
Abstract
Oculodentodigital dysplasia (ODDD) is a dominantly inherited human disorder associated with different symptoms like craniofacial anomalies, syndactyly and heart dysfunction. ODDD is caused by mutations in the GJA1 gene encoding the gap junction protein connexin43 (Cx43). Here, we have characterized four Cx43 mutations (I31M, G138R, G143S and H194P) after stable expression in HeLa cells. In patients, the I31M and G138R mutations showed all phenotypic characteristics of ODDD, whereas G143S did not result in facial abnormalities and H194P mutated patients exhibited no syndactylies. In transfected HeLa cells, these mutations led to lack of the P2 phosphorylation state of the Cx43 protein, complete inhibition of gap junctional coupling measured by neurobiotin transfer and increased hemichannel activity. In addition, altered trafficking and delayed degradation were found in these mutants by immunofluorescence and pulse-chase analyses. In G138R and G143S mutants, the increased hemichannel activity correlated with an increased half-time of the Cx43 protein. However, the I31M mutated protein showed no extended half-time. Thus, the increased hemichannel activity may be directly caused by an altered conformation of the mutated channel forming protein. We hypothesize that increased hemichannel activity may aggravate the phenotypic abnormalities in ODDD patients who are deficient in Cx43 gap junction channels.
Similar content being viewed by others

Introduction
Connexins are transmembrane proteins characterized by a cytoplasmic amino terminus, four transmembrane domains, two extracellular and one cytoplasmic loop and a regulatory carboxy terminal region. Connexins oligomerize to connexons, also called “hemichannels,” which can dock to each other in contacting plasma membranes and form intercellular conduits. These gap junction channels allow mutual intercellular diffusion of ions, second messengers or metabolites and are assumed to be necessary for normal embryogenesis and maintenance of organ functions in the adult (Bennett & Verselis, 1992).
Open hemichannels have been reported to function physiologically by release of second messengers like adenosine triphosphate (ATP), oxidized nicotinamide adenine dinucleoteide (NAD+), glutamate or prostaglandin E2, thus triggering paracrine signaling (Bruzzone et al., 2001; Stout et al., 2002; Evans, De Vuyst & Leybaert, 2006). Certain connexin mutations that enhance the opening of hemichannels can lead to cell death (Lin Liang et al., 2005) and are more or less unspecific, leaky membrane pores. However, the physiological function of hemichannels remains unclear (Spray, Ye & Ransom, 2006).
Today, at least 10 human disorders are linked to mutations in different connexin genes. Most of them lead to loss of gap junctional function. For example, mutations in human connexin 32 (Cx32) or Cx26 result in Charcot-Marie-Tooth X-linked disease (Hanemann et al., 2003) or inherited nonsyndromic deafness (Yotsumoto et al., 2003). However, recently described aberrant hemichannel properties in a Cx26 mutant causing skin disease and deafness (Gerido et al., 2007) underline the importance of hemichannel function for the maintenance of cellular homeostasis.
Oculodentodigital dysplasia (ODDD), a rare dominant human disorder characterized by ophthalmological, dental, limb and cardiac malformations, is caused by mutations in the Cx43 gene (Paznekas et al., 2003), one of the best-known members of the connexin gene family (Soehl & Willecke, 2003). More than 35 mutations in human Cx43 are known to lead to ODDD. All mutations characterized so far negatively influence gap junctional function, but only six Cx43 mutations were tested for their ability to alter the function of hemichannels (Lai et al., 2006). Three of these mutations were associated with neurological abnormalities, but all six (Y17S, G21R, A40V, F52dup, L90V and I130T), when expressed as eYFP-tagged proteins, were not able to form functional hemichannels (Lai et al., 2006).
In this study, we investigated four ODDD-causing Cx43 mutations (I31M, G138R, G143S and H194P) for their ability to form functional gap junction channels and hemichannels in stably transfected HeLa cells. Since stably transfected HeLa cell clones can express the transgene to different extents, we quantified the expression level of mutated and wild-type Cx43 protein. In this way, we found out that all of the four Cx43 mutants characterized could form open hemichannels. Three of them even exhibited >2-fold increased hemichannel activity (I31M, G138R and G143S).
