
Abstract
Aims/hypothesis
The pro-inflammatory cytokines IL-1 and IFNγ are critical molecules in immune-mediated beta cell destruction leading to type 1 diabetes mellitus. Suppressor of cytokine signalling (SOCS)-3 inhibits the cytokine-mediated destruction of insulinoma-1 cells. Here we investigate the effect of SOCS3 in primary rodent beta cells and diabetic animal models.
Methods
Using mice with beta cell-specific Socs3 expression and a Socs3-encoding adenovirus construct, we characterised the protective effect of SOCS3 in mouse and rat islets subjected to cytokine stimulation. In transplantation studies of NOD mice and alloxan-treated mice the survival of Socs3 transgenic islets was investigated.
Results
Socs3 transgenic islets showed significant resistance to cytokine-induced apoptosis and impaired insulin release. Neither glucose-stimulated insulin release, insulin content or glucose oxidation were affected by SOCS3. Rat islet cultures transduced with Socs3-adenovirus displayed reduced cytokine-induced nitric oxide and apoptosis associated with inhibition of the IL-1-induced nuclear factor-κB and mitogen-activated protein kinase (MAPK) pathways. Transplanted Socs3 transgenic islets were not protected in diabetic NOD mice, but showed a prolonged graft survival when transplanted into diabetic allogenic BALB/c mice.
Conclusions/interpretation
SOCS3 inhibits IL-1-induced signalling through the nuclear factor-κB and MAPK pathways and apoptosis induced by cytokines in primary beta cells. Moreover, Socs3 transgenic islets are protected in an allogenic transplantation model. SOCS3 may represent a target for pharmacological or genetic engineering in islet transplantation for treatment of type 1 diabetes mellitus.
Similar content being viewed by others


Introduction
The pro-inflammatory cytokines IL-1 and IFNγ are potent inducers of apoptosis and necrosis in pancreatic beta cells in vitro. During the pathogenic process preceding overt type 1 diabetes mellitus, these cytokines are secreted from activated macrophages and T helper cells infiltrating the islets of animal models [1]. Neutralisation of IL-1 and IFNγ signalling protects against type 1 diabetes mellitus in animal models [2–5]. Based on these observations, cytokines have been implicated as critical molecules in pathogenesis of type 1 diabetes [1].
The pro-apoptotic signalling initiated by IL-1 and IFNγ is complex. IL-1 signal transduction involves activation of the transcription factor nuclear factor-κB (NF-κB) pathway, which is essential for regulation of multiple pro-apoptotic genes, including inducible nitric oxide synthase [6–8]. In addition to NFκB activation, the IL-1-induced c-Jun N-terminal kinase (JNK), a member of the mitogen-activated protein kinase (MAPK) family, appears to be equally important for induction of apoptosis [9–11]. The IFNγ signalling cascade involves mainly Janus-activated kinase (JAK)-mediated activation of the transcription factor signal transducer and activator of transcription (STAT)-1, which subsequently stimulates expression of several genes including Caspase-1 [12]. In addition, IL-1 and IFNγ induce inflammatory genes/proteins in beta cells, e.g. chemokines that probably accelerate and augment the inflammatory response, enhancing local accumulation of beta cell toxins like cytokines.
The particular beta cell sensitivity towards the toxicity of cytokines may result from its specialised phenotype, leading to insufficient expression and/or regulation of protective mechanisms blocking proximal and/or distal signals following a cytokine challenge [1, 13]. Thus, understanding regulation of the signal transduction pathways activated by IL-1 and IFNγ is important. In this context, members of the suppressor of cytokine signalling (Socs) family were reported to be immediate-early response genes induced by IFNγ and subsequently suppressing IFNγ signalling, thereby constituting a negative feedback loop controlling duration and magnitude of the cellular response to certain cytokines The suppressor of cytokine signalling (SOCS) proteins contain an SH2 domain and inhibit signalling via binding between SOCS and the phosphorylated JAK-receptor complex, resulting in JAK inhibition and/or competition with downstream signalling molecules at potential receptor docking sites. Moreover, the SOCS proteins can target their bound components for ubiquitination and proteasomal degradation [14, 15]. The SOCS proteins can also inhibit signalling pathways not induced by cytokines. For example, SOCS1 and SOCS3 interact with the phosphorylated insulin receptor, thereby preventing docking and activation of IRS proteins [16, 17].
We have performed a series of investigations on the putative protective role of SOCS3 against cytokine-mediated beta cell death. In addition to suppressing IFNγ signalling, SOCS3 was found to inhibit IL-1 signalling in beta cell lines [18]. It was also found to suppress IL-1-mediated NFκB and MAPK activation, via inhibition of the \({\text{TGF}}\tilde \beta {\text{ - activated}}\) kinase, a central player in the IL-1 signalling cascade [19]. SOCS3 influences expression of several IL-1-induced inflammatory and pro-apoptotic genes in clonal beta cell lines [20].
