
Abstract
During the past decade, remarkable advances have been made in using atomic force microscopy (AFM) for measuring the forces and the dynamics of the interaction between individual ligands and receptors, providing fundamental insights into molecular recognition processes. In addition, affinity imaging using either adhesion force mapping or dynamic recognition force mapping has offered a means to localize specific binding sites on model and cellular surfaces. These single-molecule analyses provide novel insight into the structure–function relationships of molecular recognition systems. In this review, we describe the principles of molecular recognition studies using AFM and provide a flavor of recent progress made in the field.
Similar content being viewed by others
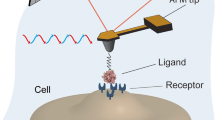


Introduction
Atomic force microscopy (AFM) has opened a wide range of novel possibilities for imaging and manipulating biological systems in their native environment. While AFM imaging offers a means to visualize surface structures at high resolution and in physiological conditions [1, 2], force spectroscopy enables researchers to measure the inter- and intramolecular interactions of biomolecules, providing new insights into the molecular bases of macromolecular elasticity [3, 4], protein folding [5], and receptor–ligand interactions [6]. In this context, AFM has recently emerged as a powerful ultrasensitive force probe for single molecular recognition studies, providing valuable information on the molecular dynamics within the complexes during association and dissociation and permitting the mapping of individual ligands and receptors on biosurfaces.
The general principle of AFM is to scan a sharp tip over the surface of a sample while sensing the so-called near-field physical interactions between the tip and the sample. This allows three-dimensional images to be generated directly in aqueous solution. The sample is mounted on a piezoelectric scanner which ensures three-dimensional positioning with high accuracy. While the tip (or sample) is being scanned in the (x, y) directions, the force interacting between tip and specimen is monitored with piconewton sensitivity. This force is measured by the deflection of a soft cantilever which is detected by a laser beam focused on the free end of the cantilever and reflected into a photodiode.
A number of different AFM imaging modes are available, which differ mainly in the way the tip is moving over the sample. In the so-called contact mode, the AFM tip is raster-scanned over the sample while the cantilever deflection, thus, the force applied to the tip is kept constant using feedback control. In dynamic imaging modes (tapping, intermittent or magnetic AC; depending on the method of excitation), an oscillating tip is scanned over the surface and the amplitude and phase of the cantilever are monitored near its resonance frequency. Because lateral forces during imaging are greatly reduced with dynamic modes, they are advantageous for imaging soft biological samples.
In force spectroscopy, the cantilever deflection is recorded as a function of the vertical displacement of the piezoelectric scanner, i.e. as the sample is pushed towards the tip and retracted. This results in a cantilever deflection (d) vs scanner displacement (z) curve, which can be transformed into a force–distance curve by converting the cantilever deflection into a force (F) using Hooke’s law (F = −k × d, where k is the cantilever spring constant) and subtracting the deflection from the scanner displacement to obtain the distance (z − d). The point of contact (zero separation distance) is determined as the position of the vertical linear parts of the curve in the contact region. Force–distance curves can be recorded either at single well-defined locations of the (x, y) plane or at multiple locations to yield a so-called “force-volume image”. In doing so, spatially resolved maps of sample properties and molecular interactions can be produced. For quantitative force measurements, calibration of the actual spring constants of the cantilevers is necessary [7].
Functionalized tips
A critical issue for measuring molecular recognition interactions is to functionalize AFM tips with biomolecules using appropriate procedures, keeping in mind the following points: (1) the forces which immobilize the molecules should be stronger than the intermolecular force being studied; (2) the attached biomolecules should have enough mobility so that they can freely interact with complementary molecules; (3) the contribution of non-specific adhesion to the measured forces should be minimized; (4) attaching biomolecules at low surface density is recommended in order to ensure single-molecule detection; (5) site-directed coupling may be desired to orientate all the interacting molecules in the same way.
