doi:10.1242/jcs.02679
Mohn JL, Alexander J, Pirone A, Palka CD, Lee SY, Mebane L, Haydon PG, Jacob MH (2014) Adenomatous polyposis coli protein deletion leads to cognitive and autism-like disabilities. Mol Psychiatry. doi:10.1038/mp.2014.61
Eom TY, Stanco A, Guo J, Wilkins G, Deslauriers D, Yan J, Monckton C, Blair J, Oon E, Perez A, Salas E, Oh A, Ghukasyan V, Snider WD, Rubenstein JR, Anton ES (2014) Differential regulation of microtubule severing by APC underlies distinct patterns of projection neuron and interneuron migration. Dev Cell 31(6):677–689. doi:10.1016/j.devcel.2014.11.022
Chen Y, Tian X, Kim WY, Snider WD (2011) Adenomatous polyposis coli regulates axon arborization and cytoskeleton organization via its n-terminus. PLoS One 6(9):e24335. doi:10.1371/journal.pone.0024335
Rosenberg MM, Yang F, Mohn JL, Storer EK, Jacob MH (2010) The postsynaptic APC multi-protein complex is required for localizing neuroligin and neurexin to neuronal nicotinic synapses in vivo. J Neurosci 30(33):11073–11085. doi:10.1523/jneurosci.0983-10.2010
Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, Burger PC, Wood PA, Taqi F, Booker SV, Petersen GM, Offerhaus GJA, Tersmette AC, Giardiello FM, Vogelstein B, Kinzler KW (1995) The Molecular Basis of Turcot’s Syndrome. N Engl J Med 332(13):839–847. doi:10.1056/NEJM199503303321302
Almuriekhi M, Shintani T, Fahiminiya S, Fujikawa A, Kuboyama K, Takeuchi Y, Nawaz Z, Nadaf J, Kamel H, Kitam A, Samiha Z, Mahmoud L, Ben-Omran T, Majewski J, Noda M (2015) Loss-of-function mutation in APC2 causes sotos syndrome features. Cell Rep 10(9):1585–1598. doi:10.1016/j.celrep.2015.02.011
Shintani T, Takeuchi Y, Fujikawa A, Noda M (2012) Directional neuronal migration is impaired in mice lacking adenomatous polyposis coli 2. J Neurosci 32(19):6468–6484. doi:10.1523/jneurosci.0590-12.2012
Shintani T, Ihara M, Tani S, Sakuraba J, Sakuta H, Noda M (2009) APC2 plays an essential role in axonal projections through the regulation of microtubule stability. J Neurosci 29(37):11628–11640. doi:10.1523/jneurosci.2394-09.2009
Bond J, Roberts E, Springell K, Lizarraga S, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, Costa SMR, Baralle D, Raponi M, Karbani G, Rashid Y, Jafri H, Bennett C, Corry P, Walsh CA, Woods CG (2005) A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 37(4):353–355. doi:10.1038/ng1539
Buchman JJ, Tseng HC, Zhou Y, Frank CL, Xie Z, Tsai LH (2010) Cdk5rap2 interacts with pericentrin to maintain the neural progenitor pool in the developing neocortex. Neuron 66(3):386–402. doi:10.1016/j.neuron.2010.03.036
Hassan MJ, Khurshid M, Azeem Z, John P, Ali G, Chishti MS, Ahmad W (2007) Previously described sequence variant in CDK5RAP2 gene in a Pakistani family with autosomal recessive primary microcephaly. BMC Med Genet 8:58. doi:10.1186/1471-2350-8-58
Lizarraga SB, Margossian SP, Harris MH, Campagna DR, Han AP, Blevins S, Mudbhary R, Barker JE, Walsh CA, Fleming MD (2010) Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 137(11):1907–1917. doi:10.1242/dev.040410
