
Key Points
-
The somatosensory system contains temperature-sensitive primary sensory neurons that convey thermal information from the skin and peripheral organs to the CNS. This sensory input enables the avoidance of prolonged contact with dangerously hot or cold objects and helps to maintain a core body temperature of ∼37 °C with minimal energy expenditure.
-
Thermal stimuli are coded by the action potential firing patterns of different types of temperature-sensitive primary sensory neurons, the cell bodies of which are located in the dorsal root and trigeminal ganglia.
-
The generation of action potentials in sensory nerve endings in response to a thermal stimulation depends on the activity of an array of temperature-sensitive ion channels. Members of the transient receptor potential (TRP) cation channel family of ion channels have been proposed as the prime thermosensors, but more recent research suggests additional important contributions from other ion channel types.
-
Several key temperature-sensitive ion channels exhibit intrinsic thermosensitivity, which implies substantial differences in enthalpy and entropy between the closed and open conformation of the channel proteins. The structural basis of this process is currently unclear.
-
Dysregulation of temperature-sensitive ion channels in sensory neurons can lead to thermal hypersensitivity and chronic pain, making these channels attractive targets for novel analgesic therapies.
-
Pharmacological inhibition of the prototype heat sensor TRPV1 and the cold sensor TRPM8 directly affects core body temperature, illustrating the important influence of peripheral thermosensation on thermoregulatory responses. Preventing unwanted changes in core body temperature is an important challenge in the future development of therapies that target temperature-sensitive ion channels.
Abstract
Our ability to perceive temperature is crucial: it enables us to swiftly react to noxiously cold or hot objects and helps us to maintain a constant body temperature. Sensory nerve endings, upon depolarization by temperature-gated ion channels, convey electrical signals from the periphery to the CNS, eliciting a sense of temperature. In the past two decades, we have witnessed important advances in our understanding of mammalian thermosensation, with the identification and animal-model assessment of candidate molecular thermosensors — such as types of transient receptor potential (TRP) cation channels — involved in peripheral thermosensation. Ongoing research aims to understand how these miniature thermometers operate at the cellular and molecular level, and how they can be pharmacologically targeted to treat pain without disturbing vital thermoregulatory processes.
Similar content being viewed by others

Main
Thermosensation, the ability to estimate temperature, is one of the most ancient sensory processes1. All organisms, from bacteria to plants and animals, have processes in place that enable them to react to changes in environmental temperature, and adequate responses to thermal challenges are absolutely crucial for survival. Indeed, temperature can have important and even detrimental effects on the structure and function of key biological macromolecules — including proteins, lipids and nucleic acids — and thus on the physiology and integrity of cells, tissues and organisms2. In mammals, changes in perceived temperature often provoke involuntary (physiological) or voluntary (behavioural) thermoregulatory actions. Inadequate thermoregulation, caused by disease, pharmacological treatment or harsh environmental conditions, can rapidly lead to harmful or even lethal hypo- or hyperthermia3,4.
In recent years, various types of ion channels, including the much-studied transient receptor potential channels (TRP channels), have been identified as highly sensitive molecular thermometers. These discoveries have fuelled detailed studies examining the molecular and cellular mechanisms of thermosensitivity and have led to the development of powerful genetic models and pharmacological tools to analyse the contributions of these channels to thermosensation in health and pathophysiology. In this article, we provide a brief overview of the cellular circuitry that underlies thermosensation and discuss current insights and controversies regarding the molecular and biophysical principles that form the basis of temperature sensing in mammals. In addition, we describe various causes and consequences of pathological thermosensation and the potential of thermosensitive ion channels as targets for pain therapy. Finally, we highlight important lacunas in our knowledge of mammalian thermosensation and provide some perspectives on further developments in this field.
Thermosensing neurons and circuits
In mammals, environmental thermal cues are primarily conveyed by afferent neurons of the somatosensory system. These neurons are individually tuned sensory cells that can convert specific thermal, but also mechanical and chemical, stimuli into electrical signals, which travel from the periphery to the CNS in the form of action potentials1,5. Mammalian sensory neurons are pseudounipolar: they contain a single axon that divides into two branches, with one branch extending to peripheral tissues such as skin, mucosa and internal organs, where it gathers information pertaining to environmental stimuli, and the other branch relaying the detected information to second-order neurons in the dorsal horn or the sensory nucleus in the brain (Fig. 1a). The cell bodies of primary sensory neurons that innervate the sensitive parts of the head and face, including the mouth, nose and eyes, are clustered in the trigeminal ganglia adjacent to the brain. By contrast, the cell bodies of sensory neurons that innervate the rest of the body are contained by the dorsal root ganglia (DRGs), which are located in the vertebral column just lateral to the spinal cord6.

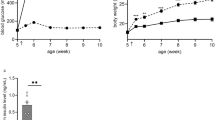
a | Pathways involved in thermal avoidance responses, thermoperception and the initiation of thermoregulatory responses. Cutaneous primary sensory neurons involved in thermosensation (green) include both non-myelinated C fibres and thinly myelinated Aδ fibres. The cell bodies of these neurons are located in the dorsal root ganglia (DRGs) and have axons with two branches. One branch extends towards the periphery, with free endings in the skin, where thermal information is coded in the form of action potentials. These action potentials propagate to the end of the other axonal branch, which forms synapses in layers I and II (for C fibres) or layers I and V (for Aδ fibres) of the dorsal horn. Activity of both Aδ and C primary sensory fibres steers three distinct neuronal pathways and ensuing responses: first, motor neurons can be activated via spinal interneurons, leading to a rapid withdrawal reflex in response to noxious temperatures (red neurons); second, thermosensory information is transmitted via second-order sensory neurons of the ascending spinothalamic tract to the thalamus and further relayed to the somatosensory cortex, where our perception of temperature is formed (blue neurons on the coronal section of the human brain); and third, thermosensory information is transmitted via lateral parabrachial (LPB) neurons, which may also receive input from the spinothalamic neurons, to the pre-optic area (POA) of the hypothalamus (blue neurons on the central sagittal section of the human brain), where thermoregulatory processes are initiated. b | Cutaneous thermosensitive neurons as shown in part a typically exhibit one of four different thermal response profiles. The first type of thermal response is related to the detection of noxious cold. These fibres are generally silent at the thermoneutral temperature, exhibit thermal thresholds at around 20–10 °C, show a linear increase in firing intensity upon cooling down to 0 °C and exhibit limited adaptation. The second type of response is observed in fibres activated by cool temperatures of 37–20 °C. The majority of these fibres exhibit ongoing activity at thermoneutral skin temperature, which disappears upon moderate warming. Discharge frequency increases upon moderate cooling, reaching a maximum firing rate between 30 and 20 °C. At temperatures of around 20 °C, fibres generally show rapid adaptation, which leads to a reduction in discharge frequency at temperatures below ∼17 °C. A similar bell-shaped temperature–frequency relationship and rapid adaptation is observed in warm-sensitive fibres. These exhibit ongoing activity at thermoneutral skin temperature, which disappears upon moderate cooling. Their discharge rate increases upon moderate heating, reaching a maximum at 40–43 °C, but these fibres rapidly fall silent upon further heating. Finally, fibres involved in sensing noxious heat are typically activated at temperatures above 43 °C, with peak discharge occurring at noxious temperatures (45–53 °C), and show little or no adaptation.
Sensory nerves are classically subdivided into four main categories on the basis of their size and conduction properties5,6,7. Aα and Aβ fibres have large cell bodies, thickly myelinated axons and high conduction velocities; Aδ fibres, by contrast, exhibit medium-sized cell bodies, thinly myelinated axons and intermediate conduction velocities; and C fibres are characterized by small cell bodies, the absence of a myelin sheath and slow conduction velocities. The latter two fibre types — the Aδ and C fibres — include the sensory nerves involved in thermosensation6,7.
The temperature changes that are encountered by visceral sensory neurons that innervate internal organs are mostly limited to a few degrees around the body's core temperature by strict homeostatic regulation8. Cutaneous sensory neurons, however, encounter and convey a much wider range of temperatures. A skin temperature of around 33 °C is perceived as thermoneutral by humans, with lower temperatures perceived to be cool or cold, and higher temperatures perceived to be warm or hot. In general, skin temperatures below ∼15 °C or above ∼45 °C are associated with pain and considered to be noxious6,9. Based on their response profiles, temperature-sensitive neurons can be classified into four main response types6,10,11,12 (Fig. 1b). Neurons that are activated by either non-noxious cool or non-noxious warm temperatures show marked basal action potential firing activity. Warm-sensitive neurons respond to moderate heating with a rapid but transient increase in firing frequency and to moderate cooling with a transient reduction of firing activity11. Cool-sensitive neurons exhibit a mirror response, with a transient decrease in firing frequency upon moderate heating and a transient increase upon moderate cooling10,13 (Fig. 1b). The transient nature of the firing response of warm- and cool-sensitive neurons is believed to underlie the typical adaptation to non-noxious temperatures that humans experience, for instance, when stepping into a warm bath or diving into a cool swimming pool. By contrast, sensory neurons that respond to noxious cold or noxious heat are mainly silent at thermoneutral temperatures but show sustained rapid action potential firing in response to prolonged noxious thermal stimuli6,12 (Fig. 1b). Accordingly, humans experience little or no adaptation to noxious temperatures14,15. Prolonged exposure to noxious temperatures also causes tissue damage, leading to the local release of various pro-algesic agents, including ATP, protons, reactive oxygen species and lipid mediators9,16. These agents can activate nociceptors and provoke pain even after removal of the painful thermal stimulus.
