
Abstract
The Cacna1a gene encodes the α1A subunit of voltage-gated CaV2.1 Ca2+ channels that are involved in neurotransmission at central synapses. CaV2.1-α1-knockout (α1KO) mice, which lack CaV2.1 channels in all neurons, have a very severe phenotype of cerebellar ataxia and dystonia, and usually die around postnatal day 20. This early lethality, combined with the wide expression of CaV2.1 channels throughout the cerebellar cortex and nuclei, prohibited determination of the contribution of particular cerebellar cell types to the development of the severe neurobiological phenotype in Cacna1a mutant mice. Here, we crossed conditional Cacna1a mice with transgenic mice expressing Cre recombinase, driven by the Purkinje cell-specific Pcp2 promoter, to specifically ablate the CaV2.1-α1A subunit and thereby CaV2.1 channels in Purkinje cells. Purkinje cell CaV2.1-α1A-knockout (PCα1KO) mice aged without difficulties, rescuing the lethal phenotype seen in α1KO mice. PCα1KO mice exhibited cerebellar ataxia starting around P12, much earlier than the first signs of progressive Purkinje cell loss, which appears in these mice between P30 and P45. Secondary cell loss was observed in the granular and molecular layers of the cerebellum and the volume of all individual cerebellar nuclei was reduced. In this mouse model with a cell type-specific ablation of CaV2.1 channels, we show that ablation of CaV2.1 channels restricted to Purkinje cells is sufficient to cause cerebellar ataxia. We demonstrate that spatial ablation of CaV2.1 channels may help in unraveling mechanisms of human disease.
Similar content being viewed by others

Introduction
CaV2.1 Ca2+ channels are plasma membrane multimeric protein complexes that play an important role in neurotransmitter release at most central brain synapses [1]. CaV2.1 channels are characterized by the presence of the pore-forming α1A subunit that is encoded by the Cacna1a gene and are widely expressed throughout the nervous system [2, 3]. CaV2.1-α1A-knockout (α1KO) mice that lack CaV2.1 channels exhibit a severe phenotype of ataxia and dystonia with a strong cerebellar component that starts around postnatal day (P)12, and die, if not given very special care, around P20 [4–7]. Histological analysis in the few “survivors” indicated that a progressive gradual loss of Purkinje cells started between P45 and P100 [5].
Cerebellar ataxia and dystonia can also be part of the phenotype of certain naturally occurring Cacna1a mouse mutants. For instance, leaner, tottering, rolling Nagoya, but not rocker, are characterized to different extent by granule and/or Purkinje cell death and chronic ataxia [8–11]. For tottering and leaner mice, it was shown that the chronic ataxia is related to irregular Purkinje cell simple spike firing caused by the loss of precision in intrinsic activity of Purkinje cells and aberrant synaptic input [12–15]. In addition to the cerebellar ataxia phenotype, tottering mice also exhibit episodic dystonia, which is related to transient low-frequency oscillations in the cerebellar cortex [16]. Notably, when tottering mutants lose their Purkinje cells on a Purkinje cell degeneration mutant background, the dystonic phenotype disappears [17]. Together these findings indicate that mutations that cause a reduction of CaV2.1-mediated Ca2+-influx affect the information processing in the cerebellar cortex and, thereby, induce chronic ataxia and episodic dystonia.
Apart from the aberrant activity in the cerebellar cortex in Cacna1a mutants, also the activity of cerebellar nuclear (CN) neurons is subject to abnormal firing patterns, as was recently shown in tottering mice [18]. Both Purkinje cells and CN neurons receive input (direct and indirect through local interneurons and/or granule cells) from mossy fibers and climbing fibers. While granule cell transmission to Purkinje cells in α1KO, tottering, leaner, and rolling Nagoya mice is abnormal, the response to climbing fiber activation is seemingly normal in these mutants [15, 19, 20]. Besides the excitatory input from climbing and mossy fibers, the neurons in the CN receive input from local interneurons and from the cerebellar cortex through Purkinje cells. Apart from the impact of these inputs, the firing pattern of CN neurons is determined by their intrinsic activities. Since Purkinje cells and CN neurons, as well as the neurons and fibers that innervate them, express CaV2.1 channels [3], the exact origin of the aberrant cerebellar activity in CaV2.1 mutants that causes cerebellar ataxia could not be resolved using the existing Cacna1a mouse mutants.
Therefore, we used the Cre-lox system to ablate CaV2.1 channels exclusively in Purkinje cells by crossing conditional Cacna1a mice that carry a “floxed” allele [21] with transgenic mice that express Cre recombinase under the control of the Purkinje cell-specific Pcp2 promoter [22]. Hereby, we were not only able to rescue the early lethality seen in α1KO mice, but also to demonstrate that lack of CaV2.1 channel function in Purkinje cells is sufficient to cause ataxia and that progressive Purkinje cell loss starts well after the onset of the ataxia.
Materials and Methods
Animals
All animal experiments were performed in accordance with guidelines of the respective universities and the national legislation. Previously, we generated conditional Cacna1a mice using a gene-targeting approach [21]. The Cacna1a allele in these mice was “floxed” by introducing loxP sites flanking exon 4 (i.e. Cacna1a flox mice) (Fig. 1a). A genomic deletion of exon 4 is achieved by crossing the conditional mice with Pcp2 Cre transgenic mice that express Cre recombinase exclusively in Purkinje cells [22]. Homozygous Cacna1a flox/flox mice were first crossed with homozygous Pcp2 Cre mice generating double heterozygous transgenic offspring. Female double transgenic mice were then crossed with Cacna1a flox/wt males to generate mice that are Cacna1a deficient (and thereby CaV2.1 channel deficient) in Purkinje cells (i.e. Purkinje cell CaV2.1-α1AKO (PCα1KO)). The same cross also produced mice in which Cacna1a was not affected (wild type (WT)). In total, we used 29 PCα1KO and 29 WT littermates. Genotyping of the Cacna1a flox allele was performed by PCR using primers P1 (5′-acctacagtctgccaggag-3′) and P2 (5′-tgaagcccagacatccttgg-3′) and genomic DNA as a template. Genomic deletion of exon 4 after Cre recombination was confirmed by PCR using primers P3 (5′-agtttctattggacagtgctggt-3′) and P4 (5′-ttgcttagcatgcacagagg-3′). For genotyping of the Cre transgene primers P5 (5′-acttagcctgggggtaactaaact-3′) and P6 (5′-ggtatctctgaccagagtcatcct-3′) were used.