Materials and Methods
Cloning of Cx43 Mutant Vectors
All ODDD mutations generated were inserted into the mouse Cx43 gene using the overlap extension polymerase chain reaction (PCR) technique. In the first PCR, the EcoRImCx43_for (CGG AAT TCA TCT TCG GCA AGT TGG GCT CG) primer carrying an EcoRI recognition site in its overhang together with a mutation-specific reverse primer were used. In the second PCR, the mCx43_rev (CAG GTG CAC GTT CTG CAA GCA CCC) primer and a mutation-specific forward primer were added. Mutation-specific primers were used as follows: I31M_for (GGT GCT CTT CAT GTT CAG AAT CC), I31M_rev (GGA TTC TGA ACA TGA AGA GCA CC), G138R_for (GAA GTT CAA GTA TCG GAT TGA AGA ACA CG), G138R_rev (CGT GTT CTT CAA TCC GAT ACT TGA ACT TC), G143S_for (GGA TTG AAG AAC ACA GCA AGG TGA AG), G143_rev (CTT CAC CTT GCT GTG TTC TTC AAT CC), H194P_for (CCC TGC CCC CCC CAG GTG GAC TGC) and H194P_rev (GCA GTC CAC CTG GGG GGG GCA GGG). The resulting amplicons from the fusion PCR were double-digested with ClaI and EcoRI, blunted and cloned into the IRES-eGFP-Zeo vector, a modified pcDNA3.1Zeo(+) vector (Invitrogen, Karlsruhe, Germany). The resulting vectors contain the Cx43 mutated genes under control of cytomegalovirus (CMV) promoter elements, followed by an internal ribosomal entry site (IRES) and DNA coding for the enhanced green fluorescent protein (eGFP). The distances between the mutated genes and IRES as well as IRES and eGFP correspond to those suggested by Attal, Theron & Houdebine (1999). Zeomycin resistance was expressed by an SV40 promoter.
Expression and Sorting of Cx43 HeLa Cell Mutants
The vectors generated were linearized and transfected into connexin-deficient HeLa cells using the lipofectamine reagent (Invitrogen). After 48 h of incubation, cells were diluted (1:10) and subjected to selection using zeocin (1 μg/ml) (Invitrogen). The resulting clones were picked, expanded and characterized for the expression of cytoplasmic GFP by in vivo imaging using a laser scanning microscope (LSM, Axioplan 2; Zeiss, Oberkochen, Germany). All heterogeneous cell populations including cells with and without GFP expression were subcloned or processed using the fluorescence-activated cell sorter (FACS; BD Biosciences, Germany) in order to obtain homozygous cells for the following analyses.
Immunofluorescence Analyses and LSM of Cx43 Mutants
All Cx43 mutated and wild-type HeLa cells were cultured to confluence, fixed with ice-cold absolute ethanol for 7 min, blocked with 5% bovine serum albumin (Sigma-Aldrich, Steinheim, Germany) and incubated with primary anti-rabbit anti-Cx43 antibody (1:1,000) for 1 h at room temperature. After washing with PBS- (phosphate-buffered saline without Ca2+ or Mg2+ ions), the secondary antibodies (goat-anti-rabbit, Alexa 594 conjugated, 1:2,000, MoBiTec, Goettingen, Germany) were added for 1 h. Then, specimens were washed with PBS- and mounted with Permaflour (Immunotech, Marseille, France). All micrographs were taken using the LSM Axioplan 2.
Microinjection Analyses and Quantitative Evaluation
Gap junctional function was tested by microinjection analyses (iontophoreses). Transfected HeLa cells, grown to confluence, were injected with neurobiotin, fixed with 1% glutaraldehyde in PBS- for 5 min, washed with PBS- and permeabilized with 2% Triton X-100 for 2 h at room temperature. The intercellular spreading of neurobiotin was indirectly determined using horseradish peroxidase-conjugated avidin D (Vector Laboratories, Burlingame, CA) for 90 min and detected with the HistoGreen POD substrate kit (Linaris, Wertheim-Bettingen, Germany) after washing with PBS- following the instructions of the manufacturer. The neurobiotin-positive cells of each HeLa cell clone tested were manually counted. At least 35 injections were carried out and statistically evaluated using Microsoft (Redmond, WA) Excel.