A pioneering immobilization strategy used the non-specific adsorption of biotinylated bovine serum albumin (BBSA). This method permitted the measurement of the interaction forces between biotin and avidin (or streptavidin) down to the single-molecule level [8–10]. The BBSA layer can also be further reacted with avidin/streptavidin to attach biotinylated molecules [11, 12]. Today, the biotin–avidin approach is rarely used because parallel breakage of multiple bonds is often observed, rendering data interpretation delicate. A better controlled approach relies on the strong binding of thiols on gold-coated tips. While proteins, oligonucleotides, and carbohydrates that bear thiol groups can directly be attached on gold [13, 14], biomolecules can also be covalently attached onto self-assembled monolayers (SAMs) of functionalized alkanethiols on gold using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and N-hydroxysuccinimide (NHS) [15]. In this context, an interesting approach is to mix long chain alkanethiols with COOH terminal functions in a matrix of shorter OH-terminated alkanethiols, which ensures a certain mobility of the attached biomolecules and minimizes non-specific adsorption [16]. A third approach uses amine-functionalization procedures to covalently anchor biomolecules on silicon tips [17, 18]. The amino-terminated surfaces are reacted with a cross-linker which provides the ligands with motional freedom and prevents their denaturation. Cross-linkers typically carry two different functional ends, e.g., an amine reactive NHS group on one end and 2-pyridyldithiopropionyl or vinyl sulfone groups, which can be covalently bound to thiols, on the other. Alternatively, aldehyde groups directly attach to proteins through their lysins [19]. Importantly, both the thiol and silane approaches make it possible to orientate the attached biomolecules via their C-terminal or N-terminal domains by linking recombinant histidine-tagged proteins onto an AFM tip coated with nitrilotriacetate groups [12, 20].
Measuring molecular recognition forces
Probing molecular recognition forces implies recording force curves between a modified tip and the sample (e.g., cells, solid surfaces) and then assessing the unbinding (or adhesion) force between complementary receptor and ligand molecules. A typical force curve obtained using an antibody-modified AFM tip and the surface of a live cell is shown in Fig. 1a [21]. If the ligand on the tip does not form a specific bond with the receptor on the cell surface, the recognition event is missing, and the retrace looks like the trace (inset in Fig. 1a). In addition, the specificity of ligand-receptor binding is usually demonstrated by blocking experiments with free ligands, which are injected into the solution to block the receptor sites on the cell surface. As a consequence, almost all specific recognition signals completely disappear and only occasional adhesion events are observed.

Measuring single molecular recognition forces. a A force–distance cycle on the surface of a living cell with an antibody-coated AFM tip shows specific interaction in the retrace (jump at 75 nm). The interaction is blocked using free antibody in solution (inset), proofing the specificity of the detected force-jump. b The probability density function (pdf) is constructed from an ensemble of forces and gives the distribution of unbinding forces, similar to a histogram. The maximum of the pdf depicts the most probable unbinding force at a given loading rate. Reprinted with permission from [21]
After acquiring hundreds of force curves, empirical probability density functions (pdf) from the detected unbinding forces can be constructed (Fig. 1b) [21]. The maximum of the distribution (Fig. 1b) reflects the most probable force upon which a single antibody–cell surface bond dissociates under the force ramp used. An overall binding probability, which is the probability to record an unbinding event, of 15% was obtained. Blocking experiments, performed by injection of free antibody in solution reduced this probability to 6%. No binding was found at all when a bare tip was used instead of an antibody-coated tip. These results strongly support the specificity of the binding events observed.
Using such force spectroscopy experiments, a variety of ligand-receptor forces have been measured at the single-molecule level including those associated with avidin/streptavidin [8, 9] antibodies [16, 17, 22], DNA [13, 23, 24], carbohydrates [15], lectins [14, 25], cadherins [26, 27], integrins [28, 29], selectins [30], and bacterial adhesins [12]. Knowledge of these forces contributes to refine our understanding of the molecular basis of molecular recognition events such as those mediating cell adhesion and immunological processes, and offers promising prospects in nanobiotechnology for the rapid detection of bio-analytes.