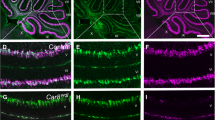
Generation of conditional Cacna1a Purkinje cell-specific knockout mice. a Schematic representation of the genomic structure of the relevant part of the floxed and recombined Cacna1a alleles. Black boxes indicate exons (E). Triangles indicate the position of loxP sites. Arrows indicate the position of the primers used for the RT-PCR. b RT-PCR of cerebellar RNA amplifies a 580-bp product from the non-recombined Cacna1a transcript and a 490-bp product from transcripts that lack exon 4. c–f Anti-CaV2.1-α1A immunostaining in cerebellar sections of P30 WT (c, e) and PCα1KO (d, f) mice. CN cerebellar nuclei, G granule cell layer, M molecular layer, PC Purkinje cell layer, WM white matter. Roman numbers indicate the cerebellar lobules
Reverse Transcription PCR
Total RNA was isolated from freshly dissected cerebellum and cerebrum of mice. For RT-PCR, first-strand cDNA was synthesized using random primers. Subsequently, Cacna1a-specific PCRs were performed using primers P7 (5′-gatgacacggaaccatac-3′) and P8 (5′-attgtagaggagatcagtcc-3′) that are located in exon 3 and 6, respectively.
Histology
Mouse brains (20 PCα1KO and 20 WT) were obtained at various ages after cardiac perfusion with PBS followed by 4% buffered paraformaldehyde (PFA). Post-fixation was performed for 1 h in 4% PFA followed by overnight incubation in 10% sucrose in 0.1 M phosphate buffer at 4°C. Subsequently, the sheet of dura mater separating cerebrum and cerebellum was removed, tissue was embedded in 10% sucrose in 10% gelatine, and the gelatin was fixed with 30% sucrose in 10% PFA for 2.5 h at room temperature. This was followed by an overnight incubation in 30% sucrose in 0.1 M phosphate buffer at 4°C. Forty-micrometer sagittal sections were made using a freezing cryotome (Leica, Bensheim, Germany). Part of the sections was processed for silver staining, which selectively stains dying neurons [23]. The remainder of the sections was processed directly for free-floating immunohistochemistry. Sections were incubated in 10% heat-inactivated normal horse serum (NHS) with 0.5% Triton-X100 in TBS, for 2 h and then incubated either with primary rabbit polyclonal anti-CaV2.1-α1A antibody (ACC-001, 1:100; Alomone Labs, Jerusalem, Israel), or with rabbit polyclonal anti-Calbindin antibody (CB38, 1:10,000; SWANT, Bellinzona, Switzerland) diluted in 1% NHS with 2% heat-inactivated fetal calf serum and 0.4% Triton-X100 in TBS at 4°C. Next, sections were washed and incubated for 2 h at room temperature with a secondary biotin-labeled goat anti-rabbit antibody (1:200; Vector Laboratories, Burlingame, CA). For protein detection, sections were incubated with the avidin-biotin-peroxidase complex (ABC kit, 1:100, Vector Laboratories) for 2 h at room temperature, and developed in 0.1 mg/ml diaminobenzidine in 0.005% H2O2. Sections were Nissl stained, air dried, and cover slipped. Paraffin-embedded cerebellar sections (5 μm) were processed for standard haematoxylin and eosin (HE) staining.
Golgi-Cox Staining
Brains from 30-day-old mice (two PCα1KO and two WT) were isolated after decapitation and processed for Golgi impregnation. Brains were rinsed briefly in Milli-Q water. Tissue was further processed for Golgi-Cox staining using FD Rapid GolgiStain™ Kit (MTR Scientific, Ijamsville, MD) with 120-μm thick sections. Light microscopy images were used to assess the dendritic arborisation of Purkinje cells using the linear Sholl analysis [24–26]. In short, 7 to 11 concentric circles increasing in size (with 15 μm intervals) were aligned with the Purkinje cell soma. The number of Purkinje cell dendrites intersecting with each circle was counted. Statistical differences between groups were determined by use of repeated measures ANOVAs with one “between-subjects” factor (genotype) and one “within-subjects” factor (i.e. number of Purkinje cell dendrites intersecting with the circle) with 11 levels. The change in the dendritic branching with increasing distance within the groups was subsequently tested by use of separate repeated measures ANOVAs for each genotype with one “within-subjects” factor with 11 levels. The assumption of normality of the distributions (as tested by Kolmogorov–Smirnov tests), and equality of error variances (as tested by Levene’s tests) were not violated. When the assumption of sphericity (as tested by Mauchly’s tests) was violated, Greenhouse–Geisser corrections were used. All statistical analyses were performed using SPSS 17.0 software (IBM corporation, Chicago, IL, USA).
Quantification Number of Purkinje Cells and Degeneration of Purkinje Cell Axons
Using 40-μm thick, calbindin-stained, transversally sectioned tissue from two PCα1KO mice and three WT littermates, we calculated the average number of Purkinje cells per 100 μm of cerebellar cortex. Five to ten pictures were taken at ×20 magnification from either the vermal parts of lobule 2, 6, or 9, or from the paramedian lobule, paraflocculus, or flocculus. In each picture, the number of Purkinje cells per 100 μm of Purkinje cell layer was determined using Metamorph (Universal Imaging, Downing Town, PA, USA). In absence of many Purkinje cells the difference in structure between the molecular and granular layers guided the observer to accurately estimate the location of the Purkinje cell layer.
The extent of degeneration of Purkinje cell axons was qualitatively examined in sagittal cerebellar sections stained with silver impregnation as described elsewhere [23]. For each mouse, photographs of white matter were taken with a ×5 magnification for overview and a ×40 magnification for detailed examination of degenerating fibers.