Temperature-sensitive primary sensory neurons from the DRG enter the spinal column through the intervertebral foramina and form glutamatergic synapses in the dorsal horn7 (Fig. 1a). Acute avoidance of noxious cold or noxious heat involves a simple spinal reflex, where primary sensory neurons signal via spinal interneurons to motor neurons, resulting in a rapid withdrawal of the challenged body part. Thermal information from cutaneous thermosensory neurons is also transmitted via the spinothalamic tract to the thalamus and then further relayed via thalamocortical radiations to the primary sensory cortex, where the perception of temperature originates and voluntary behavioural actions are initiated17 (Fig. 1a). Finally, thermosensory information from cutaneous and visceral sensory neurons is transmitted via lateral parabrachial neurons to the pre-optic area (POA; within the anterior portion of the hypothalamus). The POA itself contains intrinsically temperature-sensitive neurons, which not only receive synaptic input relaying the activity of peripheral temperature-sensitive neurons but also respond to small changes in local brain temperature17,18. Based on the integrated central and peripheral thermal information, the POA controls thermoregulatory processes, such as cutaneous vasomotion, shivering and brown adipose tissue thermogenesis9. Central mechanisms of thermosensation and thermoregulation have been reviewed in detail elsewhere17,18,19.
Ion channels as molecular thermometers
At the basis of thermosensation lies the property of specific ion channels to conduct ions in a highly temperature-dependent manner. The steepness of the temperature dependence of an ion channel's conductance can be quantified using the Q10 value (Box 1). Depending on their ionic selectivity, temperature-sensitive ion channels are either excitatory or inhibitory. In the context of a sensory nerve ending, temperature-dependent changes in ionic conductance can determine whether, and at what frequency, action potentials are generated. Over the past two decades, important advances have been made in the molecular identification and characterization of highly temperature-sensitive ion channels. In particular, several temperature-sensitive members of the TRP family of ion channels were identified and were shown to have thermal response profiles collectively covering the entire range of temperatures that mammals can discriminate20. These findings led to the proposal that these so-called thermoTRP channels function as the prime or sole molecular thermosensors.
However, for a temperature-sensitive ion channel to be considered a bona fide molecular thermosensor, evidence needs to be provided underpinning a role in thermosensation in vivo. In recent years, single- and combined-knockout mouse models have been developed to test many putative thermosensors, and these models have enabled detailed phenotyping using various behavioural assays of thermosensitivity. These assays include: acute avoidance tests, in which the latency to withdrawal from a noxious thermal stimulus applied to the tail or paw is measured; pain intensity tests, in which pain behaviours (such as jumping or licking of the paws) in response to sustained noxious stimulation are quantified; and thermotaxis tests, in which an animal's preference to migrate to a specific temperature or temperature zone is analysed21,22,23,24. Although such studies can reveal important alterations in an animal's thermosensitivity, it should be noted that altered responses in these assays may be due not only to altered thermal sensitivity of primary sensory neurons but also to altered central processing. Therefore, ex vivo assays that allow direct measurement of the activity of intact cutaneous sensory nerve endings are an important complementary approach12. Overall, knockout-mouse studies have confirmed the role of some but not all thermoTRP channels and indicate that additional TRP-independent mechanisms may play a prominent part in the transduction of thermal stimuli in the somatosensory system.
TRP channels. In mammals, the TRP superfamily consists of 28 members, which can be subdivided into 6 subfamilies (TRPC, TRPV, TRPM, TRPA, TRPML and TRPP)25. Humans only express 27 TRP channel members as, during the evolution of higher primates, the gene encoding TRPC2 became a pseudogene25. Using responsiveness to capsaicin (the hot constituent of chilli peppers) as a screening readout, vanilloid receptor 1 (VR1; now known as TRPV1) was cloned from a cDNA library that was created from rat sensory neurons26. TRPV1 is expressed in a subset of nociceptive, small-diameter Aδ and C neurons that originate from DRGs and trigeminal ganglia. Like all members of the TRP channel superfamily, TRPV1 subunits have six transmembrane domains, and four of these subunits are required to form a functional cation channel25,27. The heterologous expression of TRPV1 not only resulted in capsaicin-gated currents but also revealed that TRPV1 is strongly activated upon heating26, with Q10 values >10 (Fig. 2).

a| Members of the transient receptor potential (TRP) channel family function as excitatory cation channels and respond to cooling or heating, depending on their subtype. b | The Cl− channel anoctamin 1 (ANO1) acts as an excitatory heat-activated channel. Heating leads to only minimal activation of ORAI1 by heat-activated stromal interaction molecule 1 (STIM1) (light purple), but a large excitatory 'off' response occurs upon cooling following a heat stimulus (dark purple). c | Two-pore-domain K+ (K2P) channels and voltage-gated Na+ (Nav) channels determine the extent of sensory neurons' overall excitability by influencing the membrane's depolarization threshold above which action potentials may be fired. At cold temperatures, tetrodotoxin (TTX)-sensitive Nav channels (TTX-Nav channels) inactivate and thermosensation becomes fully dependent on Nav1.8.
TRPV1 is responsive to various noxious stimuli, including: noxious heat; acidic and basic solutions; aversive chemicals produced by certain plants and animals; and endogenous lipid signalling molecules, such as anandamide and eicosanoids28,29,30,31. The activation of temperature-sensitive ion channels by a ligand provides a molecular explanation for chemesthesis, the phenomenon whereby certain chemical compounds mimic a physical stimulus (Box 2). In support of TRPV1's role as a sensor of thermal and chemical stimuli, currents evoked by heat (>43 °C) and capsaicin were virtually absent in cultured neurons from TRPV1-deficient mice, although higher-threshold (>55 °C) heat responses were intact21,22. Moreover, in behavioural experiments, TRPV1-deficient mice showed a longer latency in their responses to noxious heat but normal latencies to painful mechanical stimuli21. The key role of TRPV1 as a noxious heat sensor becomes even more pronounced in the setting of inflamed tissue: TRPV1-deficient mice do not exhibit the prominent inflammation-associated heat hyperalgesia that is observed in wild-type animals21,22. Through the use of TRPV1 antagonists, the noxious-heat-sensing activity of TRPV1 has been further confirmed in other mammals, including humans32,33.
The residual heat-sensitivity observed in TRPV1- deficient mice suggested the existence of an additional heat sensor (or sensors), and three closely related members of the TRPV subfamily, TRPV2, TRPV3 and TRPV4, were obvious candidates. TRPV2, the closest homologue of TRPV1, has been a long-standing candidate for a high-threshold molecular sensor of noxious heat, because it was found to be expressed in a subset of medium- to large-diameter Aδ (but also Aβ) neurons, and heterologous expression of rodent TRPV2 produced a cation current that was activated at temperatures exceeding ∼52 °C34. However, a recent report demonstrated that genetic ablation of TRPV2 in mice had no discernible effect on acute pain responses to noxious heat or on inflammatory heat hyperalgesia, even when TRPV1 activity was simultaneously suppressed35, indicating that TRPV2 can be excluded as a key thermosensor in the somatosensory system.
TRPV3 and TRPV4 have been proposed to be molecular sensors that are involved in the detection of non-noxious warmth. In heterologous expression systems, these channels show a steep increase in activation in response to increases in temperature between 25 and 35 °C36,37,38,39,40. Interestingly, these two channels are abundantly and functionally expressed in epidermal and hair-follicle keratinocytes, whereas in sensory neurons, expression levels of mRNA encoding TRPV3 and TRPV4 are low or below the detection limit of northern blot or quantitative PCR analyses37,38,41,42. This expression pattern has led to the hypothesis that TRPV3 and TRPV4 could mediate the sensation and discrimination of warm temperatures by keratinocytes, which then communicate with sensory neurons by releasing diffusible messengers such as ATP or nitric oxide43,44,45. In line with this view, initial papers describing the behaviour of TRPV3- or TRPV4-deficient mice reported significant abnormalities in thermal preference in the warm temperature range24,46. In particular, TRPV4-deficient mice preferred warmer floor temperatures on a thermal gradient, whereas TRPV3-deficient mice showed a relative indifference to temperatures ranging from 20 to 35 °C24,46 However, more recent studies indicate that the alteration in temperature preference in TRPV3-deficient mice is highly dependent on the genetic background and sex of the mice44,47. On a 129S1 background, deficits in innocuous warmth discrimination were observed in TRPV3-deficient female but not male mice44. On a C57BL6 background, mice that lacked both TRPV3 and TRPV4 exhibited thermal preference behaviour that was virtually indistinguishable from that of wild-type C57BL6 mice, even when TRPV1 function was pharmacologically blocked47. Although the possibility that TRPV3 and TRPV4 have more specialized functions cannot be excluded, these results argue against a general role for TRPV3 and TRPV4 in thermosensation and stress the importance of considering the sex and genetic background of mutant mouse strains when interpreting thermosensory behaviour48.
Recently, TRPM3 was identified as an alternative noxious-heat sensor in a large subset of sensory neurons, including nociceptors, from DRGs and trigeminal ganglia. This member of the TRPM subfamily can be activated by chemical ligands, such as the neurosteroid pregnenolone sulphate, as well as by heat49,50. Heterologously expressed TRPM3 is activated by heating, with a current–temperature relationship curve that is shifted slightly towards higher temperatures compared with that of TRPV1 (Ref. 50) (Fig. 2). TRPM3-deficient mice exhibit deficits in avoidance responses to noxious heat and in the development of heat hyperalgesia in inflamed tissue50. Similarly, pharmacological inhibition of TRPM3 using citrus fruit flavanones reduced the sensitivity of mice to noxious heat51. These results suggest that TRPM3 is a bona fide thermosensor that is involved in the detection of noxious heat. Nevertheless, pharmacological inhibition of TRPV1 in TRPM3-deficient mice did not fully abrogate avoidance responses to noxious heat50, implying the existence of additional mechanisms for sensing noxious heat. Interestingly, selective ablation of TRPV1-expressing neurons in adult mice, using a diphtheria toxin-mediated strategy, leads to a complete lack of heat avoidance at temperatures up to 50 °C: a much more pronounced noxious-heat-sensing phenotype than was seen following combined elimination of TRPM3 and TRPV1 (Refs 52,53). These findings suggest that TRPV1-expressing cells are essential for heat responses but express additional heat sensors besides TRPV1 and TRPM3. This interpretation must be taken with some caution, as ablating a large proportion of sensory neurons in an adult animal might have unpredictable effects on neighbouring neurons in the sensory ganglia or dorsal horn.