Several reports showed that unbinding forces between receptors and ligands depend on the loading rate, i.e., the rate at which the force is applied to the bond (Fig. 2) [11, 31–33]. To understand this behavior, one must recall that when viewing ligand-receptor binding on the single-molecule level, the average lifetime of a ligand–receptor bond, τ(0), can be taken as the inverse of the kinetic off-rate constant, τ(0) = 1/k off. In the thermal activation model, the lifetime of a complex in solution is described by a Boltzmann equation, τ(0) = τ osc exp(E b/k B T), [34] where τ osc is the inverse of the natural oscillation frequency and E b the energy barrier for dissociation. Hence, due to the thermal energy, there is a finite probability of overcoming the energy barrier E b, which leads to the separation of the ligand–receptor complex. A force acting on a binding complex deforms the interaction energy landscape and lowers the activation energy barrier. The lifetime τ(f) of a bond loaded with a constant force f is given by τ(f) = τ osc exp(E b−fx/k B T), x being interpreted as the distance of the energy barrier from the energy minimum. Using AFM, an effective force increase or loading rate r can be deduced from r = df/dt, equal to pulling velocity times effective spring constant. The combination of the Boltzmann equation with the stochastic description of the unbinding process predicts different unbinding force distributions at different loading rates r. The maximum of each force distribution, f*(r), reflects the most probable unbinding force for the respective loading rate r. f* is related to r through f*(r) = k B T/x ln(r.x/k B T.k off). Apparently, the unbinding force f* scales linearly with the logarithm of the loading rate. For a single barrier, this would give rise to a simple linear dependence of the force on the logarithm of the loading rate. This behavior has been observed for a number of receptor–ligand interactions, including that between the clinically important vancomycin antibiotic and D–Ala–D–Ala ligands (Fig. 2a). From these dynamic force spectroscopy data, the length scale of the energy barrier, x β, can be assessed from the slope f β of the force vs ln(r) plot (e.g., for vancomycin x β ≈ 0.36 nm), while extrapolation to zero forces yields the kinetic off-rate constant of dissociation at zero force (e.g., for vancomycin k off = r F = 0 x β/k B T = 2 × 10−3 s−1). In cases where more barriers are involved along the escape path, the curve will follow a sequence of linear regimes, each of which marks a particular barrier.

Dynamics of receptor–ligand interactions. a Adhesion force measured in phosphate-buffered saline (PBS) between a vancomycin-modified tip and a D-Ala-D-Ala-modified surface as a function of the logarithm of the loading rate applied during retraction (the interaction time and approach speed were kept constant). b Adhesion frequency as a function of interaction time (the approach and retraction speeds were kept constant). Reprinted with permission from [33]
The interaction time is another parameter that may affect the force data. The adhesion probability of vascular endothelial (VE)-cadherin was shown to dramatically increase with interaction time to reach a plateau after 0.2 s [26, 27]. Similar time dependency was observed for the vancomycin/D–Ala–D–Ala interaction (Fig. 2b), pointing to a slow association rate that may involve slow molecular rearrangements. From such data, the interaction time needed for half-maximal probability of binding, t 0.5, can be estimated (for vancomycin, t 0.5 = 0.25 s), allowing then to assess the association rate constant, k on = t 0.5 −1 N A V eff (for vancomycin, k on = 5 M−1 s−1), where V eff is the effective volume explored by the tip-tethered ligand, approximated here to a half-sphere of 1 nm radius [17]. Knowledge of the rate constant values then provides an estimate of the equilibrium dissociation constant: K D = k off/k on (for vancomycin, K D = 0.4 mM).
Importantly, the interaction time may also shift the distribution of adhesion forces towards larger values. For VE-cadherins, this phenomenon was ascribed to the time-dependent association of the protein into complexes with higher order adhesion strengths [26, 27]. For the bacterial adhesin HBHA, the prolonged contact time required to establish strong interaction with heparin was suggested to reflect the time necessary for conformational changes within both molecules to allow an optimal fitting between charged groups [12]. Hence, it is essential in molecular recognition force measurements to vary the loading rate and interaction time to provide reliable data and to assess kinetic parameters of the unbinding process.
Affinity imaging
Combination of high-resolution topography imaging with single-molecule recognition provides unique possibilities for the identification and localization of specific receptors and ligands on biosurfaces such as biomimetic surfaces, cells, and membranes. To date, there are two ways to perform such affinity imaging using AFM, i.e., adhesion force mapping and dynamic recognition force mapping.