Neuronal Volume and Density Measurements
Volume measurements were bilaterally taken from the medial cerebellar nuclei (MCN), anterior interposed nuclei (AIN), posterior interposed nuclei (PIN), and lateral cerebellar nuclei (LCN) using transversal, Nissl/calbindin-stained sections from three WT and three PCα1KO mice by the investigator blinded to the genotype. Contours were drawn around the four nuclei in every other section (e.g. 80-μm intervals) using NeuroLucida (MBF Bioscience, Williston, VT, USA) to construct a 3D image. Volumes (in mm3) were then calculated using NeuroExplorer (Nex Technologies, Littleton, MA, USA). The dorsolateral hump was included in the AIN and the “interstitial cell group” was included in the PIN. Statistical differences were determined by an independent samples Student’s t test or an independent samples Mann–Whitney U test (depending on the normality of the distributions as tested by Kolmogorov–Smirnov tests). With regard to t tests, if the assumption of equality of variances was violated (as tested by Levene’s tests), corrected degrees of freedom were used.
Neuronal surface density in the cerebellar nuclei was bilaterally determined in transversal paraffin-embedded, HE-stained cerebellar sections (4 μm) of two WT and two PCα1KO mice. For each CN, sections with a 60-μm interval were photographed at ×25 magnification (five to ten pictures per nucleus). Two independent investigators, each blinded to the genotype, counted the number of cells in each picture using Meta Imaging Series 4.6 (Molecular Devices Inc., Downingtown, PA, USA). Counting was done by drawing a contour around each CN and subsequently counting all somata with a clearly visible nucleolus by contouring the neuron’s nucleus. The surface density (number of cells/mm2) was calculated for each section, after which the average density per CN was calculated. The total numbers of “small” and “large” cells were subsequently determined using a threshold of 8.5-μm diameter (see also Ref. [27]). The 8.5-μm cut-off value was constructed using a scatter plot of the neuron nucleus’ diameter from which two groups could be distinguished. Statistical differences were determined by an independent samples Student’s t test or Mann–Whitney U test (depending on the normality of the distributions as tested by Kolmogorov–Smirnov tests). With regard to t tests, if the assumption of equality of variances was violated (as tested by Levene’s tests), corrected degrees of freedom were used.
Motor Behavior Analyses
The accelerating Rotarod (UGO Basile S.R.L., Commerio VA, Italy) test was performed on a 3 cm diameter horizontal rotating rod. The rod initially rotated at a constant speed of 4 rpm and accelerated in a stepwise manner (i.e. 4 rpm every 30 s) to a maximum of 40 rpm after 270 s. The maximum time on the rod was 300 s. The latency to fall was recorded on the moment the mouse fell down or stopped walking (i.e. clinging to the rod) for three consecutive turns and subsequently converted to the “maximum rotating speed” by calculation of the rotating speed at the moment of the fall. The test was performed in a semi-dark room with a light source placed at the bottom to discourage the mice from jumping off the rod. Mice of ∼P18 (n = 3 per genotype) were subjected to three trials separated by 90 min on 1 day without pre-training. Mice of P30 and P200–250 (n = 5 per genotype, per age group) were trained on the first day (by placing the mice on the rod that rotated with a constant speed of 5 rpm for 5 min) and then subjected to two trials separated by 30 min. On days 2 to 4, the P30 and P200–250 mice were directly subjected to two trials without pre-training. The results are presented as maximal rotating speed with mean ± SD of five mice each with two trials per day over 5 days. Statistical differences between groups were determined by use of repeated measures ANOVAs with one “between-subjects” factor (genotype) and one “within-subjects” factor (i.e. rotarod performance) with three (for ∼P18 mice) or five levels (for P30 and P200–250 mice). Rotarod performance within the groups was subsequently tested for significant changes over time by use of separate repeated measures ANOVAs for each genotype with one within-subjects factor with again three or five levels. The assumption of normality of the distributions (as tested by Kolmogorov–Smirnov tests), sphericity (as tested by Mauchly’s tests) and equality of error variances (as tested by Levene’s tests) were not violated.
Motor function was also assessed in P30 and P200–250 WT (n = 4 and 5, respectively) and PCα1KO (n = 4 and 5, respectively) mice using a dyskinesia rating scale that was previously described and validated in several mouse mutants [28, 29], including mice carrying a Cacna1a mutation. At the start of the experiment, each mouse was transferred to a new cage. Mice were scored for 1 min at 10-min intervals over a 60-min period. Abnormal motor behavior within the head/neck, trunk, and limbs were recorded in terms of type, including tremor (trunk, limbs, neck, and face), tonic flexion, extension (trunk, limbs, neck, and face), or twisting (trunk and neck) and clonus (limbs and face). Uncoordinated or ataxic motor behavior not attributable to a single-body part was also recorded, including falling, listing, and stumbling. Assessments of the abnormal motor behavior were then calculated by summing the dyskinesia scores during the 60-min observation period. Mice were observed on at least two separate occasions and mean dyskinesia scores were generated for each mouse. Statistical differences were determined by Mann–Whitney U tests, because the data were non-parametric in nature.
Results
Generation of Mice That Lack CaV2.1 Channels in Purkinje Cells
Homozygous PCα1KO mice were generated by crossing Cacna1a flox/flox mice with transgenic mice expressing Cre recombinase driven by the Purkinje-specific Pcp2 gene promoter (i.e. Pcp2 Cre mice—see “Materials and Methods”) (Fig. 1a). Successful genomic deletion of exon 4 of the Cacna1a gene was assessed at the RNA level by RT-PCR on cerebral and cerebellar total RNA (Fig. 1b). Whereas in the cerebrum only non-recombined PCR product of 580 bps was observed, in the cerebellum both non-recombined and recombined (480 bps) products were present. This indicated that Cre recombination only had occurred in part of the cerebellar neuron population. Immunohistochemical staining for α1A protein showed that Cre recombination was Purkinje cell-specific as no protein signal was present in the soma of virtually any Purkinje neurons of PCα1KO mice (Fig. 1d, f). However, α1A protein expression in other cerebellar structures, e.g. the CN, appeared not different between PCα1KO and WT mice. These findings are in line with the reported Purkinje cell specificity of the Pcp2 Cre mice [22].