Srour M, Hamdan Fadi F, McKnight D, Davis E, Mandel H, Schwartzentruber J, Martin B, Patry L, Nassif C, Dionne-Laporte A, Ospina Luis H, Lemyre E, Massicotte C, Laframboise R, Maranda B, Labuda D, Décarie J-C, Rypens F, Goldsher D, Fallet-Bianco C, Soucy J-F, Laberge A-M, Maftei C, Boycott K, Brais B, Boucher R-M, Rouleau Guy A, Katsanis N, Majewski J, Elpeleg O, Kukolich Mary K, Shalev S, Michaud Jacques L (2015) Joubert syndrome in French Canadians and identification of mutations in CEP104. Am J Hum Genet 97(5):744–753. doi:10.1016/j.ajhg.2015.09.009
Larti F, Kahrizi K, Musante L, Hu H, Papari E, Fattahi Z, Bazazzadegan N, Liu Z, Banan M, Garshasbi M, Wienker TF, Ropers HH, Galjart N, Najmabadi H (2015) A defect in the CLIP1 gene (CLIP-170) can cause autosomal recessive intellectual disability. Eur J Hum Genet 23(3):331–336. doi:10.1038/ejhg.2014.13
Cheung J, Petek E, Nakabayashi K, Tsui LC, Vincent JB, Scherer SW (2001) Identification of the human cortactin-binding protein-2 gene from the autism candidate region at 7q31. Genomics 78(1–2):7–11. doi:10.1006/geno.2001.6651
Chen YK, Chen CY, Hu HT, Hsueh YP (2012) CTTNBP2, but not CTTNBP2NL, regulates dendritic spinogenesis and synaptic distribution of the striatin-PP2A complex. Mol Biol Cell 23(22):4383–4392. doi:10.1091/mbc.E12-05-0365
Chen YK, Hsueh YP (2012) Cortactin-binding protein 2 modulates the mobility of cortactin and regulates dendritic spine formation and maintenance. J Neurosci 32(3):1043–1055. doi:10.1523/jneurosci.4405-11.2012
Shih PY, Lee SP, Chen YK, Hsueh YP (2014) Cortactin-binding protein 2 increases microtubule stability and regulates dendritic arborization. J Cell Sci 127(Pt 16):3521–3534. doi:10.1242/jcs.149476
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M (2012) De novo gene disruptions in children on the autistic spectrum. Neuron 74(2):285–299. doi:10.1016/j.neuron.2012.04.009
Hsieh PC, Chiang ML, Chang JC, Yan YT, Wang FF, Chou YC (2012) DDA3 stabilizes microtubules and suppresses neurite formation. J Cell Sci 125(Pt 14):3402–3411. doi:10.1242/jcs.099150
Sweet ES, Previtera ML, Fernandez JR, Charych EI, Tseng CY, Kwon M, Starovoytov V, Zheng JQ, Firestein BL (2011) PSD-95 alters microtubule dynamics via an association with EB3. J Neurosci 31(3):1038–1047. doi:10.1523/jneurosci.1205-10.2011
Laht P, Otsus M, Remm J, Veske A (2014) B-plexins control microtubule dynamics and dendrite morphology of hippocampal neurons. Exp Cell Res 326(1):174–184. doi:10.1016/j.yexcr.2014.06.005
Nagano T, Yoneda T, Hatanaka Y, Kubota C, Murakami F, Sato M (2002) Filamin A-interacting protein (FILIP) regulates cortical cell migration out of the ventricular zone. Nat Cell Biol 4(7):495–501. doi:10.1038/ncb808
Wilson AM, Chiodo VA, Boye SL, Brecha NC, Hauswirth WW, Di Polo A (2014) Inhibitor of apoptosis-stimulating protein of p53 (iASPP) is required for neuronal survival after axonal injury. PLoS One 9(4):e94175. doi:10.1371/journal.pone.0094175
Li G, Wang R, Gao J, Deng K, Wei J, Wei Y (2011) RNA interference-mediated silencing of iASPP induces cell proliferation inhibition and G0/G1 cell cycle arrest in U251 human glioblastoma cells. Mol Cell Biochem 350(1–2):193–200. doi:10.1007/s11010-010-0698-9