Adhesion force mapping consists in recording a “force-volume image”, i.e., an array of force curves in the x, y plane on an area of given size, assessing the unbinding force values for all curves and displaying them as gray pixels [35]. So far, this method has been applied to different cell types, including red blood cells [25], osteoclasts [36], and endothelial cells [37]. The power of this approach in microbiology (bacterial pathogenesis) has also been demonstrated (Fig. 3) [12]. Bacterial infections are generally initiated by the interaction between adhesins, i.e., cell adhesion proteins, on the bacterial pathogen and specific receptors on the host cell surface. A key example is Mycobacterium tuberculosis which adheres to heparan sulfates on epithelial cells via the heparin-binding haemagglutinin adhesin (HBHA). To shed new light on the molecular bases of this interaction, M. bovis BCG cells expressing HBHA were immobilized on a polycarbonate membrane, an approach which allows live cells to be imaged without using any drying or fixation step (Fig. 3a). High-resolution images revealed a smooth and homogeneous surface (Fig. 3b), consistent with earlier scanning electron microscopy observations. Affinity maps (Fig. 3c and d) recorded on cells with a heparin-modified tip revealed adhesion events (bright pixels) in about half of the locations. The adhesion force magnitude was very close to the value expected for a single HBHA–heparin interaction, supporting the notion that single HBHA were detected. This was confirmed by showing that a mutant strain lacking HBHA did not bind the heparin tip. Interestingly, the HBHA distribution was not homogeneous, but apparently concentrated into nanodomains which may promote adhesion to target cells by inducing the recruitment of receptors within membrane rafts. In the future, these molecular recognition studies may help in the development of new drugs capable to block bacterial adhesion.

Mapping single adhesins on live bacteria. a AFM topographic image recorded in PBS showing two M. bovis BCG cells on a polymer substrate. b High-resolution image of the cell surface revealing a smooth morphology. c, d Adhesion force maps (gray scale, 100 pN) recorded in PBS with a heparin-modified AFM tip. Adhesion events (bright pixels) reflect the detection of single adhesins. The adhesin distribution is not homogeneous, but apparently concentrated into nanodomains which may play important biological functions. Reprinted with permission from [12]
More recently, antibiotic-modified tips were used to map individual binding sites on live bacteria (Fig. 4) [33]. Fluorescence microscopy with a fluorescent vancomycin probe was used to visualize D–Ala–D–Ala sites of nascent peptidoglycan in the cell wall of dividing Lactococcus lactis cells (Fig. 4a and b). Fluorescence staining of the wild-type strain was found around the septum, while no fluorescent labeling was detected for a mutant strain producing peptidoglycan precursors ending by D–Ala–D–Lac instead of D–Ala–D–Ala. AFM topographic images of L. lactis cells revealed a smooth and elongated cell morphology as well as a well-defined division septum (Fig. 4c). Ring-like structures were seen at a certain distance from the septum, presumably formed by an outgrowth of the cell wall. Notably, adhesion force maps demonstrated that binding sites were essentially located in the septum region, and more specifically on the equatorial rings (Fig. 4d), suggesting that newly formed peptidoglycan was inserted in these regions. This study shows that AFM with vancomycin tips is a complementary approach to fluorescent vancomycin to explore the architecture and assembly process of peptidoglycan during the cell cycle of Gram-positive bacteria. While fluorescence microscopy generates microscale images allowing the localization of peptidoglycan in the entire cell wall, AFM adhesion force mapping reveals the distribution of single peptidoglycan molecules on the outermost cell surface.

Imaging single antibiotic binding sites on live bacteria. Phase contrast (a) and fluorescence image (b) of Lactococcus lactis cells during the course of the division process. Fluorescent vancomycin accumulates at the division site by attaching specifically to D–Ala–D–Ala sites of cell wall peptidoglycan. c AFM image of a cell showing a well-defined division septum as well as a ring-like structure expected to be rich in nascent peptidoglycan (white box). d Adhesion force map (gray scale, 100 pN) recorded with a vancomycin tip on the septum region (highlighted by the white box in c). Adhesion events are essentially located in the septum region (red line), more specifically on the ring-like structure, suggesting that newly formed peptidoglycan is inserted. Reprinted with permission from [33]
Time resolution is an important limitation of adhesion force mapping, as the time currently required to record a map is in the order of 2–15 min depending on the acquisition parameters. This is much greater than the timescale at which dynamic processes usually occur in biology. Fortunately, this problem can be solved by using dynamic recognition imaging [38, 39] in which molecular recognition signals are detected during dynamic force microscopy imaging. Topography and recognition (abbreviated as TREC) images are acquired using tips which are magnetically oscillated during scanning and contain ligand molecules directed against the cognate receptor on the surface. For cantilevers driven at frequencies below resonance, the surface contact only affects the downward deflections (i.e., the minima) of the oscillations, whereas binding of the ligand on the tip to the receptor molecule on the surface affects only the upward deflections (i.e., the maxima) of the oscillations. Accordingly, the oscillation minima are used to record the topography image, and the oscillation maxima provide the data for the recognition image (Fig. 5a).