Motor Coordination Is Severely Affected in Purkinje Cell CaV2.1-Knockout Mice
PCα1KO mice, unlike conventional α1KO mice [4–7], have a normal life span. PCα1KO mice appear normal until P10 to P12 when they start showing an abnormal motor phenotype with a loss of balance during walking and problems righting themselves (see Supplementary video). Young PCα1KO ∼P18 mice (n = 3 per genotype) performed significantly worse on the rotarod than did their WT littermates, (F (1, 4) = 184.90, p < 0.001) (Fig. 2a). However, there was no significant genotype X times change in performance over time interaction effect (F (2, 8) = 1.00, p = 0.41) and neither PCα1KO mice nor WT mice showed an improvement over time (F (2, 4) = 2.00, p = 0.25 and F (2, 4) = 7.11, p = 0.54, respectively). This indicates that although WT mice performed significantly better on the rotarod, the young age and/or the intensity of the training (more repeats during this 1-day protocol induced fatigue in the ∼P18 WT mice as also seen with a P40 WT mouse (data not shown)) prevented a significant improvement. Rotarod experiments of PCα1KO mice tested at P30 (n = 5 per genotype) consisted of two trials per day for five consecutive days and revealed that these mice were severely impaired, in that they were not able to stay on the accelerating rod for longer than 60 ± 7.8 s on the fifth day (Fig. 2b), which corresponded to a rotating speed of 8 rpm. This maximum rotating speed was significantly slower than that in WT mice, which stayed on the accelerating rod for 272 ± 19.1 s on the fifth day, which corresponds to a rotating speed of 40 rpm (F (1, 8) = 1297.32, p < 0.001) (Fig. 2b). Additionally, there was a significant genotype X change in performance over time interaction effect (F (4, 32) = 4.89, p < 0.01), indicating that there was a significant difference in change in performance over time between the two groups. More specifically, whereas the WT mice significantly improved their performance over time (F (4, 16) = 7.14, p < 0.01), the PCα1KO mice did not (F (4, 16) = 0.48, p = 0.75). Using a similar rotarod protocol for P200–250 mice (n = 5 per genotype), we found the same extend of motor deficit (Fig. 2c). PCα1KO mice of P200–250 were not able to stay on the accelerating rotarod for more than 57 ± 14.3 s on the fifth day and fell off at a significantly lower rotating speed than did wild type mice (F (1, 8) = 908.84, p < 0.001) who managed to stay on the rotarod for 248 ± 28.4 s on the fifth day. Although the genotype X change in performance over time interaction effect was not significant, it did show a clear trend (F (4, 32) = 2.47, p = 0.064). This trend is confirmed since PCα1KO mice did not show an improvement over time (F (4, 16) = 0.87, p = 0.51), whereas WT mice did (F (4, 16) = 4.07, p < 0.02). These data should, however, be interpreted with caution in the sense that lack of improvement on the rotarod test in PCα1KO mice does not necessarily reflect a learning deficit, e.g. the ataxic phenotype may have prevented an improvement in performance in this test.

Abnormal motor behavior in Cacna1a Purkinje cell-specific knockout mice. a–c Rotarod testing revealed severe impairments in motor performance of PCα1KO mice compared to WT littermates. The latency to fall is expressed as the maximal rotating speed in rotations per min (rpm) for P18 (a), P30 (b), and P200–250 (c) PCα1KO mice and WT littermates. d, e Additional assessment of motor behavior to classify by type of movement disorder. d P30 PCα1KO mice (n = 5) but not WT littermates (n = 5) showed ataxic movements, tremors, tonic, and clonic movements. Note the predominant ataxic component. e P200-250 PCα1KO mice (n = 5) but not WT littermates (n = 5) showed ataxic movements, tremors, tonic, and clonic movements. Note the similarity in behavioral classification between P30 and P200–250 PCα1KO mice
Because it is difficult to determine the specific deficit that causes impaired rotarod performance, motor function was also assessed using a dyskinesia rating scale (see “Materials and Methods” for details). There was significantly more ataxia compared to tremor, tonic movements, or clonic movements in both P30 PCα1KO (n = 4) mice (ataxia vs. tremor: U = 16.00, p < 0.01; ataxia vs. tonic: U = 16.00, p < 0.01; ataxia vs. clonic: U = 16.00, p < 0.01) and P200–250 PCα1KO mice (n = 5; ataxia vs. tremor: U = 25.00, p < 0.01; ataxia vs. tonic: U = 25.00, p < 0.01; ataxia vs. clonic: U = 25.00, p < 0.01) (Fig. 2d, e). Ataxia, but not other movements, was significantly increased in young (P30) PCα1KO mice compared with age-matched WT mice (n = 4; U = 16.00, p < 0.01), whereas all abnormal movement types were significantly increased in old (P200–250) PCα1KO mice compared with age-matched WT littermates (n = 5; ataxia: U = 25.00, p < 0.01; tremor: U = 25.00, p < 0.01; tonic: U = 25.00, p < 0.01; clonic: U = 25.00, p < 0.01). In PCα1KO mice, there was a slight but significant increase in tremor with age (U = 20.00, p < 0.01) and a trend for an increase in ataxia and clonus with age (U s = 18 and 17, respectively), but their relative contribution to the abnormal motor behavior in mutant mice remained similar irrespective of their age.
To study whether the severe ataxia in young PCα1KO mice come about due to structural abnormalities, we studied the cytoarchitecture at P30 in more detail (Fig. 3). Both a standard HE staining and a more detailed Golgi-Cox staining did not reveal qualitative differences between PCα1KO and WT littermates. Topological analysis of the dendritic branching of Purkinje cells from two WT mice (i.e. 25 cells) and three PCα1KO mice (i.e. 30 cells) showed no significant difference (F (1, 40) = 0.00, p = 0.98) between groups, nor was there a significant genotype X change in the dendritic branching with an increasing distance interaction effect (F (3.385, 135.397) = 1.03, p = 0.39). In both the WT and PCα1KO mice, the amount of dendritic branching did significantly change with increasing distance (F (3.281, 82.024) = 42.13, p < 0.001 and F (3.002, 45.036) = 20.09, p < 0.001, respectively). These data indicate that, although PCα1KO mutants are severely ataxic already at P18, their Purkinje cells did not reveal structural abnormalities.