TRPM8, the first cold-activated TRP channel to have been identified, plays a crucial part in cold detection by the somatosensory system. In heterologous expression systems, TRPM8 activity steeply increases upon cooling (Fig. 2) and in the presence of substances that are known to produce a cooling sensation, including menthol, eucalyptol and the 'super-cooling agent' icilin54,55 (Box 2). TRPM8 is expressed in a small subpopulation of C and Aδ fibres that originate from DRGs and trigeminal ganglia. Although functional studies indicate that a small but significant proportion (approximately 20%) of TRPM8-expressing neurons respond to the TRPV1 agonist capsaicin23,54, TRPM8 shows no co-expression with typical markers of nociceptor neurons, such as calcitonin gene-related peptide (CGRP), substance P or isolectin B4 (Ref. 55). Intact sensory nerve fibres and cultured sensory neurons from TRPM8-deficient mice exhibited profoundly diminished responses to non-noxious cool temperatures56,57,58. In particular, these mice exhibited a strong reduction in the number of cold-activated C and Aδ fibres, and the remaining cold-sensitive C fibres exhibited strongly reduced basal activity at thermoneutral skin temperature56. Behaviourally, TRPM8-deficient mice exhibited a striking deficit in avoiding cool temperatures: whereas wild-type mice strongly prefer a temperature of around 30 °C to cooler temperatures, TRPM8-deficient mice showed relative indifference to temperatures between 18 and 30 °C56,57,58. Moreover, whereas mild cooling can evoke analgesia in wild-type mice, cooling-induced analgesia was absent in TRPM8-deficient mice58. Importantly, however, a subpopulation of DRG and trigeminal ganglion neurons from TRPM8-deficient mice responded to noxious cold, and these mice still showed robust avoidance behaviour in response to temperatures below 15 °C56,57,58. These data indicate that TRPM8 functions as a thermosensor in non-nociceptive Aδ and C fibres that are involved in the perception of cool temperatures. Interestingly, cold sensing by TRPM8-expressing neurons that innervate the cornea also helps to regulate basal tearing59. These neurons respond to reductions in corneal temperature as small as 1 °C, which occur upon evaporation of the tear film, and thus indirectly function as sensitive wetness sensors59. Whereas genetic deletion of TRPM8 elicits certain deficits in cold avoidance, selective ablation of TRPM8-expressing neurons in adult mice yields a stronger cold-avoidance deficit and a partial reduction in the avoidance of noxious cold52,60. These results suggest that TRPM8-expressing neurons also participate in detecting painful cold but imply the existence of additional cold sensors as well, especially for the detection of noxious cold.
TRPA1, which is the only mammalian member of the TRPA subfamily, is a prominent but controversial candidate to mediate (part of) the sensing of noxious cold. Several studies report that, in heterologous expression systems, TRPA1 is activated by cooling — with a current–temperature relationship that is shifted further towards colder temperatures compared with that of TRPM8 (Refs 23,61,62,63,64,65) (Fig. 2) — and by a plethora of chemically diverse noxious or irritant compounds62,66,67 (Box 2). TRPA1 is specifically expressed in a subset of small-diameter mammalian sensory ganglion neurons that co-express typical nociceptor markers such as CGRP, substance P and isolectin B4, as well as TRPV1 (Ref. 61). These properties are consistent with the suggestion that TRPA1 acts as a noxious-cold sensor to elicit cold-induced pain. Indeed, several in vivo studies have shown that TRPA1-null mice have marked deficiencies in the nocifensive responses to noxious cold23,68,69.
However, whereas the role of TRPA1 as a sensor for noxious chemicals is generally accepted, crucial aspects of its proposed function in (noxious) cold sensing remain controversial and highly debated. First, although several studies have demonstrated cold-induced activation of heterologously expressed human, mouse and rat TRPA1, in both whole cells and cell-free membrane patches23,61,62,63,64, there are conflicting reports showing that cold fails to activate TRPA1 from these and other mammals66,70,71. The reasons for these discrepancies are not clear. Studies showing cold-induced activation of TRPA1 report that cooling is a 10- to 30-fold less potent stimulus than chemical agonists such as mustard oil23, suggesting that differences in assay sensitivity may explain why some studies fail to detect cold-induced responses. In addition, as TRPA1 is highly sensitive to intracellular Ca2+ levels70, changes in which can potentiate or desensitize channel activity, differences in the Ca2+ content or Ca2+-buffering capacity of experimental solutions may underlie some conflicting results. Second, whereas some studies report marked deficits in acute noxious-cold sensing in TRPA1-deficient mice23,68,69, others report that TRPA1-null mice exhibit normal responses to noxious cold72,73. Furthermore, others found that Trpa1-knockout mice only show reduced cold-sensitivity under conditions in which the sensitivity of TRPA1 is enhanced by endogenous or exogenous chemicals74. Based on the available data, it is not clear whether these conflicting results reflect differences in genetic background, assays, experimental conditions, researchers or a combination thereof.
Irrespective of whether TRPA1 is involved in sensing cold, there is little doubt that additional TRPM8- and TRPA1-independent molecular mechanisms of cold sensation must exist, as mice that lack both TRPM8 and TRPA1 still avoid noxious cold73. One potential, but not yet fully explored, candidate for cold sensing within the TRP superfamily is TRPC5. In a heterologous expression system, TRPC5 channels are sensitive to cold in the temperature range of 37–25 °C. Although TRPC5 is expressed in sensory neurons, TRPC5-null mice do not exhibit obvious deficits in temperature-sensitive behaviour75. Overall, these studies illustrate that our understanding of the molecular sensors involved in cold sensation remains incomplete and suggest the existence of additional cold-activated depolarizing mechanisms in sensory neurons.
Ca2+-activated Cl− channels. Anoctamin 1 (ANO1; also known as TMEM16A), a Ca2+-activated Cl− channel76, has recently been proposed to be a potential noxious-heat sensor77. ANO1 is expressed in small-diameter sensory neurons that also express the nociceptor markers isolectin B4 and CGRP, and is steeply activated by heat in the noxious range (Fig. 2), even at low intracellular Ca2+ concentrations. At typical intracellular Cl− concentrations of ∼45 mM, the activation of ANO1 induces a depolarizing current in isolated DRG neurons that is sufficient to induce action potentials77. However, as the intracellular Cl− concentration can be dynamically modulated (for example, by inhibition of Na+–K+–Cl− cotransporter 1), situations can be envisaged wherein ANO1 activation actually counteracts the depolarization of sensory neurons. Genetic ablation or pharmacological inhibition of ANO1 in mice reduced heat-induced nociceptive behaviour77,78, suggesting that ANO1 is an additional heat sensor involved in the perception of painfully hot temperatures. ANO2, the closest homologue of ANO1, is also sensitive to heat, but its physiological relevance remains to be determined77.
STIM1–ORAI1. Another potential instrument in the repertoire of heat-sensitive ion channels is stromal interaction molecule 1 (STIM1), which has a well-established function as the endoplasmic reticulum (ER) Ca2+ sensor of store-operated Ca2+ entry. Upon ER Ca2+-store depletion, STIM1 clusters at ER–plasma membrane junctions, where it interacts with and opens Ca2+-permeable ORAI1 ion channels79. Recently, it was reported that mild heating of cells to temperatures above 35 °C induces clustering of STIM1 independently of Ca2+-store depletion80. As temperatures above 35 °C also prevent the physical interaction between STIM1 and ORAI1, this STIM1 clustering does not lead to Ca2+ entry during heat stimulation. However, subsequent cooling provokes a transient Ca2+ influx through ORAI1 channels (Fig. 2), because the ability of the STIM1 clusters to activate ORAI1 channels is temporarily restored before the clusters are disassembled80. As both STIM1 and ORAI1 are functionally expressed in sensory neurons, a potential role for them in the regulation or modulation of thermosensation can be envisaged but remains to be established.
Na+ and K+ channels. Temperature-dependent excitation of a sensory neuron depends not only on the activation of a depolarizing current, which causes depolarization of the sensory nerve ending (the receptor potential), but also on the combination of K+ channels and voltage-gated Na+ (Nav) channels that together set the threshold voltage for the generation of action potentials. Modulation of these channels can enhance or dampen the effect of activation of a depolarizing current, thereby drastically influencing the temperature sensitivity of a neuron81.
Members of the two-pore-domain K+ (K2P) channel family give rise to the background (leak) K+ conductance and thus function as brakes on excitability in many cell types82. Interestingly, three members of the TREK/TRAAK subfamily of K2P channels — namely, TREK1 (encoded by KCNK2), TRAAK (encoded by KCNK4) and TREK2 (encoded by KCNK10) — show notable heat sensitivity, with Q10 values of 5–10 in the 20–40 °C temperature range, followed by a steep drop in activity at higher temperatures83,84. These three channels are expressed in sensory neurons, in which they modulate stimulus sensitivity in a highly temperature-dependent manner82. Indeed, both TREK1- and TRAAK-deficient mice displayed an enhanced sensitivity to warmth at the transition between non-noxious and noxious heat, as well as hypersensitivity to mechanical stimuli85,86. Mice without both TREK1 and TRAAK also display a hypersensitivity to cold that is not observed in the single-knockout mice86.
The firing of action potentials in neurons requires the activity of Nav channels, and of the nine mammalian Nav channels, at least five (namely, Nav1.1 and Nav1.6–Nav1.9) are abundantly expressed in sensory neurons87. Nociceptors express both tetrodotoxin (TTX)-sensitive Nav channels (for example, Nav1.7) and the TTX-resistant Nav channels Nav1.8 and Nav1.9 (Ref. 87). At noxiously cold temperatures, most Nav channels in sensory neurons undergo slow voltage-dependent inactivation, thereby strongly reducing excitability88. Nav1.8, however, is much less sensitive to cold-induced inactivation and is thereby able to sustain action-potential initiation at noxiously cold temperatures88. As a consequence, action-potential generation at low temperatures fully relies on Nav1.8, and Nav1.8-deficient mice, as well as mice in which all sensory neurons expressing Nav1.8 are ablated, show negligible responses to noxious cold and mechanical stimulation at low temperatures88,89.