Liu X, Li F, Zhao S, Luo Y, Kang J, Zhao H, Yan F, Li S, Ji X (2013) MicroRNA-124-mediated regulation of inhibitory member of apoptosis-stimulating protein of p53 family in experimental stroke. Stroke 44(7):1973–1980. doi:10.1161/strokeaha.111.000613
Zhao WH, Wu SQ, Zhang YD (2013) Downregulation of miR-124 promotes the growth and invasiveness of glioblastoma cells involving upregulation of PPP1R13L. Int J Mol Med 32(1):101–107. doi:10.3892/ijmm.2013.1365
Bie L, Zhao G, Wang YP, Zhang B (2012) Kinesin family member 2C (KIF2C/MCAK) is a novel marker for prognosis in human gliomas. Clin Neurol Neurosurg 114(4):356–360
Falnikar A, Tole S, Baas PW (2011) Kinesin-5, a mitotic microtubule-associated motor protein, modulates neuronal migration. Mol Biol Cell 22(9):1561–1574. doi:10.1091/mbc.E10-11-0905
Haque SA, Hasaka TP, Brooks AD, Lobanov PV, Baas PW (2004) Monastrol, a prototype anti-cancer drug that inhibits a mitotic kinesin, induces rapid bursts of axonal outgrowth from cultured postmitotic neurons. Cell Motil Cytoskelet 58(1):10–16. doi:10.1002/cm.10176
Jones GE, Ostergaard P, Moore AT, Connell FC, Williams D, Quarrell O, Brady AF, Spier I, Hazan F, Moldovan O, Wieczorek D, Mikat B, Petit F, Coubes C, Saul RA, Brice G, Gordon K, Jeffery S, Mortimer PS, Vasudevan PC, Mansour S (2014) Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations. Eur J Hum Genet 22(7):881–887. doi:10.1038/ejhg.2013.263
Myers KA, Baas PW (2007) Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J Cell Biol 178(6):1081–1091. doi:10.1083/jcb.200702074
Nadar VC, Ketschek A, Myers KA, Gallo G, Baas PW (2008) Kinesin-5 Is Essential for Growth-Cone Turning. Curr Biol 18(24):1972–1977. doi:10.1016/j.cub.2008.11.021
Nadar VC, Lin S, Baas PW (2012) Microtubule redistribution in growth cones elicited by focal inactivation of kinesin-5. J Neurosci 32(17):5783–5794. doi:10.1523/jneurosci.0144-12.2012
Ostergaard P, Simpson MA, Mendola A, Vasudevan P, Connell FC, Van Impel A, Moore AT, Loeys BL, Ghalamkarpour A, Onoufriadis A, Martinez-Corral I, Devery S, Leroy JG, Van Laer L, Singer A, Bialer MG, McEntagart M, Quarrell O, Brice G, Trembath RC, Schulte-Merker S, Makinen T, Vikkula M, Mortimer PS, Mansour S, Jeffery S (2012) Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am J Hum Genet 90(2):356–362. doi:10.1016/j.ajhg.2011.12.018
Venere M, Horbinski C, Crish JF, Jin X, Vasanji A, Major J, Burrows AC, Chang C, Prokop J, Wu Q, Sims PA, Canoll P, Summers MK, Rosenfeld SS, Rich JN (2015) The mitotic kinesin KIF11 is a driver of invasion, proliferation, and self-renewal in glioblastoma. Sci Transl Med 7(304):304ra143. doi:10.1126/scitranslmed.aac6762
Yoon SY, Choi JE, Huh JW, Hwang O, Lee HS, Hong HN, Kim D (2005) Monastrol, a selective inhibitor of the mitotic kinesin Eg5, induces a distinctive growth profile of dendrites and axons in primary cortical neuron cultures. Cell Motil Cytoskelet 60(4):181–190. doi:10.1002/cm.20057
Tsai JW, Chen Y, Kriegstein AR, Vallee RB (2005) LIS1 RNA interference blocks neural stem cell division, morphogenesis, and motility at multiple stages. J Cell Biol 170(6):935–945. doi:10.1083/jcb.200505166