Overall cerebellar and Purkinje cell morphology in young Cacna1a Purkinje cell-specific knockout mice. Haematoxilin and eosin staining reveals similar gross cerebellar morphology in PCα1KO (a) and WT (b) mice at P30. G granule cell layer, PC Purkinje cell layer, M molecular layer, WM white matter. Roman numbers indicate the cerebellar lobules. Golgi-Cox-stained cerebellar sections of PCα1KO (c) and WT (d) mice at P30 reveal normal morphology of the Purkinje cells in the two genotypes; e PCα1KO mice show similar level of dendrite arborization (linear Sholl analysis, for details see “Materials and Methods”) as their WT littermates
Progressive Cell Death of Purkinje Cells in the Absence of CaV2.1 Channels
As CaV2.1 channels gate more than 90% of the high-voltage-gated Ca2+-influx in Purkinje cells, the morphology and survival of Purkinje cells in the PCα1KO mutants may be affected at later stages in their life. Therefore, we compared calbindin D28K-stained cerebellar sections of PCα1KO mice at various ages (i.e. P30, P60, P100, and P200–250) (Fig. 4). In line with the topological analysis described above, the immunostaining of the cerebellar cortex of PCα1KO mutants at P30 did not show overt differences with those of WT littermates (Fig. 4a). At later ages, however, we observed substantial Purkinje cell degeneration in the cerebellum of PCα1KO mice (Fig. 4b–d). Starting at P60, Purkinje cells showed degenerative structural abnormalities, such as somatic sprouting and axonal swellings (Fig. 4b–d). These changes progressed until P200–250, at which PCα1KO mice (n = 2) showed a significant decrease in Purkinje cell number compared with WT controls (n = 3) (lobule 2, U = 0.00, p < 0.001; lobule 6, U = 28.00, p < 0.001; lobule 9, U = 1.00, p < 0.001; paramedical lobule, U = 0.00, p < 0.001; paraflocculus, U = 179.00, p < 0.001; flocculus, U = 0.00, p < 0.001) (Fig. 4e). Although the extent of the degeneration of Purkinje cells is evident, also the variability between various lobules is apparent. Given that we did not find any anti-CaV2.1-α1A staining in the Purkinje cells of PCα1KO mice (see Fig. 1c–f), this variability in the Purkinje cell death could not be explained by inter-cellular variations in the Cre expression or the recombination efficacy of the LoxP/Cre system.

Calbindin D 28K staining of sagittal cerebellar sections of Cacna1a Purkinje cell-specific knockout mice at different ages. a Calbindin staining reveals normal foliation of the cerebellum at P30 in PCα1KO mice. Purkinje cells are organized in regular rows with no evidence of abnormal morphology. b Purkinje cell loss, particularly in the anterior lobes of PCα1KO mice at P60. Abnormalities in Purkinje cell morphology, such as axonal swellings (arrowheads in inset), can be seen. c Progressive Purkinje cell loss in PCα1KO mice at P100 is now also more prominent in posterior lobes. Axonal swellings are indicated by arrowheads. d Further evidence of the progressive nature of the Purkinje cell degeneration in PCα1KO mice at P200–250. Remaining Purkinje cells often show axonal swelling (arrowhead). Roman numbers indicate the cerebellar lobules. G granule cell layer, PC Purkinje cell layer, M molecular layer, WM white matter. e Purkinje cell degeneration in different cerebellar lobules of P200–250 PCα1KO mice. Quantification of the number of Purkinje cells (PCs) per 100 μm in different lobules in WT (blue; n = 3) and PCα1KO (green; n = 2) mice. The number of Purkinje cells is significantly decreased in all lobules of PCα1KO mice compared to WT littermates
Neurodegenerative Changes in Cerebellar Cortex
In order to visualize dying neurons, we used silver impregnation on tissue from mice at various ages (Fig. 5). At P30, PCα1KO mice showed very little argyrophylic staining (Fig. 5a, d), indicating that there are no overt degenerative changes in their cerebellum, despite their ataxia. However, at P45 the level of argyrophylic staining in PCα1KO mice dramatically increased. Many silver-stained somata, axons, and dendrites of dying Purkinje cells were randomly dispersed throughout the cerebellar cortex (Fig. 5b, e). Still, at this stage we did not observe any secondary cell death of neurons in the granular layer or molecular layer. In contrast at P200–250, both layers in PCα1KO mice contained dying neurons (Fig. 5c, f). Apparently, the loss of their target cells—the Purkinje cells—resulted in loss of part of the granule cells and molecular layer interneurons. Moreover, we observed silver-stained Purkinje cell axons in the white matter surrounding the cerebellar nuclei (Fig. 6). The density of these degenerating fibers is variable throughout the white matter, as can be expected from the alternating density of Purkinje cell axons in cerebellar white matter depending on their efferent targets [30, 31] and the non-uniform distribution of Purkinje cell degeneration (Fig. 4e).