In vivo studies have shown that diabetes is completely prevented in NOD8.3 mice with beta-cell-specific overexpression of Socs1 [21] and allograft destruction is delayed when transplanting SOCS1 transgenic islets [22]. Moreover, diabetes incidence is reduced in rat insulin promoter (RIP)-Socs1-NOD mice [23], indicating a protective role of SOCS1 in vivo. Protection of the islets correlated with inhibition of IFNγ-mediated STAT-1 activation. However, diabetes was not completely prevented, possibly due to insufficient levels of SOCS1 or redundant toxic effects not inhibited by SOCS1.
Having shown that SOCS3 can suppress IL-1- and IFNγ-induced beta cell death in vitro, we were prompted to study the potential protective effect of Socs3 expression in primary beta cells. Socs3 transgenic islets were used to investigate the ability of SOCS3 to protect transplanted islets in the NOD model and in a pure allograft model without autoimmune attack.
Methods
Animals
Generation of the transgenic RIP-Socs3 mice on the C57Bl/6J background has been described elsewhere [24]. BALB/c mice were purchased from B&K, Sollentuna, Sweden. NOD mice were obtained from a local colony at Uppsala University. Animal facilities, breeding of mice and experimentation were according to the standards stated by the Danish and Swedish authorities.
Islet isolation, culture, viral transduction and cytokine stimulation
Mouse islet isolation and cytokine stimulation
Islets from adult (>10 week old) transgenic mice and their non-transgenic littermates were isolated as previously described [25] and cultured for 5 to 7 days in RPMI 1640 glutamax-1 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% (vol./vol.) fetal bovine serum (Invitrogen) and 1% (wt/vol.) penicillin/streptomycin (Invitrogen), in an atmosphere of air + 5% (vol./vol.) CO2 at 37°C. After culture, a total of 50 islets/condition were stimulated for 72 h in 200 μl RPMI 1640 glutamax-1 with 0.5% (vol./vol.) human serum and 1% (wt/vol.) penicillin/streptomycin, in the absence or presence of a mixture of 75 pg/ml recombinant mouse IL-1 (1 × 107 U/mg; BD Pharmingen, San Diego, CA, USA) and 1 ng/ml recombinant rat IFNγ (1 × 106 U/mg; R&D Systems, Minneapolis, MN, USA).
Rat islet isolation, adenoviral transduction and cytokine stimulation
Islets from neonatal rats were isolated as described [26]. Islets used for analysis of apoptosis and nitric oxide (NO) production were dispersed into single cells using 0.2% (wt/vol.) trypsin and 10 mmol/l EDTA in 1× Hanks’ (Invitrogen). They were then seeded in 9 cm2 culture flasks (NUNC, Roskilde, Denmark) and allowed to form monolayers as described [24]. The virus titre used was found by transduction of islet cells with a green fluorescent protein (GFP)-encoding adenovirus and using a concentration giving >95% GFP-positive cells. After 2 days the cells were stimulated with cytokines. For western blot analysis rat islets were dispersed into single cells using trypsin digestion and immediately hereafter subjected to adenovirus-Luciferase or adenovirus-Socs3 exposure to a final concentration of 5 × 108 plaque-forming units per ml. After 2 days of incubation with adenovirus the cells were stimulated with 250 pg/ml of recombinant mouse IL-1. For isolation of non-beta cells islets were dissociated into single cells and beta and non-beta cells were purified by autofluorescence-activated cell sorting (FACStar, Becton-Dickinson, Sunnyvale, CA, USA) as previously described [27, 28].
Human islet stimulation
Human islets were obtained from O. Korsgren (Department of Clinical Immunology, Rudbeck Laboratory, Uppsala University Hospital, Uppsala, Sweden). Written informed consent was obtained from next of kin. Islets used in this study were from two donors: a 59-year-old woman and a 57-year-old man. Islets were cultured in RPMI 1640 containing 5.6 mmol/l glucose, 10% (vol./vol.) fetal bovine serum and 1% (wt/vol.) penicillin/streptomycin (Invitrogen), in an atmosphere of air with 5% (vol./vol.) CO2 at 37°C. Before stimulation islets were transferred to 100 mm Petri dishes (1,200 islets per dish) in RPMI 1640 supplemented with 2% (vol./vol.) human serum. The following cytokines were added at different time points: human IL-1β (1.9 × 108 U/mg; Sigma Aldrich, St Louis, MO, USA) at 150 pg/ml; human IFN-γ (3 × 107–9 × 107 U/mg; BD Pharmingen) at 20 ng/ml; and human TNF-α (2 × 107 U/mg; PeproTech, Rocky Hill, NJ, USA) at 8 ng/ml. After this RNA was isolated.
Dimethylthiazol-diphenyltetrazolium bromide assay
The dimethylthiazol-diphenyltetrazolium bromide (MTT) assay (Promega, Madison, WI, USA), was used to measure cell viability/toxicity [29].
Apoptosis assays
Life–death detection assay
This assay (Roche Diagnostics, Indianapolis, IN, USA) was performed as specified in the supplier manual.
Apoptosis detection
A TUNEL assay kit (ApopTag; Intergen, Purchase, NY, USA) was used according to the manual of the manufacturer. Rat beta cell monolayer cultures were washed in 1× PBS and fixed in 1% (wt/vol.) paraformaldehyde overnight before TUNEL analysis. In addition the fixed cells were stained with 1 µg/ml DAPI diluted in 1 × PBS to visualise the nuclei. The percentage of TUNEL-positive cells was determined by fluorescence microscopy as a fraction of the total number of DAPI-stained nuclei. A total of 1,500 cells were counted in each condition blinded to protocol.