Immunoblot Analyses of Cx43 Mutants and Wild-Type Cells
Confluent cells were lysed in Complete Triton X-100 solution (Roche, Mannheim, Germany) sonicated for 30 s and, after determination of the protein amount, separated on discontinuous sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and blotted onto a Hybond enhanced chemiluminescence (ECL) membrane (Amersham Bioscience, Aylesbury, UK). Then, the membrane was blocked with 5% milk powder for 1 h, incubated with primary antibodies (rabbit anti-Cx43, 1:2,000) (Wilgenbus et al., 1992), washed and incubated with secondary antibodies (goat anti-rabbit, horseradish peroxidase-conjugated, 1:25,000; MoBiTec, Goettingen, Germany). After a final wash step, the membranes were incubated with an ECL reagent (Perbio, Bonn, Germany) and exposed ´to X-ray film (Kodak, Rochester, NY).
Quantification of Mutated and Wild-Type Cx43 Proteins
Since transfected HeLa cells usually express different levels of the protein of interest, it was important to determine the expression level of Cx43 protein in each HeLa cell clone tested. Thus, we quantified the intensities of the immunoblot bands using “quantity one” software (Bio-Rad, Munich, Germany). The expression level of the HeLa cell clone with the highest amount of Cx43 (Cx43 wild-type transfected cells) was set to 1, and the other clones were normalized to this value. The expression level was used for quantification of ATP release.
Determination of Hemichannel Activities by ATP Release
HeLa cells were cultured to a confluence of 40–50% on six-well plates (Falcon, Becton Dickinson, Lincoln Park, NJ), stimulated with 500 μl modified Hank’s balanced salt solution (Sigma-Aldrich) without Ca2+ and Mg2+ but with 1 mm ethyleneglycoltetraacetic acid (EGTA). After incubation for 20 min at 37°C, 100 μl of the supernatants were collected and added to 100 μl of the nucleotide releasing reagent (ViaLight HS kit; Cambrex, Rockland, ME) for 20 min. Luciferase activities were determined using a Berthold (Berthold, Wildbad, Germany) Microplate LB96V luminometer, which automatically added 20 μl of the ATP monitoring reagent and measured the release for 10 s. The results were normalized to the whole amount of protein and the expression levels of the transfected Cx43 wild-type and mutant proteins.
Determination of the Half-Life of Mutated and Wild-Type Cx43 Proteins in HeLa Cells by Pulse-Chase Analyses
All proteins expressed by HeLa cells were radioactively labeled using 35S-methionine as described by Hertlein et al., (1998). After 4 h of incubation, the medium was replaced by one containing nonradioactively labeled methionine. The cells were harvested in radioimmunoprecipitation assay buffer (Hertlein et al., 1998) directly or 2, 4, 6 or 8 h after labeling. The expressed connexin proteins were immunoprecipitated using anti-Cx43 antibodies coupled to protein A-Sepharose beads (Amersham Bioscience) overnight. In order to avoid purification of proteins unspecifically binding to the Sepharose, the protein lysates were prehybridized with pure Sepharose beads before immunoprecipitation. The next day, the protein Sepharose complexes were denatured at 60°C for 5 min and separated by SDS-PAGE. The gels were fixed in a solution containing 50% methanol, 10% acetic acid and 40% water; incubated for 20 min in Amplify (Amersham Bioscience); dried; and exposed for 1–14 days. The resulting bands were quantified using Herolab software (Herolab, Wiesloch, Germany). For dosimetric evaluation, the band intensities obtained after the 0 h chase were defined as 100%. The half-life of each mutated Cx43 protein was determined as the mean of at least three independent measurements using GraphPad (San Diego, CA) Prism software, version 4.02.