TREC imaging. a Principle: the cantilever oscillation is split into lower and upper parts, resulting in simultaneously acquired topography and recognition images. b Avidin was electrostatically adsorbed to mica and imaged with a biotin-tethered tip. A good correlation between topography (left image, bright spots) and recognition (right image, dark spots) was found (solid circles). Topographical spots without recognition denote structures lacking specific interaction (dashed circle). The scan size was 500 nm. Reprinted with permission from [38]
As a test system, single avidin molecules were immobilized onto mica via electrostatic adsorption by virtue of their positive net charge at neutral pH [38]. In addition, a biotin residue was coupled to the AFM tip via a distensible poly(ethylene glycol) chain. More specifically, biotin was covalently tethered to the amino-functionalized tip in a single coupling step using “biotin–PEG–NHS” which consists of a poly(ethylene glycol) chain, a biotin on one end, and the amino-reactive N-hydroxysuccinimide ester function (NHS group) on the other end. With this configuration, two independent maps were simultaneously acquired, i.e., a topography image of the immobilized avidin molecules and a lateral map of corresponding recognition sites (Fig. 5b), both recorded at experimental times comparable to normal AFM imaging.
Figure 6 shows the application of the simultaneous imaging of topography and recognition sites on cells [39]. Real-time visualization and quantification of receptor binding sites on cell surfaces is a fundamental challenging task in cell biology. This is normally achieved by using immunofluorescence techniques such as immunostaining. Recently, the simultaneous imaging of topography and recognition sites has been applied to VE cells to localize VE-cadherin binding sites on the cell membrane [39]. In this study, topography (Fig. 6a) and recognition (Fig. 6b) images were recorded on gently fixed cells using AFM tips which were chemically functionalized with fibrinogen. Besides the prominent role of fibrinogen in hemostasis, fibrinogen molecules can bind specifically to VE-cadherins on the cell membrane. Many specific interactions were observed in the recognition image (Fig. 6b). The recognition dark spots in the recognition image reveal the locations of VE-cadherins in the topographical image with high lateral resolution and high efficiency. The specificity of the recognition process was tested by adding ethylenediaminetetraacetic acid in solution. After injection, the dark spots in the recognition image disappeared, while the topographical image did not change (not shown). Thus, the simultaneous investigation of both topography and recognition opens a wide field of applications for investigating biological structure–function relationships in native environments on the nanometer scale because the technique can map composition on top of a topographical image and can detect compositional changes occurring during biological processes.

Recognition imaging (TREC) on cells. Simultaneous recorded topography (a) and recognition (b) images on gently fixed MyEnd cells acquired with a fibrinogen-coated AFM tip. Scale bars on both images are 200 nm. The z-scales range from 0 to 25 nm (from dark brown to white) and 0 to 0.7 V for topography and recognition images, respectively
Conclusions and perspectives
Rapid advances have occurred in developing reliable protocols for attaching biomolecules on AFM tips and solid surfaces for single molecular recognition studies. Much progress has also been made in optimizing data acquisition and interpretation in single-molecule force spectroscopy measurements, allowing accurate determination of the forces and of the dynamics of a variety of receptor–ligand interactions, both on model surfaces and on cells. Remarkably, AFM used in the adhesion force or dynamic recognition force mapping modes, is the only technique capable of localizing single specific binding sites on cells.