Measurement of accumulated nitrite, insulin content, insulin release and glucose oxidation
Nitric oxide was measured as accumulated nitrite in the culture medium using the Griess reagent [30].
Accumulated insulin in the culture medium was determined by competitive ELISA assay (Novo Nordisk, Bagsværd, Denmark) as described [31]. For insulin release experiments, triplicate groups of ten islets each were transferred to glass vials containing 0.25 ml KRB supplemented with 10 mmol/l HEPES (KRBH) and 2 mg/ml BSA. During the first hour of incubation at 37°C (O2–CO2; 95:5) the medium contained 1.7 mmol/l glucose. The medium was then carefully removed and replaced by 0.25 ml KRBH supplemented with 16.7 mmol/l glucose for the second hour. After the incubation, islets were pooled and ultrasonically disrupted in 0.2 ml re-distilled water. A 50 µl fraction of the aqueous homogenate was mixed with 125 µl acid ethanol (0.18 mol/l HCl in 96% [vol./vol.] ethanol) and the insulin was extracted overnight at 4°C. Insulin concentrations were then measured by ELISA. To measure the islet glucose oxidation rate, triplicate groups of ten islets were transferred for 90 min to glass vials containing 100 µl KRBH without BSA, but supplemented with d-[U-14C]glucose (Amersham International, Amersham, UK) and non-radioactive glucose to a final concentration of 16.7 mmol/l with a specific activity of 18.5 MBq/mmol. The islet glucose oxidation rate was subsequently measured as previously described [32].
Western blotting
Western blotting was performed as described [19] with the following antibodies: Flag IgG (Sigma Aldrich), p-JNK/JNK IgG, p-p38/p38 IgG, p-extracellular regulated kinase (ERK)/ERK (Cell Signaling Technologies, Cambridge, MA, USA) and IκB IgG (Active Motif, San Francisco, CA, USA). The secondary antibodies were either rat or mouse anti-rabbit IgG Ab conjugated with horseradish peroxidase (Cell Signaling Technologies).
Real-time PCR
RNA extraction was done by the RNAzol method according to the manufacturer’s description (Cinna Biotecx, Houston, TX, USA). cDNA synthesis was performed using a kit (TaqMan Gold RT-PCR; Perkin-Elmer, Boston, MA, USA).
Primers used for the real-time PCR reaction were: rat Socs3, forward 5′-TGAGCGTCAAGACCCAGTCG-3′, reverse 5′-CACAGTCGAAGCGGGGAACT-3′, 114 bp; human SOCS3, forward 5′-ACACGCACTTCCGCACATTC-3′, reverse 5′- CGAGGCCATCTTCACGCTAA-3′, 110 bp; human GAPDH, forward 5′-GGTGAAGGTCGGAGTCAAC-3′, reverse 5′-CCATGGGTGGAATCATATTG-3′, 154 bp; and rat Sp1, forward 5′-GGCTACCCCTACCTCAAAGG-3′, reverse 5′-CACAACATACTGCCCACCAG-3′, 103 bp.
The experiments were performed as described in the sequence detector manual (ABI Prism 7700; AB Applied Biosystems, Foster City, CA, USA). Each cDNA sample was subjected to two individual PCR analyses using either the SOCS3 or the Sp1 primer pair.
Transplantation
Culture of mouse pancreatic islets was performed as described above. A total of 600 islets isolated from RIP-Socs3 transgenic or wild-type mice was packed in a braking-pipette and transplanted beneath the left kidney capsule of the recipient diabetic female NOD mice, which were anaesthetised with avertin.
Diabetes was induced in BALB/c mice by an i.v. injection of 75 mg/kg body weight alloxan (Sigma). Diabetic animals were anaesthetised with avertin and 300 islets were transplanted under the kidney capsule.
Blood glucose was measured daily on blood obtained from the tail vein of non-fasted mice using a glucose meter (Medisense, London, UK). Animals reverting to hyperglycaemia (blood glucose >11.1 mmol/l on two consecutive days) were killed and the graft removed for morphological examination.
Statistical analysis
In vitro results are presented as means ± SD and a paired t test was used for statistical analysis with significance levels at p < 0.05. The Kaplan–Meier log-rank test at p < 0.05 was used in the transplantation studies.
Results
RIP-Socs3 expression protects mouse islets against cytokine-mediated toxicity
Using the MTT viability assay, we analysed mitochondrial impairment, which correlates with viability [33] in isolated transgenic RIP-Socs3 and wild-type islets exposed to cytokines for 3 days. As evident from Fig. 1a, a mixture of cytokines (IL-1 75 pg/ml + IFNγ 1 ng/ml) significantly reduced the viability by 35% in the wild-type islets, whereas transgenic islets were unaffected by this treatment.