Temperature sensitivity of ion channels
A fascinating but largely unresolved question in the field of thermosensation is what determines the steep thermosensitivity of ion conductances in sensory neurons. Temperature mainly affects the opening and closing (gating) of the underlying channels (Box 1), and several distinct — but not mutually exclusive — mechanisms may contribute to this temperature dependence (Fig. 3).

Open channels conduct ions into or out of the cell, depending on channel type and ionic conditions. ΔT represents an activating change in temperature, which can be either negative (for cold-activated channels) or positive (for heat-activated channels). a | Temperature-dependent changes in membrane properties. Here, the channel is mechanosensitive and responds to temperature-dependent alterations in membrane tension, thickness or curvature. b | Temperature-dependent production of channel ligand. In this mechanism, an intracellular enzyme is activated by the change in temperature, leading to the production of channel-activating ligands. c | Temperature-dependent channel phosphorylation. An intracellular kinase is activated by the change in temperature, leading to phosphorylation and activation of the channel. d | Temperature-dependent activation of Ca2+-dependent Cl− channel ORAI1. The endoplasmic reticulum (ER)-resident protein stromal interaction molecule 1 (STIM1) contains a luminal Ca2+-sensing domain (CSD) and a cytosolic Ca2+-activating domain (CAD). When the temperature is below 37 °C and the ER Ca2+ content is high, Ca2+ that is bound to the CSD prevents clustering and activation of ORAI1 at the plasma membrane. Heating leads to clustering of STIM1 but prevents the interaction between the CAD and ORAI1. Subsequent cooling, however, allows the CAD domains to bind to and activate ORAI1, resulting in a transient 'heat-off' response that lasts until the STIM1 clusters are disrupted. e | Intrinsic temperature sensitivity. Here, changes in temperature directly induce conformational changes in the channel complex, leading to channel opening.
The effects of temperature on channel gating. First, changes in temperature may provoke local or global phase transitions in the plasma membrane (Fig. 3a). For instance, the cell membrane's transition between the liquid-crystalline and gel-like phases has profound and abrupt effects on its tension and/or thickness90, which in turn can affect ion channel gating. In such a model, the ion channel itself would have to be responsive to mechanical stress or to changes in membrane thickness. In this respect, it is interesting to note that some channels, in particular the K2P channels TREK1, TREK2 and TRAAK, are both mechano- and thermosensitive, which suggests that the effects of temperature on membrane tension, thickness or curvature may modulate channel gating82,84. However, given that temperature-dependent channel activation is mostly a gradual phenomenon, whereas phase transitions in membranes are much more abrupt, it is unlikely that mechanosensitivity is the prime mechanism of thermosensitive gating for most thermosensitive channel types.
Second, changes in temperature may alter the concentration of channel ligands by, for example, increasing or decreasing the activity of a temperature-dependent ligand-producing and/or ligand-degrading enzyme (Fig. 3b). Notably, in such a model, the temperature dependence of ligand metabolism does not need to be extremely steep, as long as the ligand activates the channel in a highly cooperative manner. For instance, if the ligand concentration has a Q10 of 2 (that is, a doubling of the concentration upon a 10-degree increase in temperature), and this ligand activates the channel in a cooperative manner, with a Hill coefficient of 4, then the overall channel activation has a maximal Q10 of 24 = 16. We are not aware of temperature-sensitive channels for which such an indirect, ligand-dependent pathway represents the prime activation mechanism. Nevertheless, temperature-induced changes in the cellular concentration of phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) and Ca2+ have been proposed to modulate the activity of the TRP channels TRPM8 and TRPA1 (Refs 70,91).
Third, temperature may influence channel gating by influencing post-translational modification of the channel (Fig. 3c). For instance, the activity of many channels is strongly modulated by phosphorylation by kinases such as protein kinase C and protein kinase A92, and in some cases this modulation is remarkably enhanced at increased temperatures, thus contributing to temperature dependence93. The steepness of the temperature dependence may be further increased if the effects of post-translational modification on channel gating show cooperativity, as described above for the effects of non-covalent ligands.
Fourth, temperature may alter the conformation and/or localization of a channel-regulating auxiliary subunit. A striking example is the temperature-dependent multimerization of the ER-resident protein STIM1 and the ensuing ORAI1-mediated 'off' response to a heat stimulus described above80 (Fig. 3d). This process may be a more general mechanism of thermosensitive modulation of plasma membrane channels given that STIM1 has also been proposed to regulate other membrane channels, including voltage-gated Ca2+ channels94,95 and TRPC channels96, although the lattermost interaction has been disputed94,97.
Last, the pore-forming part of the channel itself may exhibit considerable intrinsic thermal sensitivity (Fig. 3e), which manifests itself through steeply temperature-dependent changes in the channel's mean open probability Popen. Compelling evidence for such an intrinsic activation mechanism has recently been provided for cold-activated TRPM8 and heat-activated TRPV1, both of which were shown to retain high temperature sensitivity following purification and subsequent reconstitution into artificial lipid bilayers98,99. The temperature dependence of several other cold- or heat-activated TRP channels, including TRPM3, TRPM4, TRPM5 and TRPA1 (Refs 23,50,100), can be described using a unifying two-state gating model that was initially developed for TRPV1 and TRPM8 (Ref. 101). Therefore, it is reasonable to assume that these TRP channels are all intrinsically thermosensitive. In recent years, a growing number of studies have provided insight into the mechanistic basis of this steep, intrinsic thermosensitivity, which is described further below.
Basis of intrinsic thermosensitivity. Thermodynamic considerations, as explained in Box 3, dictate that the opening of a heat-activated channel (where Q10,gating >> 1) is associated with a marked increase in both enthalpy (ΔHgating >> 0) and entropy (ΔSgating >> 0), whereas cold-activated channels (Q10,gating << 1) exhibit negative values for both ΔHgating and ΔSgating. Understanding the origin of intrinsic thermosensitive gating of ion channels requires knowledge of the structural rearrangements that provoke these significant changes in enthalpy and entropy during channel gating.
Intrinsically temperature-sensitive TRP channels, such as TRPV1 and TRPM8, function as tetrameric complexes of ∼4,000 amino acids and interact with a large number of membrane lipids and water molecules. The enthalpy of a specific state of a channel complex is determined by all of the interatomic interactions in the system, which include not only covalent bonds but also weaker interactions, such as van der Waals interactions, hydrogen bonds, salt bridges (formed between acidic and basic amino-acid side chains) and cation–π interactions (formed between aromatic and basic amino-acid side chains). The disruption or formation of such weaker interactions leads to increases or decreases in enthalpy, respectively. Smaller changes in enthalpy are associated with van der Waals interactions (0.1–2 kJ mol−1) and hydrogen bonds (∼5 kJ mol−1), whereas larger amounts of energy are associated with cation–π interactions (10 kJ mol−1) and salt bridges (20 kJ mol−1)102. Concomitantly, entropy, which is a thermodynamic measure of the disorder of a system, also generally increases when weaker interactions are disrupted. In silico analysis of the recent high-resolution cryo-microscopy structure of the unliganded (and therefore most probably closed) TRPV1 channel27 indicates that one tetrameric channel complex comprises at least 70 cation–π interactions, 100 salt bridges and 1,500 hydrogen bonds. Thus, disruption of only a small percentage of these interactions upon channel opening would suffice to explain the reported values for the gating enthalpy of TRPV1 (Refs 101,103), which are in the range of 180 to 400 kJ mol−1.
In the past decade, several studies have searched for specific structural elements that underlie steep temperature dependence, in particular in TRP channels, by evaluating the consequences of channel mutagenesis on temperature-induced channel activity. On the basis of these studies, several authors have proposed the existence of confined 'thermosensor modules' in temperature-sensitive TRP channels, akin to the voltage-sensor modules found in voltage-gated cation channels104,105,106,107. In Fig. 4, the regions that have been implicated in the temperature sensitivity of TRPV channels are indicated. The observation that these regions are spread over large parts of the channel does not seem to provide strong support for the view that thermosensitivity specifically arises in conserved and/or demarcated regions but rather suggests that alterations in different parts of temperature-sensitive TRP channels can affect their response to temperature108. Conceivably, the marked difference in enthalpy between closed and open states may arise from multiple submolecular rearrangements occurring simultaneously during gating in dispersed areas of the channels. As the structural effects of point mutations and domain swaps on channel structure and gating rearrangements remain unpredictable, a detailed understanding of the structural basis of thermosensing remains some way off. However, the recent advances in high-resolution cryo-electron microscopic structural analysis, which yielded detailed pictures of TRPV1 in closed and ligand-bound states, have paved the way towards visualization of the structural rearrangements that occur during temperature-induced channel gating of ion channels27,109. This may ultimately enable calculation of the relative contributions of domains, residues and atoms to the unique thermodynamic properties that underlie thermosensitivity.

The structure of a single transient receptor potential V1 (TRPV1) subunit (left) and entire tetrameric channel (right) viewed from the side27, showing the ankyrin-repeat domain (ARD), the transmembrane domains S1–S6 and various regions implicated in the heat sensitivity of TRPV channels. These include: first, the membrane-proximal domain (MPD; between the ankyrin repeats and the first transmembrane domain), which has been put forward as a crucial modular thermal sensor in TRPV channels184; second, the pore turret, a 25-amino-acid loop that connects S5 with the pore helix, which was reported to be required for normal heat-induced but not capsaicin-induced activation of TRPV1 (Ref. 185); third, several residues in the outer pore region and in the initial part of S6, alterations to which impair heat-induced activation of TRPV1 and TRPV3 (Refs 106,186); and last, the carboxy-terminal TRP box, alterations to which were found to reduce the Q10 for heat-induced activation of TRPV1 (Ref. 187). Note that a further proximal C-terminal part, which was reported to confer heat-induced activation to the cold-sensitive TRPM8 (Refs 104,188), a more distal C-terminal region, which has been put forward as another major structural determinant of thermal sensitivity in TRPV1 (Refs 30,189), and a large part of the pore turret185 are not visible in the structure. These segments were removed to improve the biochemical stability of the expressed TRPV1 channel, which nevertheless retained sensitivity to heat in functional assays27. Based on similar structure–function experiments on TRPA1 from different animals, including Drosophila melanogaster, snakes and mammals, it was proposed that elements in the ARDs, S5 and the pore region dictate thermal sensitivity71,107,190. The RCSB Protein Data Bank identifier for TRPV1 is 3J5P.