Gambello MJ, Darling DL, Yingling J, Tanaka T, Gleeson JG, Wynshaw-Boris A (2003) Multiple dose-dependent effects of Lis1 on cerebral cortical development. J Neurosci 23(5):1719–1729
Youn YH, Pramparo T, Hirotsune S, Wynshaw-Boris A (2009) Distinct dose-dependent cortical neuronal migration and neurite extension defects in Lis1 and Ndel1 mutant mice. J Neurosci 29(49):15520–15530. doi:10.1523/jneurosci.4630-09.2009
Coquelle FM, Caspi M, Cordelières FP, Dompierre JP, Dujardin DL, Koifman C, Martin P, Hoogenraad CC, Akhmanova A, Galjart N, De Mey JR, Reiner O (2002) LIS1, CLIP-170’s key to the dynein/dynactin pathway. Mol Cell Biol 22(9):3089–3102. doi:10.1128/mcb.22.9.3089-3102.2002
Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, Ledbetter DH, McBain CJ, Wynshaw-Boris A (1998) Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet 19(4):333–339. doi:10.1038/1221
Greenwood JS, Wang Y, Estrada RC, Ackerman L, Ohara PT, Baraban SC (2009) Seizures, enhanced excitation, and increased vesicle number in Lis1 mutant mice. Ann Neurol 66(5):644–653. doi:10.1002/ana.21775
Jones DL, Baraban SC (2009) Inhibitory inputs to hippocampal interneurons are reorganized in Lis1 mutant mice. J Neurophysiol 102(2):648–658. doi:10.1152/jn.00392.2009
Hunt RF, Dinday MT, Hindle-Katel W, Baraban SC (2012) LIS1 deficiency promotes dysfunctional synaptic integration of granule cells generated in the developing and adult dentate gyrus. J Neurosci 32(37):12862–12875. doi:10.1523/jneurosci.1286-12.2012
Kawabata I, Kashiwagi Y, Obashi K, Ohkura M, Nakai J, Wynshaw-Boris A, Yanagawa Y, Okabe S (2012) LIS1-dependent retrograde translocation of excitatory synapses in developing interneuron dendrites. Nat Commun 3:722. doi:10.1038/ncomms1736
Zheng Y, Wildonger J, Ye B, Zhang Y, Kita A, Younger SH, Zimmerman S, Jan LY, Jan YN (2008) Dynein is required for polarized dendritic transport and uniform microtubule orientation in axons. Nat Cell Biol 10(10):1172–1180. doi:10.1038/ncb1777
Grabham PW, Seale GE, Bennecib M, Goldberg DJ, Vallee RB (2007) Cytoplasmic dynein and LIS1 are required for microtubule advance during growth cone remodeling and fast axonal outgrowth. J Neurosci 27(21):5823–5834. doi:10.1523/jneurosci.1135-07.2007
Sudarov A, Gooden F, Tseng D, Gan WB, Ross ME (2013) Lis1 controls dynamics of neuronal filopodia and spines to impact synaptogenesis and social behaviour. EMBO Mol Med 5(4):591–607. doi:10.1002/emmm.201202106
Fleck MW, Hirotsune S, Gambello MJ, Phillips-Tansey E, Suares G, Mervis RF, Wynshaw-Boris A, McBain CJ (2000) Hippocampal abnormalities and enhanced excitability in a murine model of human lissencephaly. J Neurosci 20(7):2439–2450
Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH (1993) Isolation of a Miller–Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 364(6439):717–721. doi:10.1038/364717a0
Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH (1993) Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA 270(23):2838–2842
Pilz DT, Kuc J, Matsumoto N, Bodurtha J, Bernadi B, Tassinari CA, Dobyns WB, Ledbetter DH (1999) Subcortical band heterotopia in rare affected males can be caused by missense mutations in DCX (XLIS) or LIS1. Hum Mol Genet 8(9):1757–1760