Results
Cloning and Stable Expression of Cx43 Mutants in HeLa Cells
In order to characterize the Cx43 mutations I31M, G138R, G143S and H194P, we mutated the mouse Cx43 coding DNA by overlap extension PCR and cloned the DNA amplicons into the IRES-eGFP-Zeo vector. The resulting constructs allowed the CMV promoter-driven overexpression of the Cx43 mutated genes and discrete cytoplasmic expression of eGFP reporter molecules in transfected connexin-deficient HeLa cells. In this way, cells that express the mutated Cx43 isoform could be easily detected by eGFP fluorescence.
FACS of Cells Expressing Cx43 Mutations
For the purpose of analyzing the Cx43 mutants, it was necessary to enrich cell populations that homogeneously expressed the mutated proteins. Use of eGFP as the reporter molecule allowed in vivo identification of transfected cells expressing the Cx43 mutated proteins and establishment of highly homogeneous cultures after subcloning or FACS of the transfectants. Thus, after a successful FACS, all cells of each sorted clone showed GFP expression (Fig. 1).

Establishment of homogenous HeLa cell cultures by FACS. Representative GFP fluorescence before (a) and after (b) successful sorting of transfected HeLa cell clones is shown. After sorting, all cells showed eGFP expression. (c) Granulation as indicated by the sideward scatter (SSC) and cell size as indicated by the forward scatter (FSC) were used to remove dead cells. (d) Overview of cell fractioning: blue, eGFP-positive cells selected for sorting; green, cells without eGFP (approximately 70%). Sorting efficiency was determined by comparing the number of eGFP-positive cells before (e) and after (f) sorting. Scale bars = 20 μm
Cellular Localization and Tracer Transfer of Mutated and Wild-Type Cx43 Protein
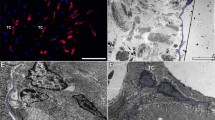
In order to investigate the trafficking of the mutated Cx43 proteins, immunofluorescence analyses with fixed HeLa cell transfectants were performed in comparison to cells expressing wild-type Cx43 (Fig. 2). Thus, Cx43 I31M, G138R and G143S mutants yielded immunosignals in membranes but also showed Cx43-positive staining in the cytoplasm. The I31M mutant showed less immunosignal in plasma membranes and increased expression in the endoplasmic reticulum region adjacent to the nucleus. In G138R and G143S mutants, only little Cx43-positive staining was expressed in the cytoplasm but on the contact membrane staining was observed to same extent as in Cx43 wild-type expressing cells. However, no punctate Cx43 staining in the membranes of Cx43 H194P mutants could be observed, indicating absence of gap junction plaques and a possible trafficking defect in this mutant. Here, Cx43-positive signals were detected only in the cytoplasm. Furthermore, the ability of the mutants to generate functional gap junctions was tested by neurobiotin transfer (Fig. 3). All Cx43 mutants analyzed showed no spreading of the injected dye, suggesting complete lack of intercellular communication.