Cytokine-mediated mitochondrial impairment, insulin release and apoptosis is inhibited in RIP-Socs3 transgenic mouse islets. Wild-type (white bars) and RIP-Socs3 transgenic (black bars) mouse islets were stimulated with no cytokine (Control) or a mixture of cytokines (IL-1 75 pg/ml + IFNγ 1 ng/ml) for 72 h. The islets were subjected to the MTT assay (a) and the accumulated insulin release in the medium was analysed by ELISA (b). Apoptosis induction was analysed by life–death detection assay (c). Results are presented as mean±SD of three to five individual experiments, *p < 0.05; **p < 0.01. d Western blot analysis of SOCS3 levels in RIP-Socs3 transgenic (Tg) and wild-type (Wt) islets. Equal amounts of protein (10 µg) were loaded in each lane and analysed using a monoclonal Flag antibody directed against recombinant flag-tagged SOCS3
Analysis of accumulated insulin levels in medium containing 16.7 mmol/l glucose following cytokine treatment (Fig. 1b) demonstrated an impairment of insulin release (65% decrease) in wild-type islets, an effect that was completely counteracted by Socs3 expression in transgenic islets after the same treatment.
We next examined whether the transgenic islets were protected from cytokine-induced apoptosis (Fig. 1c). A 13-fold induction of apoptosis in wild-type islets after exposure to cytokine mixture for 3 days (IL-1 75 pg/ml + IFNγ 1 ng/ml) was observed. Compared with this, only a fourfold increase in apoptosis was observed in the transgenic islets.
Transgenic expression of Soc3 does not affect islet function
As shown in Fig. 2a, transgenic and wild-type islets subjected to low glucose (1.7 mmol/l) and a glucose challenge (16.7 mmol/l) displayed similar levels of glucose-stimulated insulin release. Also, the insulin content of the islets was not different (Fig. 2b). Likewise, the ability to metabolise glucose during incubation in 16.7 mmol/l glucose was unaffected by the transgene (Fig. 2c). Thus the data suggest that Socs3 expression at the level obtained in these studies does not alter islet secretory function.

RIP-Socs3 transgene expression does not interfere with glucose-stimulated insulin release, total insulin content or glucose oxidation in isolated mouse islets. For measurement of glucose-stimulated insulin release (a) RIP-Socs3 transgenic (Tg; black bars) and wild-type (Wt; white bars) mouse islets were incubated for 60 min at 1.7 mmol/l glucose followed by a second 60 min incubation at 16.7 mmol/l glucose. After the incubations the islet were homogenised and insulin content was measured (b). c Glucose oxidation rate at 16.7 mmol/l glucose of wild-type and transgenic islets. The results are presented as mean ± SD of six individual experiments
Inhibition of cytokine-induced apoptosis and NO production in adenovirus-Socs3 transduced rat beta cell cultures
To further characterise SOCS3 as an inhibitor of cytokine-induced apoptosis in primary cells, we used adenoviral transduction to induce Socs3 expression in primary rat islet cells grown in monolayer. Immunohistochemical staining for SOCS3 showed that >95% of the beta cells produced SOCS3 after transduction with the Socs3-adenovirus (data not shown). Following 20 h exposure of non- or luciferase-transduced islets to cytokines (IL-1 250 pg/ml + IFNγ 10 ng/ml), an increase in the number of TUNEL-positive cells (Fig. 3a) was observed. Compared with this, the induction of apoptosis was reduced by approximately 50% when the rat islets were transduced with the adenovirus-Socs3-encoding construct.

Cytokine-mediated apoptosis and NO generation is inhibited in Socs3 adenovirus-transduced primary rat beta cells. Dispersed rat islet cultures were transduced as follows: no adenovirus (white bars), Luciferase virus (hatched bars) and Socs3 adenovirus (black bars). After 48 h of viral transduction the cells were left either unstimulated (Control), or exposed to IL-1 (250 pg/ml) or a mixture of cytokines (IL-1 250 pg/ml + IFNγ 10 ng/ml) for 20 h. Apoptosis was determined (a) using TUNEL analysis and positive (apoptotic) cells were determined by fluorescence microscopy. b Accumulated NO in the media. Results are shown as mean ± SD of three individual experiments, *p < 0.05
In parallel to the apoptotic measurements, we analysed NO production after 20 h of exposure to either IL-1 250 pg/ml or the cytokine mixture (Fig. 3b). Control- or luciferase-transduced islets responded similarly to IL-1 with increased NO production. In islets transduced with Socs3, NO production was reduced by 60% after IL-1 or IL-1 plus IFNγ compared with control islets.
Adenovirus-Socs3 expression inhibits IL-1-induced JNK, p38 and ERK activation and suppresses IκB degradation in rat islets
Phosphorylation of the MAPKs ERK, p38 and JNK after 15 min of exposure to IL-1 (250 pg/ml IL-1) was analysed by western blotting. As shown in Fig. 4, IL-1 induced phosphorylation of all three MAPKs in control and luciferase-tranduced islets, an effect that was inhibited in the presence of SOCS3. The NF-κB inhibitory protein IκB was completely degraded following 15 min of IL-1 exposure. However, in the presence of SOCS3, the IκB degradation was markedly inhibited.