Ka M, Kim WY (2015) Microtubule-actin crosslinking factor 1 is required for dendritic arborization and axon outgrowth in the developing brain. Mol Neurobiol. doi:10.1007/s12035-015-9508-4
Jorgensen LH, Mosbech MB, Faergeman NJ, Graakjaer J, Jacobsen SV, Schroder HD (2014) Duplication in the microtubule-actin cross-linking factor 1 gene causes a novel neuromuscular condition. Sci Rep 4:5180. doi:10.1038/srep05180
Hekimi S, Kershaw D (1993) Axonal guidance defects in a Caenorhabditis elegans mutant reveal cell-extrinsic determinants of neuronal morphology. J Neurosci 13(10):4254–4271
Coy JF, Wiemann S, Bechmann I, Bächner D, Nitsch R, Kretz O, Christiansen H, Poustka A (2002) Pore membrane and/or filament interacting like protein 1 (POMFIL1) is predominantly expressed in the nervous system and encodes different protein isoforms. Gene 290(1–2):73–94. doi:10.1016/s0378-1119(02)00567-x
Peeters PJ, Baker A, Goris I, Daneels G, Verhasselt P, Luyten WHML, Geysen JJGH, Kass SU, Moechars DWE (2004) Sensory deficits in mice hypomorphic for a mammalian homologue of unc-53. Dev Brain Res 150(2):89–101. doi:10.1016/j.devbrainres.2004.03.004
Muley PD, McNeill EM, Marzinke MA, Knobel KM, Barr MM, Clagett-Dame M (2008) The atRA-responsive gene neuron navigator 2 functions in neurite outgrowth and axonal elongation. Dev Neurobiol 68(13):1441–1453. doi:10.1002/dneu.20670
Martinez-Lopez MJ, Alcantara S, Mascaro C, Perez-Branguli F, Ruiz-Lozano P, Maes T, Soriano E, Buesa C (2005) Mouse neuron navigator 1, a novel microtubule-associated protein involved in neuronal migration. Mol Cell Neurosci 28(4):599–612. doi:10.1016/j.mcn.2004.09.016
McNeill EM, Klockner-Bormann M, Roesler EC, Talton LE, Moechars D, Clagett-Dame M (2011) Nav2 hypomorphic mutant mice are ataxic and exhibit abnormalities in cerebellar development. Dev Biol 353(2):331–343. doi:10.1016/j.ydbio.2011.03.008
Marzinke MA, Mavencamp T, Duratinsky J, Clagett-Dame M (2013) 14-3-3epsilon and NAV2 interact to regulate neurite outgrowth and axon elongation. Arch Biochem Biophys 540(1–2):94–100. doi:10.1016/j.abb.2013.10.012
Carlsson E, Krohn K, Ovaska K, Lindberg P, Hayry V, Maliniemi P, Lintulahti A, Korja M, Kivisaari R, Hussein S, Sarna S, Niiranen K, Hautaniemi S, Haapasalo H, Ranki A (2013) Neuron navigator 3 alterations in nervous system tumors associate with tumor malignancy grade and prognosis. Genes Chromosom Cancer 52(2):191–201. doi:10.1002/gcc.22019
Van Haren J, Boudeau J, Schmidt S, Basu S, Liu Z, Lammers D, Demmers J, Benhari J, Grosveld F, Debant A, Galjart N (2014) Dynamic microtubules catalyze formation of navigator-TRIO complexes to regulate neurite extension. Curr Biol 24(15):1778–1785. doi:10.1016/j.cub.2014.06.037
Tomasoni R, Repetto D, Morini R, Elia C, Gardoni F, Di Luca M, Turco E, Defilippi P, Matteoli M (2013) SNAP-25 regulates spine formation through postsynaptic binding to p140Cap. Nat Commun 4:1–13. doi:10.1038/ncomms3136
Yang Y, Wei M, Xiong Y, Du X, Zhu S, Yang L, Zhang C, Liu JJ (2015) Endophilin A1 regulates dendritic spine morphogenesis and stability through interaction with p140Cap. Cell Res 25(4):496–516. doi:10.1038/cr.2015.31
Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G, Ludolph AC (2004) Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63(4):724–726
Kwinter DM, Lo K, Mafi P, Silverman MA (2009) Dynactin regulates bidirectional transport of dense-core vesicles in the axon and dendrites of cultured hippocampal neurons. Neuroscience 162(4):1001–1010. doi:10.1016/j.neuroscience.2009.05.038
Bell N, Hann V, Redfern CP (1833) Cheek TR (2013) Store-operated Ca(2+) entry in proliferating and retinoic acid-differentiated N- and S-type neuroblastoma cells. Biochim Biophys Acta 3:643–651. doi:10.1016/j.bbamcr.2012.11.025
Shim S, Zheng JQ, Ming GL (2013) A critical role for STIM1 in filopodial calcium entry and axon guidance. Mol Brain 6:51. doi:10.1186/1756-6606-6-51
Zhang M, Wu Y, Song JN, Zhao YL, Fu ZF, Ma XD, Zhai HC (2013) The role of store-operated calcium channels in early brain injury after subarachnoid hemorrhage. J Xi’an Jiaotong Univ 34(6):710–716. doi:10.7652/jdyxb201306003
Li Y, Song J, Liu X, Zhang M, An J, Sun P, Li D, Jin T, Wang J (2013) High expression of STIM1 in the early stages of diffuse axonal injury. Brain Res 1495:95–102. doi:10.1016/j.brainres.2012.12.005
Hao B, Lu Y, Wang Q, Guo W, Cheung KH, Yue J (2014) Role of STIM1 in survival and neural differentiation of mouse embryonic stem cells independent of Orai1-mediated Ca2+ entry. Stem cell research 12(2):452–466. doi:10.1016/j.scr.2013.12.005
Kawamata H, Ng SK, Diaz N, Burstein S, Morel L, Osgood A, Sider B, Higashimori H, Haydon PG, Manfredi G, Yang Y (2014) Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J Neurosci 34(6):2331–2348. doi:10.1523/jneurosci.2689-13.2014
Vigont VA, Zimina OA, Glushankova LN, Kolobkova JA, Ryazantseva MA, Mozhayeva GN, Kaznacheyeva EV (2014) STIM1 protein activates store-operated calcium channels in cellular model of Huntington’s disease. Acta Nat 6(4):40–47
Su Q, Cai Q, Gerwin C, Smith CL, Sheng ZH (2004) Syntabulin is a microtubule-associated protein implicated in syntaxin transport in neurons. Nat Cell Biol 6(10):941–953. doi:10.1038/ncb1169
Cai Q, Gerwin C, Sheng ZH (2005) Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J Cell Biol 170(6):959–969. doi:10.1083/jcb.200506042
Cai Q, Pan PY, Sheng ZH (2007) Syntabulin-kinesin-1 family member 5B-mediated axonal transport contributes to activity-dependent presynaptic assembly. J Neurosci 27(27):7284–7296. doi:10.1523/jneurosci.0731-07.2007
Ma H, Cai Q, Lu W, Sheng ZH, Mochida S (2009) KIF5B motor adaptor syntabulin maintains synaptic transmission in sympathetic neurons. J Neurosci 29(41):13019–13029. doi:10.1523/jneurosci.2517-09.2009
Yang YT, Wang CL, Van Aelst L (2012) DOCK7 interacts with TACC3 to regulate interkinetic nuclear migration and cortical neurogenesis. Nat Neurosci 15(9):1201–1210. doi:10.1038/nn.3171
Xie Z, Moy LY, Sanada K, Zhou Y, Buchman JJ, Tsai LH (2007) Cep120 and TACCs control interkinetic nuclear migration and the neural progenitor pool. Neuron 56(1):79–93. doi:10.1016/j.neuron.2007.08.026