Immunofluorescence analyses of HeLa cells expressing Cx43WT, Cx43I31M, Cx43G138R, Cx43G143S and Cx43G194P mutant proteins reveal the localization of the mutant proteins within the transfected cells. The occurrence of gap junction plaques is suggested by punctate Cx43 immunosignals in the plasma membrane (white arrows). Furthermore, Cx43-positive staining was also detected in the cytoplasm of some cells (blue arrows). Scale bars = 20 μm

Microinjection of neurobiotin. The connexin-deficient HeLa wild-type cells (a) as well as all Cx43 mutants (c-f) show no neurobiotin transfer, in contrast to Cx43 wild-type expressing cells (b). Neurobiotin transfer in Cx43 wild-type expressing cells was detected in about 130 cells (standard error of the mean = 12.1). Injected cells are marked by an arrow. Scale bars = 20 μm
ODDD Mutations Lead to Decreased Cx43 Phosphorylation
Wild-type Cx43 protein is characterized after electrophoretic separation by three phosphorylated isoforms, the P0, P1 and P2 protein bands, which can be detected after electrophoretic separation (Fig. 4). All tested mutants showed changes in their phosphorylation pattern (Fig. 4). Loss of the P2 phosphorylation band, which has been suggested to be necessary for gap junctional function (Oh et al., 1993), was found in the G138R mutant, whereas the mutations I31M, G143S and H194P expressed neither the P2 nor the P1 band. Some Cx43 mutations express functional hemichannels. It was previously reported that Cx43 hemichannels and gap junction channels are differentially regulated (De Vuyst et al., 2007) and that some Cx43 mutations lead to decreased hemichannel activity (Lai et al., 2006). Thus, we analyzed the hemichannel activity in Cx43 transfected HeLa cells under Ca2+-free conditions by determination of the extracellular ATP concentration normalized to the whole amount of protein and the amount of the Cx43 protein. In order to compare hemichannel activities of the stable HeLa cell transfectants, the expression level of Cx43 in each clone was determined by quantitative immunoblot analyses (Fig. 5). The ATP release studies revealed that Cx43 I31M, G138R and G143S mutants released more ATP into the extracellular space relative to Cx43 wild type (Fig. 6), suggesting enhanced activity of open hemichannels (Table 1). The hemichannel activity measured in the Cx43 H194P mutant was similar to that in cells expressing wild-type Cx43 protein.

Immunoblot analysis of all Cx43 transfectants. Cx43-positive signals were detected in all clones. Wild-type Cx43-expressing cells showed all known phospho-isoforms (P0, P1 and P2). In contrast, the I31M-, G143S- and H194P-expressing cells revealed a lack of the P1 and P2 bands, whereas the G138R transfectants did not express the P2 isoform but the P1 band was detected

Quantification of Cx43 expression in transfected cells. The band intensities seen on anti-Cx43 immunoblot (Fig. 4) were quantitatively compared to each other relative to the most intensive band (Cx43 wild-type expressing cell clone). The relative Cx43 expression allows comparison of each clone with any other clone and was used for the determination of relative ATP release

ATP release from Cx43 mutants. The I31M, G138R and G143S mutants release significantly more ATP into the extracellular solution than Cx43 wild-type cells, but the H194P mutant shows similar ATP release to Cx43 wild-type transfectants
Some Cx43 Mutated Proteins Exhibit Extended Half-Life
A possible explanation for the observed increased hemichannel activity of some mutants could be a disturbed turnover of the proteins. Thus, pulse-chase experiments followed by Cx43 immunoprecipitations with lysates from mutant and wild-type Cx43-expressing cells were performed. The Cx43 G138R and G143S mutations showed an increased half-time (t 1/2, G138R = 4.8 h, t 1/2, G143S = 3.7 h) (Fig. 7). However, the I31M mutation, which also exhibited increased hemichannel activity, did not show an obvious turnover defect (t 1/2, I31M = 2.8 h), similar to the H194P mutation without altered hemichannel activity, the Cx43 H194P mutation (t 1/2, H194P = 2.2 h). The half-life of Cx43 wild-type protein was confirmed in our study to be 2.7 h.

Pulse-chase analyses indicate degradation defects in some ODDD mutants. The G138R as well as G143S mutants showed a half-life increase from 2.7 h in Cx43WT (a) to 4.8 h in G138R (c) and 3.7 h in G143S (d) mutants, suggesting a degradation problem of these proteins. The I31M (b) and H194P (e) mutated proteins showed no obvious turnover alterations, as indicated by the similar half-lives (2.8 and 2.2 h, respectively)
Discussion
In this study, we characterized four ODDD-associated Cx43 mutations (I31M, G138R, G143S and H194P) in stable and homogeneous HeLa cell cultures. All these mutations inhibited gap junctional coupling, as indicated by neurobiotin transfer, but interestingly did not disturb and, in some cases, even showed enhanced hemichannel activity. These results could be due to a change in phosphorylation, trafficking and degradation of the mutated Cx43 protein analyzed by immunoblot, immunofluorescence and half-life determination.