Silver impregnation of cerebellar sections in Cacna1a Purkinje cell-specific knockout mice. a, d No apparent neurodegeneration is seen in the cerebellum of PCα1KO mice at P30. b, e Silver-stained Purkinje cell bodies as well as dendrites and axons indicating dying cells in the cerebellum of PCα1KO mice at P45. Arrows indicate dying Purkinje cells in (e). c, f No Purkinje cells are present the respective areas. Silver-stained cells can be found in the granular and molecular layers indicating some granule and interneuron loss at P200–250. G granule cell layer, PC Purkinje cell layer, M molecular layer, WM white matter. Roman numbers indicate the cerebellar lobules. Dotted boxes in (a–c) correspond to regions shown in (d–f)

Silver impregnation of cerebellar section reveal stained axons surrounding the medial cerebellar nuclei in Cacna1a Purkinje cell-specific knockout mice. a–d Silver-stained Purkinje cells and their axons are found in PCα1KO mice at P60 (c, d) but not in tissue from WT littermates (a, b). Dotted boxes in (a, c) correspond to regions shown in (b, d)
Neurodegenerative Changes in Cerebellar Nuclei
The loss of Purkinje cells in PCα1KO may not only have an effect on upstream neurons in the cerebellar cortex, but also on downstream neurons in the cerebellar nuclei. To test for loss of Purkinje cell target neurons, we calculated the volume (in mm3) and surface density (in number of cells/mm2) of the MCN, AIN, PIN, and LCN in PCα1KO and WT mice at age P200–250. The volume quantification revealed pronounced volume reductions in all cerebellar nuclei of PCα1KO mice compared to those of WT mice, (LCN: t (10) = 8.63, p < 0.001; AIN: t (10) = 8.86, p < 0.001; PIN: U = 3.00, p < 0.02; MCN: U = 0.00, p < 0.01; total volume: U = 0.00, p < 0.01) (Table 1) (Fig. 7a–c). The surface density was significantly increased in all cerebellar nuclei of PCα1KO mice compared to those of WT mice (LCN: t (6) = −3.43, p < 0.02; AIN: t (6) = −4.99, p < 0.01; PIN: t (6) = −2.94, p < 0.03; MCN: t (3.354) = −4.19, p < 0.03) (Table 1) (Fig. 7d). The increased surface density also appeared in the total density of “small cells” (nucleus, <8.5 μm in diameter) averaged for all nuclei (M = 262.5 ± 47.8 for WT and 509.9 ± 111.3 for PCα1KO mice, t (6) = −4.09, p < 0.01) (Fig. 7e) but not for the “large cells” (nucleus, >8.5 μm in diameter; M = 178.3 ± 21.1 for WT and 198.0 ± 11.4 for PCα1KO mice, t (6) = −1.65, p = 0.151) (Fig. 7f). Thus, a general decrease in volume and an increase in cell density can be seen with the exception of an unchanged “large cell” density.

Changes in volume and neuronal density in cerebellar nuclei of P200–250 PCα1KO and WT mice. a–c Quantification of the volume of the cerebellar nuclei in WT (blue; n = 3) and PCα1KO (green; n = 3) mice. a Example of a bilateral 3D reconstruction of the cerebellar nuclei of one WT (top) and one PCα1KO mouse (bottom). Light blue lateral cerebellar nucleus (LCN), dark blue anterior interposed nucleus (AIN), purple posterior interposed nucleus (PIN) and gray medial cerebellar nucleus (MCN). b An average decrease of 54% in the total volume of the cerebellar nuclei in PCα1KO mice is shown. c Quantification of the volume of individual cerebellar nuclei reveals a pronounced volume reduction in all four nuclei. d Overall neuronal surface density of CN neurons per individual CN. e Surface density of small cells (<8.5 μm in diameter). f Surface density of big cells (>8.5 μm in diameter). *p < 0.05; **p < 0.001 (significant differences among groups)
Discussion
Here we used conditional gene targeting to ablate CaV2.1 calcium channels selectively in Purkinje cells in vivo in mice. This approach allowed us to study the specific contribution of these channels in Purkinje cells to the development of ataxia that is associated with mutations in the Cacna1a gene. Our cell type-specific knockout strategy using conditional Cacna1a-knockout mice [21] succeeded in circumventing early postnatal lethality that is a very prominent feature in Cacna1a-knockout mice that lack CaV2.1 channels in all neurons [4–7]. In line with the well-defined expression of Cre recombinase in Purkinje cells in Pcp2 Cre mice [22], Cre recombination affected α1A expression in almost all Purkinje neurons, as is evident by the lack of anti-α1A staining in these cells at P30 while α1A expression seemed preserved in the rest of the brain (data not shown). We could demonstrate that lack of CaV2.1 channels in Purkinje cells is sufficient to cause an ataxic phenotype since the ataxia occurred prior to structural abnormalities. This finding adds to existing lines of evidence [8–11], which indicate that the cerebellar ataxia associated with abnormal CaV2.1 channel function can be caused by Purkinje cell dysfunction.
Interestingly, our PCα1ΚΟ mutants exhibited a predominantly ataxic phenotype without episodic motor attacks during the first months of life. Notably, recently developed Cacna1a purk(−/−) mice, which lack functional CaV2.1 channels due to a Purkinje cell specific deletion of exon 1 of the Cacna1a gene, were reported to have dyskinesia attacks from 3 weeks on [32]. Intrinsically generated low-frequency oscillations in the cerebellum of tottering mice are thought to be the source of the episodic motor attacks in these mutants [16]. Further evidence for this hypothesis came from microinjections of the non-selective voltage-sensitive K+-channel blocker 3,4 DiAP in the cerebellum of tottering mice that abolished the episodic attacks of dyskinesia [33]. Future experiments will have to show whether similar mechanisms in the cerebellum of PCα1KO underlie the episodic nature of dyskinesia.
Visual inspection of the mice revealed that the ataxia in PCα1KO mice started at P10–12, around the same age as in conventional α1KO mice that lack CaV2.1 channels in all neurons [4–7]. Likewise, loss of CaV2.1 function in Purkinje cells did not affect normal development of the cerebellar cortex, similar to what was shown for conventional CaV2.1KO mice [4, 5] and Cacna1a purk(−/−) mice [32]. These findings are in line with the fact that during the first postnatal week CaV1 (L-type) channels are the dominant voltage-gated Ca2+ channel type in Purkinje cell somata [34]. At this time, however, we cannot rule out the possibility that other high-voltage-gated calcium channels, such as CaV2.2 (N-type) and CaV2.3 (R-type), are upregulated during later stages in PCα1KO mice and thereby rescue partly the detrimental effects of CaV2.1 absence. Similar compensatory mechanisms involving CaV2.2, CaV2.3, and CaV1 have been described in other Cacna1a mutant mice at the level of neurotransmission [15, 17, 32, 35].