Pathophysiology of thermosensation
The thermal sensitivity of sensory neurons, including nociceptors, is determined by the balance of the activities of a blend of depolarizing and repolarizing ion channels. Unbalanced activity of one or more of these channels causes a distorted perception of thermal stimuli, which may result in thermal dysaesthesias, pain or improper thermal homeostasis.
Genetic factors influencing thermosensation. Reduced acuity of thermosensation and/or thermoregulation is among the symptoms of various pleiotropic disorders — including certain hereditary forms of neuropathies, ataxia and congenital insensitivity to pain — all of which are associated with more generalized sensory disturbances110. Even in healthy humans, there is substantial interindividual variation in thermosensitivity. Multiple factors, including age, sex, body-mass index, prior temperature experience and psychological state, are known to influence an individual's sensitivity and response to cold or heat as well as the subjective experience of pain evoked by a noxious thermal stimulus111,112. In addition, there is evidence for a genetic influence on temperature sensitivity, such as the strong variation in pain response to noxious temperatures between ethnic groups97. Studies investigating whether genetic variants in the genes encoding temperature-sensitive ion channels can contribute to this interindividual variability have reported that variations in the gene encoding TRPA1 account for a small part of the variation in cold-withdrawal latency in healthy subjects113 and that variants in the genes encoding TRPA1 and TRPV1 strongly correlate with thermosensory abnormalities in individuals with neuropathic pain114. The full consequences of these mutations on channel expression and function remain to be explored.
A more striking example of genetic variation in a temperature-sensitive TRP channel is provided by the rare autosomal dominant disease familial episodic pain syndrome (FEPS), which has been characterized in a single Colombian family65. The FEPS-associated mutant TRPA1 channel exhibits a gain of function, including an increased sensitivity to cold and chemical agonists. Individuals with FEPS show normal thresholds for cold- or heat-evoked pain but experience episodes of debilitating upper-body pain that can be triggered by fasting or cold stimuli, such as swimming in cold water, and terminated by eating and warming65.
Acquired thermal hypersensitivity. Several acquired pathologies are characterized by large shifts in temperature sensitivity. For instance, inflamed or wounded tissue typically exhibits hypersensitivity to both mechanical and thermal stimuli7,9. Thermal hypersensitivity is characterized by lower thresholds for both cold- and heat-evoked pain (thermal allodynia) as well as by the exacerbation of pain responses to noxious thermal stimuli (thermal hyperalgesia)9. Whereas hyperalgesia and allodynia have a useful purpose — namely, to warn and protect already-injured tissue from further damage — hypersensitivity can become chronic and debilitating. Both peripheral sensitization, which occurs when local mediators alter the function and sensitivity of thermal sensors at the sensory nerve ending, and phenotypic switching, which involves changes in gene expression owing to pathological status, have been shown to contribute to hypersensitivity101. Central-sensitization mechanisms, whereby hyperalgesia and allodynia result from enhanced processing of thermal input from the somatosensory system in the CNS, have been discussed elsewhere9,115.
A large body of evidence links inflammatory heat hyperalgesia to TRPV1 activity. Initial studies on TRPV1-deficient mice showed that these animals fail to develop heat hyperalgesia in response to experimental local inflammation induced by injection of complete Freund's adjuvant (CFA) or carrageenan21,22. Inflamed tissue, including skin injured by ultraviolet B irradiation, releases various extracellular factors, including bradykinin, lipid mediators, ATP, nerve growth factor (NGF) and protons (collectively called the inflammatory soup), which can all sensitize or potentiate TRPV1-mediated heat responses116,117. Whereas protons can directly bind to the extracellular part of TRPV1, resulting in sensitization to heat118, other factors act through their cognate membrane receptors119, which subsequently activate diverse intracellular signalling pathways.
First, the heat-sensitizing effects of NGF and bradykinin have been proposed to reflect relief of tonic inhibition of TRPV1 by PtdIns(4,5)P2, as the binding of NGF or bradykinin to their respective receptors leads to the activation of the PtdIns(4,5)P2-hydrolysing enzyme phospholipase C (PLC)119,120. Although the inhibitory effect of PtdIns(4,5)P2 on TRPV1 activity was recently confirmed in reconstitution experiments in artificial lipid bilayers99, several other studies have indicated that PtdIns(4,5)P2 can also potentiate TRPV1 activity and that its breakdown correlates with channel desensitization121,122. Second, diacylglycerol — which is formed upon PLC-mediated hydrolysis of PtdIns(4,5)P2 — activates protein kinase C, which in turn phosphorylates TRPV1 on multiple sites, leading to the sensitization of the channel to heat123,124. Similar sensitization is observed following TRPV1 phosphorylation by other serine/threonine kinases, including protein kinase A and Ca2+/calmodulin-dependent protein kinase II125,126. Third, there is evidence that, within minutes, receptor activation can increase the number of functional TRPV1 channels in the plasma membrane. In the F-11 cell line, which mimics rat DRG neurons, activation of the NGF receptor TRKA (also known as NTRK1) promotes the exocytotic insertion of TRPV1 through a PLC-independent mechanism that involves phosphatidylinositol 3-kinase121. In addition, several chronic painful pathologies — including chronic inflammation, nerve injury, diabetic neuropathy and bone cancer — can lead to increased expression of TRPV1 (Refs 127,128,129). Indeed, there is evidence for both transcriptional and post-transcriptional regulation of TRPV1 expression under these conditions, which can result in prolonged heat hypersensitivity127,128,130.
In addition to TRPV1, several other temperature-sensitive channels have been linked to heat hyperalgesia. Most strikingly, similarly to TRPV1-deficient mice, TRPM3-null mice fail to develop heat hyperalgesia in inflamed tissue after injection of CFA50. In addition, mice that lack expression of ANO1 in DRG neurons show reduced heat hyperalgesia under conditions of inflammation or nerve injury77,78, whereas TREK1-deficient mice exhibit exacerbated heat hyperalgesia85. Although it remains to be established how the activity and/or expression of these channels is modulated during inflammation or other pathological conditions, it is becoming clear that TRPV1 should no longer be regarded as the sole mediator of heat hyperalgesia.
Cold allodynia and cold hyperalgesia are severe dysaesthesias that are common following chemotherapy or are associated with conditions such as diabetes mellitus, herpes virus infections, inflammation and nerve compression9,131. Recent research has highlighted the role of temperature-sensitive ion channels in the development of cold hypersensitivity. TRPM8 expression and activity are augmented in rat DRG neurons affected by nerve injury, leading to increases in both the proportion of cold-sensitive neurons and their responsiveness to cold or menthol131. Moreover, genetic disruption or pharmacological inhibition of TRPM8 was found to strongly reduce cold hypersensitivity caused by inflammation or nerve constriction57,132. The mechanisms that modulate TRPM8 expression and function under pathophysiological conditions are not fully understood. Several studies provide in vitro evidence that TRPM8 activity is reduced after activation of PLC-coupled receptors, an effect that has been attributed to either depletion of PtdIns(4,5)P2 or activation of protein kinase C133,134,135,136. Surprisingly, however, one study provided evidence that inhibition of TRPM8 function following the activation of bradykinin and histamine receptors was independent of PLC, PtdIns(4,5)P2 depletion and protein kinase C and was instead mediated though direct binding of Gαq to the channel137.
There is evidence that TRPA1 is involved in the development of cold hypersensitivity following nerve injury or inflammation, but whether this is due to increased expression or sensitivity of TRPA1 remains controversial74,138,139. TRPA1 also has a crucial role in the cold allodynia that occurs after intoxication with ciguatoxins, a class of fat-soluble compounds that are produced by microalgae and that accumulate in large tropical fish140. These toxins shift the voltage dependence of Nav channels in TRPA1-expressing cold-activated nociceptors such that these neurons fire action potentials at non-noxious cool temperatures, resulting in severe pain141. Finally, TRPA1 is involved in the development of cold hypersensitivity associated with the use of anticancer drugs such as paclitaxel and oxaliplatin142,143,144. In addition to sensitizing TRPA1, oxaliplatin has been shown to reduce the expression of the temperature-sensitive K2P channels TREK1 and TRAAK, and to increase the expression of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, thus greatly increasing the excitability of cold-sensitive sensory neurons145.
TRP channels as therapeutic targets
An estimated one in every five adults experiences moderate to severe chronic pain, and about one half of these individuals report inadequate pain control146. The elucidation of the essential role of temperature-sensitive TRP channels in various forms of acute and chronic pain has created the opportunity to investigate their potential as molecular targets for novel, more specific and/or effective analgesic drugs147.
Inspired by the finding that TRPV1-deficient mice are generally healthy but fail to develop thermal hyperalgesia, intensive efforts have been made to develop TRPV1 antagonists as novel analgesic drugs32. This has resulted in multiple classes of highly potent and selective compounds that effectively inhibit TRPV1 activity both in vitro and in vivo32. Several of these antagonists have shown encouraging analgesic effects in animal models of inflammatory, neuropathic, bone-cancer and post-operative pain148,149,150, as well as in some limited studies of post-operative pain in humans33,151. However, the first generation of TRPV1 antagonists was associated with mild to severe hyperthermia (up to 40 °C) in humans and other animals33,148,151,152. This hyperthermia is a general, on-target effect, as it is observed in rodents, dogs, primates and humans but not in TRPV1-deficient mice153,154,155. Mechanistically, inhibition of TRPV1 in the peripheral sensory neurons that innervate the skin and viscera causes the thermoregulatory centres in the brain to underestimate the actual core and environmental temperatures, leading to the unnecessary activation of thermoregulatory processes such as skin vasoconstriction and thermogenesis that are aimed at increasing body temperature154. In line herewith, humans dosed with TRPV1 antagonists regularly describe feeling cold and exhibit visible shivering before the onset of hyperthermia155. This unwanted hyperthermia has resulted in the termination of several drug development programmes.