To avoid the influence of a tagged reporter protein on mutant proteins, we used a construct coding for an eGFP reporter protein 5´ downstream of IRES and the mutated Cx43 gene. Thus, we were able to enrich by FACS eGFP-positive cells which express the mutant protein detected by its immunofluorescence. Here, the Cx43 I31M mutation showed decreased immunosignal and increased Cx43-positive staining in regions of endoplasmic reticulum and Golgi apparatus, indicating disturbed, but not abolished, trafficking. Thus, it is possible that in this mutant only the Golgi apparatus-dependent transport of Cx43 is impaired and the alternative, Golgi-independent, transport is still functioning, as already described for the I28L mutation (Martin et al., 2001). The G138R and G143S mutations yielded gap junction plaques and a slight deposition in the cytoplasm, most probably due to overexpression of the mutant proteins. An obvious difference between the two mutants and the wild-type protein could not be observed. A clear deficit in trafficking was seen in cells expressing the Cx43 H194P mutant, where no immunosignal in the plasma membrane could be seen. In this case Cx43-positive staining could only be detected in the cytoplasm. The H194P mutation is localized in the second extracellular loop of Cx43, a domain important for docking to a hemichannel in the adjacent cell and thus for generation of a gap junctional channel (Foote et al., 1998; Dahl et al., 1992). The mutation altered the histidine residue at position 194 to proline, which strongly inhibits ß-sheet conformation in proteins. This could impair correct folding of the Cx43 protein and abolish the generation of disulfide bonds between adjacent cysteine residues at positions 192 and 198 with those in the first extracellular loop.
Gap junction-mediated transfer of neurobiotin was inhibited by the Cx43 mutations. Cx43 I31M is localized in the first transmembrane domain, which is assumed to be a part of the gap junctional pore (Fleishmann et al., 2004) and an important determinant of conductance of gap junction channels (Hu, Ma & Dahl, 2006). An altered secondary structure of Cx43 could also be a reason for the disrupted gap junctional coupling in G138R- and G143S-expressing cells. Both mutations flank the α-helix observed between amino acid residues at positions 138 and 144 at pH 5.8 and 7°C (Duffy et al., 2002) and change the α-helix braking glycine residues to amino acids without direct influence on the structure. Thus, it is possible that the α-helical region is extended by these two mutations, favoring binding to the carboxy-terminal tail and thus causing the closure of the mutated channel. However, enhanced binding of the cytoplasmic loop to the carboxy terminus needs to be demonstrated. The disturbed neurobiotin transfer seen in the H194P mutant is most probably due to the mislocalization of the mutant protein and a lack of gap junction plaques observed in the immunofluorescence studies.
In order to compare the influence of the mutated proteins on hemichannel function, expression was quantified by immunoblot analyses and used to normalize the results obtained by ATP release studies. Thus, all mutants showed functional hemichannels, the I31M, G138R and G143S mutants even revealing enhanced ATP release. Our results regarding the H194P mutant are similar to those reported for the Cx43 cysteine-less mutant (Bao et al., 2004), which also revealed a lack of gap junction plaques but still indicated functional hemichannels. The expression of increased hemichannel function by Cx43 mutants as seen in I31M, G138R and G143S cells has not been yet reported but is similar to that found with other connexin isoforms, such as F235C (Lin Liang et al., 2005) and G45E (Stong et al., 2006) mutants, resulting in “leaky” hemichannels. There is only one study describing an influence of ODDD-associated Cx43 mutations on hemichannel function (Y17S, G21R, A40V, F52dup, L90V and I130T; Lai et al., 2006). Here, eGFP fusion proteins were analyzed by propidium iodide uptake, which revealed loss of hemichannel activity in these mutants. In our study, the enhanced hemichannel activity of some mutants (G138R and G143S) can be explained by the obvious degradation defect seen in pulse-chase analyses. The increased hemichannel function observed in the I31M mutant showing a normal half-time cannot be explained by an increased amount of Cx43 hemichannels in the membrane. Thus, it is possible that the I31M mutation localized in the first transmembrane domain, which is an important determinant of conductance (Hu et al., 2006), changes the properties of Cx43 hemichannels. However, it has recently been controversially discussed whether dye uptake or ATP release results are due to hemichannel activity (Spray et al., 2006). We cannot exclude that other mechanisms influenced by Cx43 mutations could be responsible for the effects linked to connexin hemichannels.