Yet, a number of technological challenges remain to be solved for exploiting the full potential of AFM in physiology. A first issue is the alteration of soft cellular samples by the scanning tip during imaging, a problem that is usually solved by gentle fixation of the cells. Because fixation may change the structural, mechanical, and diffusion properties of cell membranes, the relevance of the data remain questionable. Progress in sample preparation procedures and recording conditions are needed to solve this problem. Time resolution is another crucial factor limiting the use of AFM in live cell studies. To date, recording a molecular recognition image takes several minutes (TREC image), which is much greater than the timescale at which dynamic processes usually occur in cell physiology. Interestingly, remarkable advances have been made in developing scanning probe instruments with increased imaging rates, giving access to unprecedented timescales (millisecond resolution) [40–42]. These ultrafast techniques open up fascinating new perspectives to explore molecular and cellular dynamics. Another exciting challenge for functional studies on cells will be to combine AFM molecular recognition imaging with advanced optical techniques, particularly stimulated emission depletion microscopy which currently reaches resolutions of a few tens of nanometers on cells [43].
In the future, we anticipate that single molecular recognition studies will be increasingly used to probe receptors sites on cell surfaces in connection with medical and physiological issues. In this context, the AFM was recently employed to elucidate a genetic disease, cystic fibrosis (CF), which can cause enormous obstructive airway functions and hypoxia [44]. Cystic fibrosis transmembrane regulator (CFTR) molecules on membrane patches of CFTR were imaged and counted using AFM. Their number density approximately doubled after stimulation with cAMP and, in addition, cluster formation was observed, consistent with the concept that CFTR function involves interaction with itself or other proteins. For the identification of CFTR in the heterogeneous membrane patches, either gold particles functionalized with antibodies directed against CFTR as specific markers, or alternatively, TREC imaging using a tip functionalized with the same antibody, were applied. It was found that there is a lack of CFTR in the red blood cells (RBC) of CF patients, which influences the cell stiffness, as probed in AFM-based force-indentation cycles. The twofold increase in stiffness for RBCs from patients with CF might contribute to the development of pulmonary hypertension and hemolytic amentia. Along the same line, we expect that AFM-based single-molecule studies should help in elucidating the factors determining the strength and specificity of cellular interactions, such as those mediated by cadherins and integrins [45, 46].
References
Engel A, Muller DJ (2000) Observing single biomolecules at work with the atomic force microscope. Nat Struct Biol 7:715–718
Jena BP, Hörber JK (2002) Atomic force microscopy in cell biology, methods in cell biology vol 68. Academic, San Diego
Clausen-Schaumann H, Seitz M, Krautbauer R, Gaub HE (2000) Force spectroscopy with single bio-molecules. Curr Opin Chem Biol 4:524–530
Fisher TE, Marszalek PE, Fernandez JM (2000) Stretching single molecules into novel conformations using the atomic force microscope. Nat Struct Biol 7:719–724
Janovjak H, Kedrov A, Cisneros DA, Sapra KT, Struckmeier J, Müller DJ (2006) Imaging and detecting molecular interactions of single transmembrane proteins. Neurobiol Aging 27:546–561
Hinterdorfer P, Dufrêne YF (2006) Detection and localization of single molecular recognition events using atomic force microscopy. Nat Methods 3:347–355
Burnham NA, Chen X, Hodges CS, Matei GA, Thoreson EJ, Roberts CJ, Davies MC, Tendler SJB (2003) Comparison of calibration methods for atomic-force microscopy cantilevers. Nanotechnology 14:1–6