Adenovirus-mediated SOCS3 production inhibits IL-1-induced JNK, p38 and ERK activity and prevents IκB degradation in primary rat beta cells. Dispersed rat islet cultures were transduced as follows: no adenovirus (Control), Luciferase virus (Luc) and Socs3 adenovirus. After 2 days the cultures were either left unstimulated or exposed for 15 min to IL-1 (250 pg/ml). Cell lysates (10 μg protein/lane) were subjected to western blotting using specific antibodies against phospho-/total MAPKs (JNK, p38 and ERK) and IκBα. SOCS3 production was confirmed using an antibody towards flag-tagged SOCS3. The figure is representative of three individual experiments. Adv, adenovirus
RIP-Socs3 overexpression prolongs islet graft survival in BALB/c mice, but not in NOD mice
Groups of 600 Socs3 transgenic or wild-type islets were transplanted into spontaneously diabetic NOD mice (Fig. 5). The mice became normoglycaemic within 1 to 2 days. However, from day 4 and onwards up to day 20 post-transplantation, all animals reverted to hyperglycaemia. Analysis of the curves for graft survival, using the Kaplan–Meier log-rank test, showed no difference between the groups in islet graft survival (p > 0.05). Next, transgenic and wild-type islets were transplanted into alloxan-diabetic BALB/c mice (Fig. 6). In these experiments only 300 islets were needed to achieve normoglycaemia. Moreover, in this situation the Socs3 transgenic transplanted mice remained normoglycaemic longer than mice grafted with wild-type islets (p < 0.05). Thus, Socs3 beta cell overexpression was able to prolong islet survival in diabetic mice, but when autoimmunity was also superimposed (NOD mice) the protective effect was not sufficient.

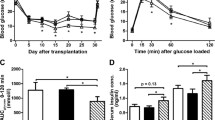
Socs3 transgenic islets are not protected in diabetic NOD mice. A total of 600 Socs3 transgenic (dotted line) and 600 wild-type islets (continuous line) were transplanted under the kidney capsule of diabetic NOD mice (n = 6, n = 9 respectively). The blood glucose was followed to study recurrence of diabetes, defined as blood glucose >11.1 mmol/l on 2 consecutive days

Prolonged graft function in diabetic BALB/c mice transplanted with Socs3 transgenic islets. Diabetes was induced in BALB/c mice by injecting 75 mg/kg alloxan. Diabetic BALB/c mice were transplanted with 300 wild-type (continuous line; n = 8 animals) or Socs3 transgenic (dotted line; n = 10 animals) islets and the blood glucose followed in order to observe graft function. Kaplan–Meier survival curve with p < 0.05 (log-rank test)
Socs3 is upregulated in rat and human islets following cytokine stimulation
We analysed the expression of Socs3 mRNA in rat islets following exposure to 250 pg/ml of IL-1 and/or 10 ng/ml IFNγ by real-time PCR. Figure 7a shows that IL-1 induced the expression of Socs3 mRNA significantly (20-fold) only after 4 h, after which expression declined to a level not significantly different from the control level 24 h after stimulation. IFNγ alone did not significantly induce Socs3 (hatched bars). The effect of combined stimulation with IL-1 and IFNγ on Socs3 expression was additive (black bars), being detectable and significant after 1 h of stimulation and reaching maximal level of induction after 4 h. In sorted non-beta islet cells the kinetics of Socs mRNA induction by IL-1 was accelerated. Within 2 h significant induction was observed (Fig. 7b) and after 6 h no more IL-1 stimulation of Socs3 mRNA was observed. In human islets both IL-1 alone and a mixture of IL-1, IFN-γ and TNF-α also induced SOCS3 mRNA in a time-dependent manner (Fig. 7b). A maximal fourfold stimulation was observed after 4 h of IL-1 stimulation, with SOCS3 mRNA levels declining to control levels after 24 h. A mixture of IL-1, IFN-γ and TNF-α induced SOCS3 sixfold after 4 to 6 h and mRNA levels remained elevated about fourfold after 24 h of stimulation.

Cytokines upregulate SOCS3 mRNA in human and rat islets. a RNA from rat islets subjected to 1, 2, 4, 6 or 24 h of exposure to 250 pg/ml IL-1 (white bars) or 10 ng/ml IFNγ (hatched bars) or a mixture of the two (black bars). b Sorted islet non-beta cells exposed to 250 pg/ml IL-1 (white bars) or 10 ng/ml IFNg (hatched bars). c Human islets exposed to 150 pg/ml IL-1 (white bars) or a mixture of 150 pg/ml IL-1, 20 ng/ml IFN-γ and 8 ng/ml TNF-α (black bars). Analysis (a–c) was by real-time PCR with specific primers for SOCS3. Data are expressed as fold induction of SOCS3 mRNA relative to control mRNA as indicated. Data are presented as (a, b) means ± SD of four and three individual experiments respectively, *p < 0.05 compared with respective controls, and (c) mean from two experiments, with individual data points from each experiment represented by dots
Discussion
The identification of beta cell protective proteins is of considerable interest with respect to the development of novel treatment strategies for type 1 diabetes mellitus. We previously found SOCS3 to be an endogenously expressed protective protein in a beta cell line [18]. We hypothesised that insufficient SOCS3 gene expression upon cytokine exposure may contribute to beta cell hypersensitivity to cytokines. Since this study was performed in insulinoma (INS)-1 tumour cells in vitro, the physiological role of SOCS3 in mouse and rat islets as well as in transplantation studies is addressed here.