More recent evidence suggests that modality-specific antagonists, which inhibit TRPV1 activity induced by heat or capsaicin but not by protons, do not markedly affect body temperature156. However, whether such compounds are equally effective analgesics remains to be established. As an alternative, compounds that inhibit sensitization of TRPV1 — for instance, by disrupting its interaction with the scaffolding protein AKAP79 (A-kinase anchor protein 79) — have shown analgesic effects in inflammatory pain in mice without causing hyperthermia157. Interestingly, the hyperthermic effects of TRPV1 antagonists are mirrored by the hypothermia that is induced by TRPV1 agonists such as capsaicin, which fool the body into believing that it is hot, resulting in thermoregulatory responses such as cutaneous vasodilation and sweating that are aimed at increasing body-heat loss. It has been proposed that mild, TRPV1 agonist-induced hypothermia could be of therapeutic use in, for example, reducing neurological injury in survivors of cardiac arrest158,159.
TRPM8-activating stimuli, including cold temperatures or natural products such as menthol or eucalyptol, have long been used for topical pain relief, and recent experiments using TRPM8-deficient mice confirm that the analgesic effects of such treatments are fully dependent on TRPM8 activation58,160. These findings suggest that TRPM8-expressing, non-nociceptive sensory neurons can exert inhibitory control over nociceptive signalling160. More recently, TRPM8 antagonists have been developed as potential drugs to treat cold hypersensitivity and cold-related pain. The first results in rodent models confirm that TRPM8 is a promising target to treat cold hyperalgesia associated with inflammation and peripheral nerve injury but also show that, in contrast to the hyperthermic effects of TRPV1 antagonists, systemic inhibition of TRPM8 causes mild and transient hypothermia132,161. TRPM8 antagonists cause a reduction in the activity of cool-sensitive sensory neurons innervating the skin, which leads to an overestimation of the environmental temperature, resulting in the inhibition of cold-avoidance behaviour, cold-induced cutaneous vasoconstriction and non-shivering thermogenesis161. The inverse is seen with TRPM8 agonists such as icilin or menthol, which provoke warm-seeking behaviour and shivering, leading to hyperthermia132,162.
Thus, whereas TRPV1 and TRPM8 antagonists have demonstrated their effectiveness in reducing thermal hypersensitivity, they have also revealed that their target channels have a central role in thermal homeostasis, acting as molecular thermometers that provide essential information about the environmental and core body temperature to the body's thermostat. This may put constraints on these antagonists' future therapeutic use, especially under conditions in which thermal homeostasis is already compromised, such as fever or severe climatological circumstances. Pharmacological targeting of TRPM3 and TRPA1, which detect more extreme temperatures (Fig. 2), may provide a safer alternative. Initial preclinical tests indeed suggest that antagonists of these channels can inhibit certain forms of thermal pain without affecting core body temperature51,163.
Conclusion and perspectives
Over the past two decades, we have witnessed the identification of various ion channels as molecular thermometers that translate environmental and internal thermal cues into electrical activity in the somatosensory system. Advanced approaches towards the dissection of the molecular and cellular logic of mammalian thermosensation have provided genetic and pharmacological tools to modulate thermosensitivity in vivo and have opened the way to the development of novel drugs to treat various forms of acute or chronic pain. Nevertheless, it should be pointed out that our understanding of the fundamentals of thermosensation is still fragmentary for the following reasons.
First, whereas for some time it was believed that temperature-sensitive TRP channels could fully explain the ability of the mammalian somatosensory system to detect and discriminate temperatures covering the 0–60 °C range, more recent research calls the universal role of TRP channels into question. In particular, responses to noxious cold and noxious heat are partially blunted, but by no means abrogated, following combined elimination of the established cold- and heat-sensitive TRP channels50,73. This indicates the existence of multiple redundant molecular mechanisms to detect these potentially life-threatening stimuli, including thermosensitive elements that remain to be identified. In recent years, several novel classes of ion channels (for example, the mechanosensitive PIEZOs164, the channels related to the Ca2+-activated Cl− channel ANO1 (Refs 165,166,167) and the store-operated ORAI channels168,169) have been identified using powerful techniques such as expression profiling, genome-wide or rational RNAi screens, expression cloning and bioinformatics. Similar approaches may lead to the discovery of additional novel classes of ion channels representing the currently missing links in thermosensation.
Second, despite numerous structure–function studies, we are still far from understanding the mechanisms that underlie the striking temperature sensitivity of the several ion channels that are involved in thermosensation. Well-studied activating mechanisms for ion channels, such as transmembrane voltage changes or ligand binding, act on clearly delineated channel domains. By contrast, temperature seems to be particularly intractable, because it affects all atoms in the channel and its surroundings, and alterations in various parts of temperature-sensitive channels affect temperature dependence. Therefore, a full understanding of the basis of the steep thermosensitivity in ion channels will most probably require atomic-detail structural information of the conformational changes that occur during gating. Although crystallization of entire TRP channels has proven to be extremely difficult, recent advances in structure determination using single-particle cryo-electron microscopy may hold the potential to obtain detailed structures of different channel conformations during temperature-dependent gating27,109.
Last, initial enthusiasm for the use of antagonists of the prototype heat sensor TRPV1 as potent new analgesics has been tempered by the finding that such compounds can dangerously disturb normal temperature homeostasis32. This unforeseen consequence of tampering with temperature-sensitive channels illustrates our limited knowledge of the processes that steer thermoregulation in mammals but has also fuelled investigations that have revealed important aspects of the neuronal pathways that translate thermosensory information into thermoregulatory responses17,18. Furthermore, powerful tools that will undoubtedly expand our knowledge of the physiology of thermoregulation further have been developed, including genetic techniques that enable controlled elimination of specific thermosensitive neurons in vivo52 and genetically encoded sensors that enable visualization of thermogenesis with subcellular resolution170. A more comprehensive understanding of the physiology of thermoregulation, in combination with deeper insights into the molecular and submolecular aspects of thermosensation in normal and pathophysiological conditions, may provide the basis for the development of more efficacious and safer therapies that target temperature-sensitive channels to treat thermal hypersensitivity and pain.
Accession codes
References
Damann, N., Voets, T. & Nilius, B. TRPs in our senses. Curr. Biol. 18, R880–R889 (2008).
Tattersall, G. J. et al. Coping with thermal challenges: physiological adaptations to environmental temperatures. Compr. Physiol. 2, 2151–2202 (2012).
Bouchama, A. & Knochel, J. P. Heat stroke. N. Engl. J. Med. 346, 1978–1988 (2002).
Brown, D. J., Brugger, H., Boyd, J. & Paal, P. Accidental hypothermia. N. Engl. J. Med. 367, 1930–1938 (2012).
Delmas, P., Hao, J. & Rodat-Despoix, L. Molecular mechanisms of mechanotransduction in mammalian sensory neurons. Nature Rev. Neurosci. 12, 139–153 (2011).
Kandel, E. R., Schwartz, J. H., Jessell, T. M., Siegelbaum, S. A. & Hudspeth, A. J. (eds) Principles of Neural Science (McGraw-Hill Companies, 2013).
Dubin, A. E. & Patapoutian, A. Nociceptors: the sensors of the pain pathway. J. Clin. Invest. 120, 3760–3772 (2010).
Romanovsky, A. A. Thermoregulation: some concepts have changed. Functional architecture of the thermoregulatory system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R37–R46 (2007).
Basbaum, A. I., Bautista, D. M., Scherrer, G. & Julius, D. Cellular and molecular mechanisms of pain. Cell 139, 267–284 (2009).
Campero, M. & Bostock, H. Unmyelinated afferents in human skin and their responsiveness to low temperature. Neurosci. Lett. 470, 188–192 (2010).
Hensel, H. & Iggo, A. Analysis of cutaneous warm and cold fibres in primates. Pflugers Arch. 329, 1–8 (1971).
Zimmermann, K. et al. Phenotyping sensory nerve endings in vitro in the mouse. Nature Protoc. 4, 174–196 (2009).
Nilius, B. & Voets, T. Neurophysiology: channelling cold reception. Nature 448, 147–148 (2007).
Frolich, M. A., Bolding, M. S., Cutter, G. R., Ness, T. J. & Zhang, K. Temporal characteristics of cold pain perception. Neurosci. Lett. 480, 12–15 (2010).
Torebjork, H. E., LaMotte, R. H. & Robinson, C. J. Peripheral neural correlates of magnitude of cutaneous pain and hyperalgesia: simultaneous recordings in humans of sensory judgments of pain and evoked responses in nociceptors with C-fibers. J. Neurophysiol. 51, 325–339 (1984).
Piomelli, D. & Sasso, O. Peripheral gating of pain signals by endogenous lipid mediators. Nature Neurosci. 17, 164–174 (2014).
Romanovsky, A. A. et al. The transient receptor potential vanilloid-1 channel in thermoregulation: a thermosensor it is not. Pharmacol. Rev. 61, 228–261 (2009).
Nakamura, K. Central circuitries for body temperature regulation and fever. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1207–R1228 (2011).
Clapham, J. C. Central control of thermogenesis. Neuropharmacology 63, 111–123 (2012).
Voets, T., Talavera, K., Owsianik, G. & Nilius, B. Sensing with TRP channels. Nature Chem. Biol. 1, 85–92 (2005).
Caterina, M. J. et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313 (2000).