Lee GU, Kidwell DA, Colton RJ (1994) Sensing discrete streptavidin–biotin interactions with atomic force microscopy. Langmuir 10:354–357
Florin EL, Moy VT, Gaub HE (1994) Adhesion forces between individual ligand–receptor pairs. Science 264:415–417
Lo YS, Huefner ND, Chan WS, Stevens F, Harris JM, Beebe TP Jr (1999) Specific interactions between biotin and avidin studied by atomic force microscopy using the Poisson statistical analysis method. Langmuir 15:1373–1382
Fritz J, Katopodis AG, Kolbinger F, Anselmetti D (1998) Force-mediated kinetics of single P-selectin/ligand complexes observed by atomic force microscopy. Proc Natl Acad Sci U S A 95:12283–12288
Dupres V, Menozzi FD, Locht C, Clare BH, Abbott NL, Cuenot S, Bompard C, Raze D, Dufrêne YF (2005) Nanoscale mapping and functional analysis of individual adhesins on living bacteria. Nat Methods 2:515–520
Lee GU, Chrisey LA, Colton RJ (1994) Direct measurement of the forces between complementary strands of DNA. Science 266:771–773
Touhami A, Hoffmann B, Vasella A, Denis FA, Dufrêne YF (2003) Probing specific lectin–carbohydrate interactions using atomic force microscopy imaging and force measurements. Langmuir 19:1745–1751
Dammer U, Popescu O, Wagner P, Anselmetti D, Güntherodt HJ, Misevic GN (1995) Binding strength between cell adhesion proteoglycans measured by atomic force microscope. Science 267:1173–1175
Berquand A, Xia N, Castner DG, Clare BH, Abbott NL, Dupres V, Adriaensen Y, Dufrêne YF (2005) Antigen binding forces of single antilysozyme Fv fragments explored by atomic force microscopy. Langmuir 21:5517–5523
Hinterdorfer P, Baumgartner W, Gruber HJ, Schilcher K, Schindler H (1996) Detection and localization of individual antibody-antigen recognition events by atomic force microscopy. Proc Natl Acad Sci U S A 93:3477–3481
Allen S, Chen X, Davies J, Davies MC, Dawkes AC, Edwards JC, Roberts CJ, Sefton J, Tendler SJB, Williams PM (1997) Detection of antigen–antibody binding events with the atomic force microscope. Biochemistry 36:7457–7463
Bonanni B, Kamruzzahan AS, Bizzarri AR, Rankl C, Gruber HJ, Hinterdorfer P, Cannistraro S (2005) Single molecule recognition between Cytochrome C 551 and gold-immobilized Azurin by force spectroscopy. Biophys J 89:2783–2791
Kienberger F, Kada G, Gruber HJ, Pastushenko VPh, Riener C, Trieb M, Knaus HG, Schindler H, Hinterdorfer P (2000) Recognition force spectroscopy studies of the NTA–His6 bond. Single Molecules 1:59–65
Pfister G, Stroh CM, Perschinka H, Kind M, Knoflach M, Hinterdorfer P, Wick G (2005) Detection of HSP60 on the membrane surface of stressed human endothelial cells by atomic force and confocal microscopy. J Cell Sci 118:1587–1594
Ros R, Schwesinger F, Anselmetti D, Kubon M, Schäfer R, Plückthun A, Tiefenauer L (1998) Antigen binding forces of individually addressed single-chain Fv antibody molecules. Proc Natl Acad Sci U S A 95:7402–7405
Strunz T, Oroszalan K, Schäfer R, Güntherodt HJ (1999) Dynamic force spectroscopy of single DNA molecules. Proc Natl Acad Sci U S A 96:11277–11282
Schumakovitch I, Grange W, Strunz T, Bertoncini P, Güntherodt HJ, Hegner M (2002) Temperature dependance of unbinding forces between complementary DNA strands. Biophys J 82:517–521
Grandbois M, Dettmann W, Benoit M, Gaub HE (2000) Affinity imaging of red blood cells using an atomic force microscopy. J Histochem Cytochem 48:719–724
Baumgartner W, Hinterdorfer P, Ness W, Raab A, Vestweber D, Schindler H, Drenckhahn D (2000) Cadherin interaction probed by atomic force microscopy. Proc Natl Acad Sci U S A 97:4005–4010
Baumgartner W, Gruber HJ, Hinterdorfer P, Drenckhahn D (2000) Affinity of trans-interacting VE-cadherin determined by atomic force microscopy. Single Mol 1:119–122
Zhang X, Wojcikiewicz E, Moy VT (2002) Force spectroscopy of the leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction. Biophys J 83:2270–2279
Li F, Redick SD, Erickson HP, Moy VT (2003) Force measurements of the α5β1 integrin–fibronectin interaction. Biophys J 84:1252–1262