In the current study we found that cultured RIP-Socs3 mouse islets are resistant to cytokine-mediated apoptosis and impaired insulin secretion. Expression of Socs3 in islets from wild-type and Socs3 transgenic mice has been described previously [24]. The pattern of expression is heterogeneous among beta cells in islets from SOCS3 transgenic mice, both at the mRNA and protein levels. In islets from wild-type mice, no expression of Socs3 mRNA or protein could be detected. To be potentially useful in islet transplantation, SOCS3 expression must not interfere with the glucose sensitivity/insulin secretion of the beta cell. To address this issue, we examined whether beta-cell-specific Socs3 expression influenced glucose-stimulated insulin release, insulin content and glucose oxidation in vitro. As none of these parameters were affected by SOCS3, constitutive Socs3 expression by itself, at least at the level obtained in the present study, does not seem to interfere with the complex signalling pathways involved in insulin secretion, regulation or glucose sensitivity. Moreover, RIP-Socs3 transgenic mice have identical fasting blood glucose levels to their non-transgenic littermates [24] and RIP-Socs3 transgenic islets are able to maintain normal blood glucose when transplanted into syngeneic high-dose streptozotocin diabetic mice (data not shown). In summary, although SOCS proteins may influence signalling pathways other than the inflammatory-induced ones (e.g. insulin signalling) in the beta cell, Socs3 overexpression does not seem to influence normal beta cell function.
Genetic engineering of islets/beta cells before transplantation would require ex vivo manipulation to introduce the transgene. To mimic this situation and at the same time investigate the effect of SOCS3 following one possible ex vivo expression approach, we expressed Socs3 by using a Socs3-encoding adenoviral construct. In this model we found that Socs3 was expressed in more than 95% of beta cells and that the expression level of Socs3 mRNA was 10 to 20 times higher than in non-transduced islet cells. In agreement with the finding in mouse islets, SOCS3 was found to protect rat islets from cytokine-induced NO production and apoptosis. These data suggest that this transduction approach makes it possible to produce SOCS3 in primary rat islet beta cells and to inhibit cytokine-induced signalling and death as effectively as observed in RIP-Socs3 transgenic mouse islets.
We have previously reported that SOCS3 inhibits NFκB and MAPK activation in INS-1 cells [19, 20]. By means of an adenovirus approach we here extend these studies to primary beta cells and show a pronounced inhibitory effect of SOCS3 on IL-1-induced IκB degradation and MAPK activation, suggesting that SOCS3 is indeed capable of inhibiting these proximal IL-1 signalling pathways documented to be critically involved in IL-1-induced apoptosis [10, 11, 30, 34].
Taken together, the data obtained from both transgenic RIP-Socs3 islets and adenovirus-Socs3 transduced rat islets demonstrate that SOCS3 is a potent inhibitor of cytokine-mediated signalling and death in primary rodent beta cells by similar mechanisms to those previously found in INS beta cells [18].
Beta-cell-specific Socs1 expression in NOD mice significantly reduced the incidence of spontaneous diabetes [21, 23]. Interestingly, islets from these transgenic animals had a similar lymphocytic infiltration to islets from wild-type littermates indicating protection at the beta cell level. We have previously observed by microarray analysis in INS-1 cells [20] that Socs3 expression inhibits expression of multiple IL-1-induced inflammatory mediators such as homing receptors and chemokines. Thus, SOCS3, in contrast to SOCS1, may also be able to counteract the level of infiltration. When RIP-Socs3 transgenic or wild-type islets were transplanted into diabetic NOD mice no difference in disease recurrence was observed. One explanation of this could be an insufficient expression level of Socs3 in the transgenic islets. However, a comparable study showed that SOCS1 is also not able to protect islets transplanted into diabetic NOD mice [22], suggesting that enhanced expression of one of the SOCS proteins is not adequate to protect beta cells against the massive inflammatory response seen in NOD mice. However, in an exclusively allogenic transplantation model, we found graft function was prolonged in diabetic BALB/c mice transplanted with RIP-Socs3 islets, a finding in line with a prior study in which Socs1 transgenic islets prolonged graft survival in an allogenic model [22]. Our in vivo studies demonstrate that overexpression of Socs1 cannot circumvent the autoimmune response seen in the NOD model, but that Socs3 can protect islets against the allogenic MHC response seen after transplantation.