The first signs of neurodegenerative changes in PCα1KO mice were observed around P45. At this stage, silver impregnation revealed clear argyrophylic staining in the Purkinje cell and molecular layers. From P60 on, Purkinje cell loss was clearly visible and many of the remaining Purkinje cells showed abnormalities such as torpedo’s or axonal swellings. At older ages (P100 and P200–250), Purkinje cell loss became progressively worse and ultimately also affected the survival of neurons in the granular layer and molecular layer. In lurcher and Purkinje cell degeneration (Pcd) mice, which both show near-complete Purkinje cell loss in adulthood, a progressive postnatal Purkinje cell degeneration is also followed by large scale death of granule cells after the loss of Purkinje cells [36–38]. Thus, the fact that three completely different mutations lead to the same sequence of cerebellar events (for lurcher see Ref. [39]; for Pcd see Ref. [40]) indicates that the degeneration of granule cells and molecular layer interneurons is probably a common, secondary effect to the loss of Purkinje cells.
Downstream Effects of Widespread Purkinje Cell Loss
The loss of Purkinje cells also affected the cerebellar nuclei in PCα1KO mice. The extent of volume reduction in PCα1KO mice was similar to that reported in lurcher and Pcd mouse mutants [27, 41]. In lurcher mice deafferentation of the CN resulted in a ∼60% reduction in the volume of all individual CN [27]. We also found a widespread increase in neuronal surface densities in all CN. However, it appeared that the increase in neuronal density is more pronounced in small cells than in large cells. Despite the fact that our results do not allow a firm statement about the total number of cells per CN, our data seems to indicate a possible reduction in the number of large neurons. The data presented here shows a somewhat different pattern from lurcher mutants with regard to cell density [27]. Future experiments should provide a more detailed picture of the total number of neurons per cell type per CN.
Behavioral Effects of Widespread Purkinje Cell Loss
The abnormal motor behavior in both young and old PCα1KO mice was dominated by ataxic movements. Since the behavioral abnormalities were already visible before the degeneration occurred (i.e. in young P18 and P30 PCα1KO mice), this indicates that the lack of functional CaV2.1 channels in Purkinje cells can induce the same behavioral aberrations as an absence of Purkinje cells (i.e. in old P200–250 PCα1KO mice). The severity of the ataxic symptoms in PCα1KO mice was also reflected by the fact that, unlike the WT controls, the mutants did not show an increase in their performance on the rotarod over time. Whether there is cerebellar motor learning deficit in the PCα1KO mice could not be assessed since motor learning (i.e. showing improved performance on the rod over time) may have been obscured by the severity of the ataxic phenotype. The severe effect on motor coordination in PCα1KO mice is likely to be due to the disrupted Purkinje cell output and the subsequent deregulation of activity patterns downstream [18, 42], which might be relatively normal, at least initially, as was described for lurcher mice [43].
Summary
This report represents a cell type-specific ablation of CaV2.1 channels leading to a disease phenotype. We demonstrate that specific ablation of these channels in Purkinje cells is sufficient to cause cerebellar ataxia that precedes progressive Purkinje cell loss. Conditional Cacna1a mice appear especially useful for dissecting the cell-specific role of CaV2.1 channels in neuronal circuitries such as in the cerebellum and in revealing the mechanisms underlying human disease.
References
Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1999;9:85–95.
Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, et al. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–18.
Kulik A, Nakadate K, Hagiwara A, Fukazawa Y, Luján R, Saito H, et al. Immunocytochemical localization of the alpha 1A subunit of the P/Q-type calcium channel in the rat cerebellum. Eur J Neurosci. 2004;19:2169–78.
Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, et al. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci USA. 1999;96:15245–50.
Fletcher CF, Tottene A, Lennon VA, Wilson SM, Dubel SJ, Paylor R, et al. Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB J. 2001;15:1288–90.
Kaja S, van de Ven RC, Broos LA, Frants RR, Ferrari MD, van den Maagdenberg AM, et al. Characterization of acetylcholine release and the compensatory contribution of non-Ca(v)2.1 channels at motor nerve terminals of leaner Ca(v)2.1-mutant mice. Neuroscience. 2007;144:1278–87.
Katoh A, Jindal JA, Raymond JL. Motor deficits in homozygous and heterozygous P/Q-type calcium channel mutants. J Neurophysiol. 2007;97:1280–7.
Herrup K, Wilczynski SL. Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience. 1982;7:2185–96.
Rhyu IJ, Oda S, Uhm CS, Kim H, Suh YS, Abbott LC. Morphologic investigation of rolling mouse Nagoya (tg(rol)/tg(rol)) cerebellar Purkinje cells: an ataxic mutant, revisited. Neurosci Lett. 1999;266:49–52.
Sawada K, Kalam Azad A, Sakata-Haga H, Lee NS, Jeong YG, Fukui Y. Striking pattern of Purkinje cell loss in cerebellum of an ataxic mutant mouse, tottering. Acta Neurobiol Exp. 2009;69:138–45.
Zwingman TA, Neumann PE, Noebels JL, Herrup K. Rocker is a new variant of the voltage-dependent calcium channel gene Cacna1a. J Neurosci. 2001;21:1169–78.
Hoebeek FE, Stahl JS, van Alphen AM, Schonewille M, Luo C, Rutteman M, et al. Increased noise level of Purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron. 2005;45:953–65.
Walter JT, Alviña K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–97.
Zhou YD, Turner TJ, Dunlap K. Enhanced G protein-dependent modulation of excitatory synaptic transmission in the cerebellum of the Ca2+ channel-mutant mouse, tottering. J Physiol. 2003;547:497–507.
Matsushita K, Wakamori M, Rhyu IJ, Arii T, Oda S, Mori Y, et al. Bidirectional alterations in cerebellar synaptic transmission of tottering and rolling Ca2+ channel mutant mice. J Neurosci. 2002;22:4388–98.