Zhang X, Bogorin DF, Moy VT (2004) Molecular basis of the dynamic strength of the sialyl Lewis X-selectin interaction. ChemPhysChem 5:175–182
Merkel R, Nassoy P, Leung A, Ritchie K, Evans E (1999) Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature 397:50–53
Nevo R, Stroh C, Kienberger F, Kaftan D, Brumfeld V, Elbaum M, Reich Z, Hinterdorfer P (2003) A molecular switch between alternative conformational states in the complex of Ran and importin β1. Nat Struct Biol 10:553–557
Gilbert Y, Deghorain M, Wang L, Xu B, Pollheimer PD, Gruber HJ, Errington J, Hallet B, Haulot X, Verbelen C, Hols P, Dufrêne YF (2007) Single-molecule force spectroscopy and imaging of the Vancomycin/D–Ala–D–Ala interaction. Nano Lett 7:796–801
Bell GI (1978) Models for the specific adhesion of cells to cells. Science 200:618–627
Ludwig M, Dettmann W, Gaub HE (1997) Atomic force microscope imaging contrast based on molecular recognition. Biophys J 72:445–448
Lehenkari PP, Charras GT, Nykänen A, Horton MA (2000) Adapting atomic force microscopy for cell biology. Ultramicroscopy 82:289–295
Almqvist N, Bhatia R, Primbs G, Desai N, Banerjee S, Lal R (2004) Elasticity and adhesion force mapping reveals real-time clustering of growth factor receptors and associated changes in local cellular rheological properties. Biophys J 86:1753–1762
Ebner A, Kienberger F, Kada G, Stroh C, Geretschlaeger M, Kamruzzahan ASM, Wildling L, Johnson WT, Ashcroft B, Nelson J, Lindsay SM, Gruber H, Hinterdorfer P (2005) Localization of single avidin–biotin interactions using simultaneous topography and molecular recognition imaging. Chem Phys Chem 6:897–900
Chtcheglova LA, Waschke J, Wildling L, Drenckhahn D, Hinterdorfer P (2007) Nano-scale dynamic recognition imaging on vascular endothelial cells. Biophys J 93:L11–L13
Viani MB, Pietrasanta LI, Thompson JB, Chand A, Gebeshuber IC, Kindt JH, Richter M, Hansma HG, Hansma PK (2000) Probing protein–protein interactions in real time. Nat Struct Biol 7:644–647
Ando T, Kodera N, Takai E, Maruyama D, Saito K, Toda A (2001) A high-speed atomic force microscope for studying biological macromolecules. Proc Natl Acad Sci U S A 98:12468–12472
Humphris ADL, Hobbs JK, Miles MJ (2003) Ultrahigh-speed scanning near-field optical microscopy capable of over 100 frames per second. Appl Phys Lett 83:6–8
Willig KI, Kellner RR, Medda R, Hein B, Jakobs S, Hell SW (2006) Nanoscale resolution in GFP-based microscopy. Nat Methods 3:721–723
Nikova D, Lange T, Oberleithner H, Schillers H, Ebner A, Hinterdorfer P (2006) Atomic force microscopy in nanomedicine. In: Bhushan B, Fuchs H (eds) Applied scanning probe methods III. Springer, Berlin, pp 1–27
Patel SD, Chen CP, Bahna F, Honig B, Shapiro L (2003) Cadherin-mediated cell–cell adhesion: sticking together as a family. Curr Opin Struct Biol 13:690–698
Zamir E, Katz M, Posen Y, Erez N, Yamada KM, Katz BZ, Lin S, Lin DC, Bershadsky A, Kam Z, Geiger B (2000) Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts. Nat Cell Biol 2:191–196
Acknowledgments
Our work is supported by Belgian Funds (National Foundation for Scientific Research (FNRS), Fonds Spéciaux de Recherche (Université Catholique de Louvain), Région wallonne, Interuniversity Poles of Attraction Programme (Federal Office for Scientific, Technical and Cultural Affairs), Concerted Research Action (Research Department of the Communauté Française de Belgique) by the Austrian National Science Fund and the FP6 of EU. Y.F.D. is a Research Associate of the FNRS. We are indebted to C. Stroh, A. Ebner, L. Chtcheglova, and F. Kienberger, V. Dupres, Y. Gilbert for their performance of the experiments and enlightening discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dufrêne, Y.F., Hinterdorfer, P. Recent progress in AFM molecular recognition studies. Pflugers Arch - Eur J Physiol 456, 237–245 (2008). https://doi.org/10.1007/s00424-007-0413-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-007-0413-1