To elucidate whether SOCS3 is an endogenous, but perhaps insufficiently regulated component of the downstream signalling network initiated by IL-1 and IFNγ, we investigated endogenous Socs3 expression following cytokine exposure in rat islets. Interestingly, IL-1 alone significantly induced Socs3 mRNA after 4 h of stimulation, but at earlier time points an effect of the combination of IL-1 and IFNγ was evident. As SOCS3 has the ability to inhibit IL-1 and IFNγ signalling, the kinetics by which they induce Socs3 expression could be critical. IFNγ alone is not toxic to rat islets, possibly due to the rapid albeit modest induction of Socs3. Compared to this, rat islets are sensitive to the toxic effect of IL-1, which could be explained by the slower induction of Socs3 by this cytokine and the resulting insufficient or delayed negative feedback. Despite the kinetics of Socs3 expression induced by combined treatment with IL-1 and IFNγ, neither mouse nor rat beta cells were protected from apoptosis induced by this treatment. This may be because the major fraction of Socs3 mRNA is not expressed until 4 h after cytokine exposure, which is too late to rescue the cells from the devastating effects of IL-1 and IFNγ. Interestingly, in a non-beta cell fraction of rat islets, a more rapid induction of Socs3 mRNA in response to IL-1 was evident. This could explain why beta cells are killed selectively by cytokines, since non-beta cells are able to upregulate expression of Socs3 rapidly, thus preventing prolonged cytokine action and induction of apoptosis. We also observed an increase in SOCS3 mRNA after 2 h of stimulation with IL-1 alone in human islets from two donors. Human beta cells require exposure to IL-1, IFN-γ and TNF-α in combination for induction of apoptosis. When human islets were exposed to this mixture of cytokines, SOCS3 mRNA was induced six- to tenfold after 4 to 6 h, suggesting a relatively slow induction and further supporting the hypothesis of a delayed and insufficient negative feedback of cytokine signalling in beta cells.
Studies have demonstrated that beta cell death induced by high glucose exposure, a proposed mechanism of beta cell failure in type 2 diabetes mellitus, is mediated by glucose-induced expression of IL-1 and executed via autocrine signalling [35]. This suggests that beta cell apoptosis during type 1 and type 2 diabetes mellitus may be mediated by cytokines [36, 37]. Thus, SOCS3 may not only be an interesting molecule in type 1 diabetes, but may be of equally relevant function in type 2 diabetes. In addition to the perspective of beta cell mass preservation in type 1 diabetes mellitus and an improved survival rate of transplanted islets, information from this study may be valuable for stem cell therapy strategies and in the development of engineered cytokine-resistant cell lines suitable for therapeutic use in human diseases other than type 1 diabetes. Finally, detailed knowledge of the pro-apoptotic signalling mechanisms influenced by SOCS3 may also provide new targets for pharmacological intervention in the beta cell destroyed in type 1 diabetes mellitus.
Abbreviations
- ERK:
-
extracellular regulated kinase
- GFP:
-
green fluorescent protein
- INS:
-
insulinoma
- JAK:
-
Janus-activated kinase
- JNK:
-
c-Jun N-terminal kinase
- KRBH:
-
KRB supplemented with 10 mmol/l HEPES
- MAPK:
-
mitogen-activated protein kinase
- MTT:
-
dimethylthiazol-diphenyltetrazolium bromide
- NFκB:
-
nuclear factor-κB
- NO:
-
nitric oxide
- RIP:
-
rat insulin promoter
- SOCS:
-
suppressor of cytokine signalling
- STAT:
-
signal transducer and activator of transcription
References
Eizirik DL, Mandrup-Poulsen T (2001) A choice of death—the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 44:2115–2133
Nicoletti F, Dimarco R, Barcellini W et al (1994) Protection from experimental autoimmune diabetes in the nonobese diabetic mouse with soluble interleukin-1 receptor. Eur J Immunol 24:1843–1847
Cailleau C, Diu-Hercend A, Ruuth E, Westwood R, Carnaud C (1997) Treatment with neutralizing antibodies specific for IL-1 beta prevents cyclophosphamide-induced diabetes in nonobese diabetic mice. Diabetes 46:937–940
Nicoletti F, Zaccone P, Dimarco R et al (1997) Prevention of spontaneous autoimmune diabetes in diabetes-prone bb rats by prophylactic treatment with antirat interferon-gamma antibody. Endocrinology 138:281–288
Wang B, Andre I, Gonzalez A et al (1997) Interferon-gamma impacts at multiple points during the progression of autoimmune diabetes. Proc Natl Acad Sci USA 94:13844–13849
Cardozo AK, Kruhoffer M, Leeman R, Orntoft T, Eizirik DL (2001) Identification of novel cytokine-induced genes in pancreatic beta-cells by high-density oligonucleotide arrays. Diabetes 50:909–920
Cardozo AK, Heimberg H, Heremans Y et al (2001) A comprehensive analysis of cytokine-induced and nuclear factor-kappa B-dependent genes in primary rat pancreatic beta-cells. J Biol Chem 276:48879–48886
Kwon G, Corbett JA, Rodi CP, Sullivan P, McDaniel ML (1995) Interleukin-1-beta-induced nitric-oxide synthase expression by rat pancreatic beta-cells—evidence for the involvement of nuclear factor kappa-b in the signaling mechanism. Endocrinol 136:4790–4795
Ammendrup A, Maillard A, Nielsen K et al (2000) The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 49:1468–1476
Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF (2001) Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50:77–82