The obvious change in trafficking, coupling and hemichannel function of the mutated proteins could be due to their disturbed phosphorylation. In our immunoblot analyses, absence of the P2 phosphorylation band and loss of the P1 band in all mutants except G138R were observed, indicating severe dysregulation of the mutant proteins. It had been reported that phosphorylation of Cx43 induces closure, changes the permeant size selectivity of its hemichannels (Saez et al., 2005; Bao et al., 2007) and influences the regulation of gap junction channel selectivity (Lampe et al., 2006). The dephosphorylation of the G138R and G143S mutated proteins can be explained by enhanced binding of the carboxy-terminal region to the mutated cytoplasmic loop, making the carboxy terminus partially accessible to kinases. Otherwise, the lack of the P1 and P2 phosphorylation bands in I31M and H194P mutants can be explained by their disturbed trafficking. Phosphorylation of connexin proteins in the endoplasmic reticulum and Golgi apparatus is necessary for correct trafficking to the membrane and the generation of functional gap junction plaques (Laird, Castillo & Kasprzak, 1995). Our data suggest that some ODDD-associated Cx43 mutations can exhibit enhanced hemichannel activity. This may aggravate the phenotypical consequences due to strongly decreased Cx43 gap junctional channels in ODDD mutants.
References
Attal J, Theron MC, Houdebine LM (1999) The optimal use of IRES (internal ribosome entry site) in expression vectors. Genet Anal 15:161–165
Bao X, Chen Y, Reuss L, Altenberg GA (2004) Functional expression in Xenopus oocytes of gap-junctional hemichannels formed by a cysteine-less connexin 43. J Biol Chem 279:9689–9692
Bao X, Lee SC, Reuss L, Altenberg GA (2007) Change in permeant size selectivity by phosphorylation of connexin 43 gap-junctional hemichannels by PKC. Proc Natl Acad Sci USA 104:4919–4924
Bennett MVL, Verselis VK (1992) Biophysics of gap junctions. Semin Cell Biol 3:29–47
Bruzzone S, Guida L, Zocchi E, Franco L, De Flora A (2001) Connexin 43 hemichannels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J 15:10–12
Dahl G, Werner R, Levine E, Rabadan-Diehl C (1992) Mutational analysis of gap junction formation. Biophys J 62:172–180
De Vuyst E, Decrock E, De Bock M, Yamasaki H, Naus CC, Evans WH, Leybaert L (2007) Connexin hemichannels and gap junction channels are differentially influenced by lipopolysaccharide and basic fibroblast growth factor. Mol Biol Cell 18:34–46
Duffy HS, Sorgen PL, Girvin ME, O’Donnell P, Coombs W, Taffet SM, Delmar M, Spray DC (2002) pH-dependent intramolecular binding and structure involving Cx43 cytoplasmic domains. J Biol Chem 277:36706–36714
Evans WH, De Vuyst E, Leybaert L (2006) The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J 397:1–14
Fleishman SJ, Unger VM, Yeager M, Ben-Tal N (2004) A Calpha model for the transmembrane alpha helices of gap junction intercellular channels. Mol Cell 15:879–888
Foote CI, Zhou L, Zhu X, Nicholson BJ (1998) The pattern of disulfide linkages in the extracellular loop regions of connexin 32 suggests a model for the docking interface of gap junctions. J Cell Biol 140:1187–1197
Gerido DA, Derosa AM, Richard G, White TW (2007) Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am J Physiol Cell Physiol 293:337–345
Hanemann CO, Bergmann C, Senderek J, Zerres K, Sperfeld AD (2003) Transient, recurrent, white matter lesions in X-linked Charcot-Marie-Tooth disease with novel connexin 32 mutation. Arch Neurol 60:605–609