Davis, J. B. et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405, 183–187 (2000). The first description (together with reference 21) of TRPV1-deficient mice, establishing the channel's role in heat sensation and heat hyperalgesia.
Karashima, Y. et al. TRPA1 acts as a cold sensor in vitro and in vivo. Proc. Natl Acad. Sci. USA 106, 1273–1278 (2009).
Moqrich, A. et al. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science 307, 1468–1472 (2005).
Gees, M., Owsianik, G., Nilius, B. & Voets, T. TRP channels. Compr. Physiol. 2, 563–608 (2012).
Caterina, M. J. et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824 (1997). A groundbreaking study identifying the capsaicin receptor (now known as TRPV1) as the first heat-activated cation channel in the somatosensory system.
Liao, M., Cao, E., Julius, D. & Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 (2013). This paper reports the first high-resolution structure of a TRP channel involved in thermosensation.
Tominaga, M. et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543 (1998).
Zygmunt, P. M. et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400, 452–457 (1999).
Hwang, S. W. et al. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc. Natl Acad. Sci. USA 97, 6155–6160 (2000).
Vriens, J., Nilius, B. & Vennekens, R. Herbal compounds and toxins modulating TRP channels. Curr. Neuropharmacol. 6, 79–96 (2008).
Szallasi, A. & Sheta, M. Targeting TRPV1 for pain relief: limits, losers and laurels. Expert Opin. Investig. Drugs 21, 1351–1369 (2012).
Schaffler, K. et al. An oral TRPV1 antagonist attenuates laser radiant-heat-evoked potentials and pain ratings from UVB-inflamed and normal skin. Br. J. Clin. Pharmacol. 75, 404–414 (2013).
Caterina, M. J., Rosen, T. A., Tominaga, M., Brake, A. J. & Julius, D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398, 436–441 (1999).
Park, U. et al. TRP vanilloid 2 knock-out mice are susceptible to perinatal lethality but display normal thermal and mechanical nociception. J. Neurosci. 31, 11425–11436 (2011).
Guler, A. D. et al. Heat-evoked activation of the ion channel, TRPV4. J. Neurosci. 22, 6408–6414 (2002).
Peier, A. M. et al. A heat-sensitive TRP channel expressed in keratinocytes. Science 296, 2046–2049 (2002).
Smith, G. D. et al. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 418, 186–190 (2002).
Watanabe, H. et al. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J. Biol. Chem. 277, 47044–47051 (2002).
Xu, H. et al. TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature 418, 181–186 (2002).
Chung, M. K., Lee, H. & Caterina, M. J. Warm temperatures activate TRPV4 in mouse 308 keratinocytes. J. Biol. Chem. 278, 32037–32046 (2003).
Vandewauw, I., Owsianik, G. & Voets, T. Systematic and quantitative mRNA expression analysis of TRP channel genes at the single trigeminal and dorsal root ganglion level in mouse. BMC Neurosci. 14, 21 (2013).
Mandadi, S. et al. TRPV3 in keratinocytes transmits temperature information to sensory neurons via ATP. Pflugers Arch. 458, 1093–1102 (2009).
Miyamoto, T., Petrus, M. J., Dubin, A. E. & Patapoutian, A. TRPV3 regulates nitric oxide synthase-independent nitric oxide synthesis in the skin. Nature Commun. 2, 369 (2011).
Mihara, H., Boudaka, A., Sugiyama, T., Moriyama, Y. & Tominaga, M. Transient receptor potential vanilloid 4 (TRPV4)-dependent calcium influx and ATP release in mouse oesophageal keratinocytes. J. Physiol. 589, 3471–3482 (2011).
Lee, H., Iida, T., Mizuno, A., Suzuki, M. & Caterina, M. J. Altered thermal selection behavior in mice lacking transient receptor potential vanilloid 4. J. Neurosci. 25, 1304–1310 (2005).
Huang, S. M., Li, X., Yu, Y., Wang, J. & Caterina, M. J. TRPV3 and TRPV4 ion channels are not major contributors to mouse heat sensation. Mol. Pain 7, 37 (2011).
Lariviere, W. R., Chesler, E. J. & Mogil, J. S. Transgenic studies of pain and analgesia: mutation or background genotype? J. Pharmacol. Exp. Ther. 297, 467–473 (2001).
Wagner, T. F. et al. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic β cells. Nature Cell Biol. 10, 1421–1430 (2008).
Vriens, J. et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 70, 482–494 (2011). This study demonstrates that TRPM3 functions as a receptor for noxious heat in the somatosensory system and is essential for the development of inflammatory heat hyperalgesia.
Straub, I. et al. Flavanones that selectively inhibit TRPM3 attenuate thermal nociception in vivo. Mol. Pharmacol. 84, 736–750 (2013).
Pogorzala, L. A., Mishra, S. K. & Hoon, M. A. The cellular code for mammalian thermosensation. J. Neurosci. 33, 5533–5541 (2013).
Mishra, S. K. & Hoon, M. A. Ablation of TrpV1 neurons reveals their selective role in thermal pain sensation. Mol. Cell. Neurosci. 43, 157–163 (2010).
McKemy, D. D., Neuhausser, W. M. & Julius, D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416, 52–58 (2002).
Peier, A. M. et al. A TRP channel that senses cold stimuli and menthol. Cell 108, 705–715 (2002). This study (together with reference 54) identifies TRPM8 as a sensor for cold and menthol.
Bautista, D. M. et al. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 448, 204–208 (2007).
Colburn, R. W. et al. Attenuated cold sensitivity in TRPM8 null mice. Neuron 54, 379–386 (2007).
Dhaka, A. et al. TRPM8 is required for cold sensation in mice. Neuron 54, 371–378 (2007). The first description (together with references 56 and 57) of TRPM8-deficient mice, establishing the channel's role in non-noxious cold sensation.
Parra, A. et al. Ocular surface wetness is regulated by TRPM8-dependent cold thermoreceptors of the cornea. Nature Med. 16, 1396–1399 (2010). This study shows that the cold sensor TRPM8 can act as a wetness sensor in the eye, thereby regulating basal tearing.
Knowlton, W. M. et al. A sensory-labeled line for cold: TRPM8-expressing sensory neurons define the cellular basis for cold, cold pain, and cooling-mediated analgesia. J. Neurosci. 33, 2837–2848 (2013).
Story, G. M. et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112, 819–829 (2003).
Bandell, M. et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41, 849–857 (2004).
Sawada, Y., Hosokawa, H., Hori, A., Matsumura, K. & Kobayashi, S. Cold sensitivity of recombinant TRPA1 channels. Brain Res. 1160, 39–46 (2007).
Fajardo, O., Meseguer, V., Belmonte, C. & Viana, F. TRPA1 channels mediate cold temperature sensing in mammalian vagal sensory neurons: pharmacological and genetic evidence. J. Neurosci. 28, 7863–7875 (2008).
Kremeyer, B. et al. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron 66, 671–680 (2010). The first example of an inherited disease, FEPS, caused by a gain-of-function mutation in the gene encoding a channel implicated in thermosensation.
Jordt, S. E. et al. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427, 260–265 (2004).
Talavera, K. et al. Nicotine activates the chemosensory cation channel TRPA1. Nature Neurosci. 12, 1293–1299 (2009).
Kwan, K. Y. et al. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron 50, 277–289 (2006).
Gentry, C., Stoakley, N., Andersson, D. A. & Bevan, S. The roles of iPLA2, TRPM8 and TRPA1 in chemically induced cold hypersensitivity. Mol. Pain 6, 4 (2010).
Zurborg, S., Yurgionas, B., Jira, J. A., Caspani, O. & Heppenstall, P. A. Direct activation of the ion channel TRPA1 by Ca2+. Nature Neurosci. 10, 277–279 (2007).
Chen, J. et al. Species differences and molecular determinant of TRPA1 cold sensitivity. Nature Commun. 4, 2501 (2013).
Bautista, D. M. et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124, 1269–1282 (2006).
Knowlton, W. M., Bifolck-Fisher, A., Bautista, D. M. & McKemy, D. D. TRPM8, but not TRPA1, is required for neural and behavioral responses to acute noxious cold temperatures and cold-mimetics in vivo. Pain 150, 340–350 (2010).
del Camino, D. et al. TRPA1 contributes to cold hypersensitivity. J. Neurosci. 30, 15165–15174 (2010).
Zimmermann, K. et al. Transient receptor potential cation channel, subfamily C, member 5 (TRPC5) is a cold-transducer in the peripheral nervous system. Proc. Natl Acad. Sci. USA 108, 18114–18119 (2011).
Huang, F., Wong, X. & Jan, L. Y. International Union of Basic and Clinical Pharmacology. LXXXV: calcium-activated chloride channels. Pharmacol. Rev. 64, 1–15 (2012).
Cho, H. et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nature Neurosci. 15, 1015–1021 (2012).
Lee, B. et al. Anoctamin 1 contributes to inflammatory and nerve-injury induced hypersensitivity. Mol. Pain 10, 5 (2014).
Cahalan, M. D. STIMulating store-operated Ca2+ entry. Nature Cell Biol. 11, 669–677 (2009).
Xiao, B., Coste, B., Mathur, J. & Patapoutian, A. Temperature-dependent STIM1 activation induces Ca2+ influx and modulates gene expression. Nature Chem. Biol. 7, 351–358 (2011).
Viana, F., de la Pena, E. & Belmonte, C. Specificity of cold thermotransduction is determined by differential ionic channel expression. Nature Neurosci. 5, 254–260 (2002).
Honore, E. The neuronal background K2P channels: focus on TREK1. Nature Rev. Neurosci. 8, 251–261 (2007).
Maingret, F. et al. TREK-1 is a heat-activated background K+ channel. EMBO J. 19, 2483–2491 (2000).
Kang, D., Choe, C. & Kim, D. Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J. Physiol. 564, 103–116 (2005).
Alloui, A. et al. TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 25, 2368–2376 (2006).
Noel, J. et al. The mechano-activated K+ channels TRAAK and TREK-1 control both warm and cold perception. EMBO J. 28, 1308–1318 (2009).