Bonny C, Oberson A, Steinmann M, Schorderet DF, Nicod P, Waeber G (2000) IB1 reduces cytokine-induced apoptosis of insulin-secreting cells. J Biol Chem 275:16466–16472
Karlsen AE, Pavlovic D, Nielsen K et al (2000) Interferon-gamma induces interleukin-1 converting enzyme expression in pancreatic islets by an interferon regulatory factor-1-dependent mechanism. J Clin Endocrinol Metab 85:830–836
Nerup J, Mandrup-Poulsen T, Helqvist S et al (1994) On the pathogenesis of IDDM. Diabetologia 37(Suppl 2):S82–S89
Krebs DL, Hilton DJ (2000) SOCS: physiological suppressors of cytokine signaling. J Cell Sci 113:2813–2819
Johnston JA, O’Shea JJ (2003) Matching SOCS with function. Nat Immunol 4:507–509
Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E (2000) SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem 275:15985–15991
Ueki K, Kondo T, Kahn CR (2004) Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol 24:5434–5446
Karlsen AE, Ronn SG, Lindberg K et al (2001) Suppressor of cytokine signaling 3 (SOCS-3) protects beta- cells against interleukin-1 beta- and interferon-gamma-mediated toxicity. Proc Natl Acad Sci USA 98:12191–12196
Frobose H, Groth Rønn S, Heding PE et al (2006) Suppressor of cytokine signaling-3 inhibits interleukin-1 signaling by targeting the TRAF-6/TAK1 complex. Mol Endocrinol 20:1587–1596
Karlsen AE, Heding PE, Frobose H et al (2004) Suppressor of cytokine signalling (SOCS)-3 protects beta cells against IL-1 beta-mediated toxicity through inhibition of multiple nuclear factor-kappa B-regulated proapoptotic pathways. Diabetologia 47:1998–2011
Chong MM, Chen Y, Darwiche R et al (2004) Suppressor of cytokine signaling-1 overexpression protects pancreatic beta cells from CD8+ T cell-mediated autoimmune destruction. J Immunol 172:5714–5721
Solomon M, Flodstrom-Tullberg M, Sarvetnick N (2005) Differences in suppressor of cytokine signaling-I (SOCS-1) expressing islet allograft destruction in normal BALB/c and spontaneously-diabetic NOD recipient mice. Transplantation 79:1104–1109
Flodstrom-Tullberg M, Yadav D, Hagerkvist R et al (2003) Target cell expression of suppressor of cytokine signaling-1 prevents diabetes in the NOD mouse. Diabetes 52:2696–2700
Lindberg K, Ronn SG, Tornehave D et al (2005) Regulation of pancreatic beta-cell mass and proliferation by SOCS-3. J Mol Endocrinol 35:231–243
Lernmark A, Nathans A, Steiner DF (1976) Preparation and characterization of plasma membrane-enriched fractions from rat pancreatic islets. J Cell Biol 71:606–623
Brunstedt J (1980) Rapid isolation of functionally intact pancreatic islets from mice and rats by percollTM gradient centrifugation. Diabetes Metab 6:87–89
Pipeleers DG, In’t Veld PA, Van de Winkel M et al (1985) A new in vitro model for the study of pancreatic A and B cells. Endocrinology 117:806–816
Rasschaert J, Ladriere L, Urbain M et al (2005) Toll-like receptor 3 and STAT-1 contribute to double-stranded RNA+ interferon-γ-induced apoptosis in primary pancreatic β-cells. J Biol Chem 280:33984–33991
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR (1982) Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal Biochem 126:131–138
Nielsen K, Sparre T, Larsen MR et al (2004) Protein expression changes in a cell system of beta-cell maturation reflect an acquired sensitivity to IL-1beta. Diabetologia 47:62–74
Andersson A, Sandler S (1983) Viability tests of cryopreserved endocrine pancreatic cells. Cryobiology 20:161–168
Nielsen K, Karlsen AE, Deckert M et al (1999) Beta-cell maturation leads to in vitro sensitivity to cytotoxins. Diabetes 48:2324–2332
Heimberg H, Heremans Y, Jobin C et al (2001) Inhibition of cytokine-induced NF-kappaB activation by adenovirus-mediated expression of a NF-kappaB super-repressor prevents beta-cell apoptosis. Diabetes 50:2219–2224
Maedler K, Sergeev P, Ris F et al (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110:851–860
Donath MY, Storling J, Maedler K, Mandrup-Poulsen T (2003) Inflammatory mediators and islet beta-cell failure: a link between type 1 and type 2 diabetes. J Mol Med 81:455–470
Larsen CM, Faulenbach M, Vaag A et al (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356:1517–1526
Acknowledgements
We thank A. Hellgren, H. Foght, S. Munch and H. Fjordvang for excellent technical assistance. The Nordic Network for Clinical Islet Transplantation, O. Korsgren and the Department of Clinical Immunology, Rudbeck Laboratory, Uppsala University Hospital, Sweden are thanked for providing human islets. P. E. Heding was supported by the Juvenile Diabetes Research Foundation International (no. 1-2001-706) and in part by the Swedish Research Council, the Swedish Diabetes Association and Novo Nordisk. S. G. Rønn and C. Bruun were supported by the Juvenile Diabetes Research Foundation International (no. 1-2004-736). A. Börjesson was supported by Novo Nordisk.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. G. Rønn and A. Börjesson contributed equally to this work.
Rights and permissions
About this article
Cite this article
Rønn, S.G., Börjesson, A., Bruun, C. et al. Suppressor of cytokine signalling-3 expression inhibits cytokine-mediated destruction of primary mouse and rat pancreatic islets and delays allograft rejection. Diabetologia 51, 1873–1882 (2008). https://doi.org/10.1007/s00125-008-1090-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1090-0