Hertlein B, Butterweck A, Haubrich S, Willecke K, Traub O (1998) Phosphorylated carboxy terminal serine residues stabilize the mouse gap junction protein connexin45 against degradation. J Membr Biol 162:247–257
Hu X, Ma M, Dahl G (2006) Conductance of connexin hemichannels segregates with the first transmembrane segment. Biophys J 90:140–150
Lai A, Le D-N, Paznekas WA, Gifford WD, Wang Jabs E, Charles AC (2006) Oculodentodigital dysplasia connexin43 mutations result in non-functional connexin hemichannels and gap junctions in C6 glioma cells. J Cell Sci 119: 532–541
Laird DW, Castillo M, Kasprzak L (1995) Gap junction turnover, intracellular trafficking, and phosphorylation of connexin43 in brefeldin A-treated rat mammary tumor cells. J Cell Biol 131:1193–1203
Lampe PD, Cooper CD, King TJ, Burt JM (2006) Analysis of connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci 119:3435–3442
Lin Liang SG, de Miguel M, Gomez-Hernandez JM, Glass JD, Scherer SS, Mintz M, Barrio LC, Fischbeck KH (2005) Severe neuropathy with leaky connexin32 hemichannels. Ann Neurol 57:749–754
Martin PE, Blundell G, Ahmad S, Errington RJ, Evans WH (2001) Multiple pathways in the trafficking and assembly of connexin 26, 32 and 43 into gap junction intercellular communication channels. J Cell Sci 114:3845–3855
Oh SY, Dupont E, Madhukar BV, Briand JP, Chang CC, Beyer E, Trosko JE (1993) Characterization of gap junctional communication-deficient mutants of a rat liver epithalial cell line. Eur J Cell Biol 60:250–255
Paznekas WA, Boyadjiev SA, Shapiro RE, et al. (2003) Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet 72:408–418
Saez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV (2005) Connexin-based gap junction hemichannels: gating mechanisms. Biochim Biophys Acta 1711:215–224
Soehl G, Willecke K (2003) An update on connexin genes and their nomenclature in mouse and man. Cell Commun Adhes 10:173–180
Spray DC, Ye ZC, Ransom BR (2006) Functional connexin “hemichannels”: a critical appraisal. Glia 547:758–773
Stong BC, Chang Q, Ahmad S, Lin X (2006) A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope 116:2205–2210
Stout CE, Costantin JL, Naus CCG, Charles AC (2002) Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem 12:10482–10488
Wilgenbus KK, Kirkpatrick CJ, Knuechel R, Willecke K, Traub O (1992) Expression of Cx26, Cx32 and Cx43 gap junction proteins in normal and neoplastic human tissues. Int J Cancer 51:522–529
Yotsumoto S, Hashiguchi T, Chen X, Ohtake N, Tomitaka A, Akamatsu H, Matsunaga K, Shiraishi S, Miura H, Adachi J, Kanzaki T (2003) Novel mutations in GJB2 encoding connexin-26 in Japanese patients with keratitis-ichthyosis-deafness syndrome. Br J Dermatol 148:649–653
Acknowledgment
This work was supported by a grant of the German Research Association through SFB 645, project B2 (to K. W.). We thank Dr. Elmar Endl (Institute of Molecular Medicine, University of Bonn) for advice on the use of the FACS instrument.
Author information
Authors and Affiliations
Corresponding author
Additional information
Radoslaw Dobrowolski and Annette Sommershof contributed equally to this work.
Rights and permissions
About this article
Cite this article
Dobrowolski, R., Sommershof, A. & Willecke, K. Some Oculodentodigital Dysplasia-Associated Cx43 Mutations Cause Increased Hemichannel Activity in Addition to Deficient Gap Junction Channels. J Membrane Biol 219, 9–17 (2007). https://doi.org/10.1007/s00232-007-9055-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-007-9055-7