Wurdak H, Zhu S, Min KH, Aimone L, Lairson LL, Watson J, Chopiuk G, Demas J, Charette B, Weerapana E, Cravatt BF, Cline HT, Peters EC, Zhang J, Walker JR, Wu C, Chang J, Tuntland T, Cho CY, Schultz PG (2010) A small molecule accelerates neuronal differentiation in the adult rat. Proc Natl Acad Sci USA 107(38):16542–16547. doi:10.1073/pnas.1010300107
Nwagbara BU, Faris AE, Bearce EA, Erdogan B, Ebbert PT, Evans MF, Rutherford EL, Enzenbacher TB, Lowery LA (2014) TACC3 is a microtubule plus end-tracking protein that promotes axon elongation and also regulates microtubule plus end dynamics in multiple embryonic cell types. Mol Biol Cell 25(21):3350–3362. doi:10.1091/mbc.E14-06-1121
DeGeer J, Boudeau J, Schmidt S, Bedford F, Lamarche-Vane N, Debant A (2013) Tyrosine phosphorylation of the Rho guanine nucleotide exchange factor Trio regulates netrin-1/DCC-mediated cortical axon outgrowth. Mol Cell Biol 33(4):739–751. doi:10.1128/mcb.01264-12
O’Brien SP, Seipel K, Medley QG, Bronson R, Segal R, Streuli M (2000) Skeletal muscle deformity and neuronal disorder in Trio exchange factor-deficient mouse embryos. Proc Natl Acad Sci USA 97(22):12074–12078. doi:10.1073/pnas.97.22.12074
Newsome TP, Schmidt S, Dietzl G, Keleman K, Asling B, Debant A, Dickson BJ (2000) Trio combines with dock to regulate Pak activity during photoreceptor axon pathfinding in Drosophila. Cell 101(3):283–294
Bateman J, Shu H, Van Vactor D (2000) The guanine nucleotide exchange factor trio mediates axonal development in the Drosophila embryo. Neuron 26(1):93–106
Liebl EC, Forsthoefel DJ, Franco LS, Sample SH, Hess JE, Cowger JA, Chandler MP, Shupert AM, Seeger MA (2000) Dosage-sensitive, reciprocal genetic interactions between the Abl tyrosine kinase and the putative GEF trio reveal trio’s role in axon pathfinding. Neuron 26(1):107–118
Awasaki T, Saito M, Sone M, Suzuki E, Sakai R, Ito K, Hama C (2000) The Drosophila trio plays an essential role in patterning of axons by regulating their directional extension. Neuron 26(1):119–131
Briancon-Marjollet A, Ghogha A, Nawabi H, Triki I, Auziol C, Fromont S, Piche C, Enslen H, Chebli K, Cloutier JF, Castellani V, Debant A, Lamarche-Vane N (2008) Trio mediates netrin-1-induced Rac1 activation in axon outgrowth and guidance. Mol Cell Biol 28(7):2314–2323. doi:10.1128/mcb.00998-07
Peng YJ, He WQ, Tang J, Tao T, Chen C, Gao YQ, Zhang WC, He XY, Dai YY, Zhu NC, Lv N, Zhang CH, Qiao YN, Zhao LP, Gao X, Zhu MS (2010) Trio is a key guanine nucleotide exchange factor coordinating regulation of the migration and morphogenesis of granule cells in the developing cerebellum. J Biol Chem 285(32):24834–24844. doi:10.1074/jbc.M109.096537
Lund H, Cowburn RF, Gustafsson E, Stromberg K, Svensson A, Dahllund L, Malinowsky D, Sunnemark D (2013) Tau-tubulin kinase 1 expression, phosphorylation and co-localization with phospho-Ser422 tau in the Alzheimer’s disease brain. Brain Pathol 23(4):378–389. doi:10.1111/bpa.12001
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
van de Willige, D., Hoogenraad, C.C. & Akhmanova, A. Microtubule plus-end tracking proteins in neuronal development. Cell. Mol. Life Sci. 73, 2053–2077 (2016). https://doi.org/10.1007/s00018-016-2168-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2168-3