Chen G, Popa LS, Wang X, Gao W, Barnes J, Hendrix CM, et al. Low-frequency oscillations in the cerebellar cortex of the tottering mouse. J Neurophysiol. 2009;101:234–45.
Campbell DB, North JB, Hess EJ. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268–78.
Hoebeek FE, Khosrovani S, Witter L, De Zeeuw CI. Purkinje cell input to cerebellar nuclei in tottering: ultrastructure and physiology. Cerebellum. 2008;7:547–58.
Miyazaki T, Hashimoto K, Shin HS, Kano M, Watanabe M. P/Q-type Ca2+ channel alpha1A regulates synaptic competition on developing cerebellar Purkinje cells. J Neurosci. 2004;24:1734–43.
Liu S, Friel DD. Impact of the leaner P/Q-type Ca2+ channel mutation on excitatory synaptic transmission in cerebellar Purkinje cells. J Physiol. 2008;586:4501–15.
Todorov B, van de Ven RC, Kaja S, Broos LA, Verbeek SJ, Plomp JJ, et al. Conditional inactivation of the Cacna1a gene in transgenic mice. Genesis. 2006;44:589–94.
Barski JJ, Dethleffsen K, Meyer M. Cre recombinase expression in cerebellar Purkinje cells. Genesis. 2000;28:93–8.
Nadler JV, Evenson DA. Use of excitatory amino acids to make axon-sparing lesions of hypothalamus. Methods Enzymol. 1983;103:393–400.
Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406.
Espinosa JS, Wheeler DG, Tsien RW, Luo L. Uncoupling dendrite growth and patterning: single-cell knockout analysis of NMDA receptor 2B. Neuron. 2009;62:205–17.
Van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, et al. High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann Neurol. 2010;67:85–98.
Heckroth JA. Quantitative morphological analysis of the cerebellar nuclei in normal and lurcher mutant mice. I. Morphology and cell number. J Comp Neurol. 1994;343:173–82.
Devanagondi R, Egami K, LeDoux MS, Hess EJ, Jinnah HA. Neuroanatomical substrates for paroxysmal dyskinesia in lethargic mice. Neurobiol Dis. 2007;27:249–57.
Shirley TL, Rao LM, Hess EJ, Jinnah HA. Paroxysmal dyskinesias in mice. Mov Disord. 2008;23:259–64.
Buisseret-Delmas C, Angaut P, Compoint C, Diagne M, Buisseret P. Brainstem efferents from the interface between the nucleus medialis and the nucleus interpositus in the rat. J Comp Neurol. 1998;402:264–75.
Apps R, Hawkes R. Cerebellar cortical organization: a one-map hypothesis. Nat Rev Neurosci. 2009;10:670–81.
Mark MD, Maejima T, Kuckelsberg D, Yoo JW, Hyde RA, Shah V, et al. Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J Neurosci. 2011;31:4311–26.
Weisz CJ, Raike RS, Soria-Jasso LE, Hess EJ. Potassium channel blockers inhibit the triggers of attacks in the calcium channel mouse mutant tottering. J Neurosci. 2005;25:4141–5.
Liljelund P, Netzeband JG, Gruol DL. L-Type calcium channels mediate calcium oscillations in early postnatal Purkinje neurons. J Neurosci. 2000;20:7394–403.
Qian J, Noebels JL. Presynaptic Ca(2+) influx at a mouse central synapse with Ca(2+) channel subunit mutations. J Neurosci. 2000;20:163–70.
Caddy KW, Biscoe TJ. Structural and quantitative studies on the normal C3H and Lurcher mutant mouse. Philos Trans R Soc Lond B Biol Sci. 1979;287:167–201.
Mullen RJ, Eicher EM, Sidman RL. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci USA. 1976;73:208–12.
Ghetti B, Truex L, Sawyer B, Strada S, Schmidt M. Exaggerated cyclic AMP accumulation and glial cell reaction in the cerebellum during Purkinje cell degeneration in pcd mutant mice. J Neurosci Res. 1981;6:789–901.
Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature. 1997;388:769–73.
Fernandez-Gonzalez A, La Spada AR, Treadaway J, Higdon JC, Harris BS, Sidman RL, et al. Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy-induced gene, Nna1. Science. 2002;295:1904–6.
Roffler-Tarlov S, Beart PM, O’Gorman S, Sidman RL. Neurochemical and morphological consequences of axon terminal degeneration in cerebellar deep nuclei of mice with inherited Purkinje cell degeneration. Brain Res. 1979;168:75–95.
Tara E, Khodakhah K (2010) Altered purkinje cell activity during dyskinesia attacks in a mouse model of Episodic Ataxia. FENS [abstract 047.4]
Linnemann C, Sultan F, Pedroarena CM, Schwarz C, Thier P. Lurcher mice exhibit potentiation of GABA(A)-receptor-mediated conductance in cerebellar nuclei neurons in close temporal relationship to Purkinje cell death. J Neurophysiol. 2004;91:1102–7.
Acknowledgments
The authors are grateful to E. Galliano for constructive discussion and help with the rotarod experiments and T.J.H. Ruigrok for the excellent assistance with the quantification of the histological data. This work was supported by a grant from the Centre for Medical Systems Biology within the framework of the Netherlands Genomics Initiative/Netherlands Organisation for Scientific Research (NWO; A.v.d.M.), Erasmus Fellowship (F.E.H.), NWO-ALW (F.E.H., C.D.Z.), NWO-ZON-MW (F.E.H., C.D.Z.), Neuro-BSIK (C.D.Z.), Prinses Beatrix fonds (C.D.Z.), and National Institutes of Health R01 NS33592 (E.J.H.).
Conflicts of interest
The authors declare that there are no potential conflicts.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Boyan Todorov and Lieke Kros equally contributed to this work.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM 1
(MP4 1560 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Todorov, B., Kros, L., Shyti, R. et al. Purkinje Cell-Specific Ablation of CaV2.1 Channels is Sufficient to Cause Cerebellar Ataxia in Mice. Cerebellum 11, 246–258 (2012). https://doi.org/10.1007/s12311-011-0302-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-011-0302-1