Catterall, W. A. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J. Physiol. 590, 2577–2589 (2012).
Zimmermann, K. et al. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature 447, 855–858 (2007). This study demonstrates that the voltage-gated Na+ channel Nav1.8 remains active at noxiously low temperatures and is crucial for the transmission of painful stimuli in the cold.
Abrahamsen, B. et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321, 702–705 (2008).
Heimburg, T. Mechanical aspects of membrane thermodynamics. Estimation of the mechanical properties of lipid membranes close to the chain melting transition from calorimetry. Biochim. Biophys. Acta 1415, 147–162 (1998).
Yudin, Y., Lukacs, V., Cao, C. & Rohacs, T. Decrease in phosphatidylinositol 4,5-bisphosphate levels mediates desensitization of the cold sensor TRPM8 channels. J. Physiol. 589, 6007–6027 (2011).
Levitan, I. B. Signaling protein complexes associated with neuronal ion channels. Nature Neurosci. 9, 305–310 (2006).
Walsh, K. B. & Kass, R. S. Regulation of a heart potassium channel by protein kinase A and C. Science 242, 67–69 (1988).
Wang, Y. et al. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330, 105–109 (2010).
Park, C. Y., Shcheglovitov, A. & Dolmetsch, R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330, 101–105 (2010).
Yuan, J. P., Zeng, W., Huang, G. N., Worley, P. F. & Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nature Cell Biol. 9, 636–645 (2007).
DeHaven, W. I. et al. TRPC channels function independently of STIM1 and Orai1. J. Physiol. 587, 2275–2298 (2009).
Zakharian, E., Cao, C. & Rohacs, T. Gating of transient receptor potential melastatin 8 (TRPM8) channels activated by cold and chemical agonists in planar lipid bilayers. J. Neurosci. 30, 12526–12534 (2010).
Cao, E., Cordero-Morales, J. F., Liu, B., Qin, F. & Julius, D. TRPV1 channels are intrinsically heat sensitive and negatively regulated by phosphoinositide lipids. Neuron 77, 667–679 (2013).
Talavera, K. et al. Heat activation of TRPM5 underlies thermal sensitivity of sweet taste. Nature 438, 1022–1025 (2005).
Voets, T. et al. The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature 430, 748–754 (2004). This study provides a unifying thermodynamic model to explain the temperature sensitivity of TRP channels.
Jackson, M. B. Molecular and Cellular Biophysics (Cambridge Univ. Press, 2006).
Yao, J., Liu, B. & Qin, F. Kinetic and energetic analysis of thermally activated TRPV1 channels. Biophys. J. 99, 1743–1753 (2010).
Brauchi, S., Orta, G., Salazar, M., Rosenmann, E. & Latorre, R. A hot-sensing cold receptor: C-terminal domain determines thermosensation in transient receptor potential channels. J. Neurosci. 26, 4835–4840 (2006).
Latorre, R., Brauchi, S., Orta, G., Zaelzer, C. & Vargas, G. ThermoTRP channels as modular proteins with allosteric gating. Cell Calcium 42, 427–438 (2007).
Grandl, J. et al. Pore region of TRPV3 ion channel is specifically required for heat activation. Nature Neurosci. 11, 1007–1013 (2008).
Cordero-Morales, J. F., Gracheva, E. O. & Julius, D. Cytoplasmic ankyrin repeats of transient receptor potential A1 (TRPA1) dictate sensitivity to thermal and chemical stimuli. Proc. Natl Acad. Sci. USA 108, E1184–E1191 (2011).
Voets, T. Quantifying and modeling the temperature-dependent gating of TRP channels. Rev. Physiol. Biochem. Pharmacol. 162, 91–119 (2012).
Cao, E., Liao, M., Cheng, Y. & Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 (2013).
Tan, P. L. & Katsanis, N. Thermosensory and mechanosensory perception in human genetic disease. Hum. Mol. Genet. 18, R146–R155 (2009).
Cruz-Almeida, Y., Riley, J. L. & Fillingim, R. B. Experimental pain phenotype profiles in a racially and ethnically diverse sample of healthy adults. Pain Med. 14, 1708–1718 (2013).
Hollins, M. Somesthetic senses. Annu. Rev. Psychol. 61, 243–271 (2010).
Kim, H., Mittal, D. P., Iadarola, M. J. & Dionne, R. A. Genetic predictors for acute experimental cold and heat pain sensitivity in humans. J. Med. Genet. 43, e40 (2006).
Binder, A. et al. Transient receptor potential channel polymorphisms are associated with the somatosensory function in neuropathic pain patients. PLoS ONE 6, e17387 (2011).
Woolf, C. J. Central sensitization: implications for the diagnosis and treatment of pain. Pain 152, S2–S15 (2011).
Julius, D. & Basbaum, A. I. Molecular mechanisms of nociception. Nature 413, 203–210 (2001).
Sisignano, M. et al. Synthesis of lipid mediators during UVB-induced inflammatory hyperalgesia in rats and mice. PLoS ONE 8, e81228 (2013).
Jordt, S. E., Tominaga, M. & Julius, D. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl Acad. Sci. USA 97, 8134–8139 (2000).
Chuang, H. H. et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411, 957–962 (2001).
Prescott, E. D. & Julius, D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 300, 1284–1288 (2003).
Stein, A. T., Ufret-Vincenty, C. A., Hua, L., Santana, L. F. & Gordon, S. E. Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J. Gen. Physiol. 128, 509–522 (2006).
Yao, J. & Qin, F. Interaction with phosphoinositides confers adaptation onto the TRPV1 pain receptor. PLoS Biol. 7, e46 (2009).
Premkumar, L. S. & Ahern, G. P. Induction of vanilloid receptor channel activity by protein kinase C. Nature 408, 985–990 (2000).
Bhave, G. et al. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1). Proc. Natl Acad. Sci. USA 100, 12480–12485 (2003).
Bhave, G. et al. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 35, 721–731 (2002).
Bonnington, J. K. & McNaughton, P. A. Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J. Physiol. 551, 433–446 (2003).
Hudson, L. J. et al. VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur. J. Neurosci. 13, 2105–2114 (2001).
Zakir, H. M. et al. Expression of TRPV1 channels after nerve injury provides an essential delivery tool for neuropathic pain attenuation. PLoS ONE 7, e44023 (2012).
Hong, S. & Wiley, J. W. Early painful diabetic neuropathy is associated with differential changes in the expression and function of vanilloid receptor 1. J. Biol. Chem. 280, 618–627 (2005).
Bishnoi, M., Bosgraaf, C. A., Abooj, M., Zhong, L. & Premkumar, L. S. Streptozotocin-induced early thermal hyperalgesia is independent of glycemic state of rats: role of transient receptor potential vanilloid 1 (TRPV1) and inflammatory mediators. Mol. Pain 7, 52 (2011).
Xing, H., Chen, M., Ling, J., Tan, W. & Gu, J. G. TRPM8 mechanism of cold allodynia after chronic nerve injury. J. Neurosci. 27, 13680–13690 (2007).
Knowlton, W. M., Daniels, R. L., Palkar, R., McCoy, D. D. & McKemy, D. D. Pharmacological blockade of TRPM8 ion channels alters cold and cold pain responses in mice. PLoS ONE 6, e25894 (2011).
Rohacs, T., Lopes, C. M., Michailidis, I. & Logothetis, D. E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nature Neurosci. 8, 626–634 (2005).
Fujita, F., Uchida, K., Takaishi, M., Sokabe, T. & Tominaga, M. Ambient temperature affects the temperature threshold for TRPM8 activation through interaction of phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 33, 6154–6159 (2013).
Liu, B. & Qin, F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25, 1674–1681 (2005).
Premkumar, L. S., Raisinghani, M., Pingle, S. C., Long, C. & Pimentel, F. Downregulation of transient receptor potential melastatin 8 by protein kinase C-mediated dephosphorylation. J. Neurosci. 25, 11322–11329 (2005).
Zhang, X. et al. Direct inhibition of the cold-activated TRPM8 ion channel by Gαq . Nature Cell Biol. 14, 851–858 (2012).
Obata, K. et al. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J. Clin. Invest. 115, 2393–2401 (2005).
Katsura, H. et al. Antisense knock down of TRPA1, but not TRPM8, alleviates cold hyperalgesia after spinal nerve ligation in rats. Exp. Neurol. 200, 112–123 (2006).
Voets, T. TRP channel blamed for burning cold after a tropical fish meal. EMBO J. 31, 3785–3787 (2012).
Vetter, I. et al. Ciguatoxins activate specific cold pain pathways to elicit burning pain from cooling. EMBO J. 31, 3795–3808 (2012).
Zhao, M. et al. Acute cold hypersensitivity characteristically induced by oxaliplatin is caused by the enhanced responsiveness of TRPA1 in mice. Mol. Pain 8, 55 (2012).
Materazzi, S. et al. TRPA1 and TRPV4 mediate paclitaxel-induced peripheral neuropathy in mice via a glutathione-sensitive mechanism. Pflugers Arch. 463, 561–569 (2012).
Nassini, R. et al. Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 152, 1621–1631 (2011).
Descoeur, J. et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med. 3, 266–278 (2011).
Reid, K. J. et al. Epidemiology of chronic non-cancer pain in Europe: narrative review of prevalence, pain treatments and pain impact. Curr. Med. Res. Opin. 27, 449–462 (2011).
Moran, M. M., McAlexander, M. A., Biro, T. & Szallasi, A. Transient receptor potential channels as therapeutic targets. Nature Rev. Drug Discov. 10, 601–620 (2011).
Garcia-Martinez, C. et al. Attenuation of thermal nociception and hyperalgesia by VR1 blockers. Proc. Natl Acad. Sci. USA 99, 2374–2379 (2002).
Ghilardi, J. R. et al. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J. Neurosci. 25, 3126–3131 (2005).
Honore, P. et al. A-425619 [1-isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)-urea], a novel transient receptor potential type V1 receptor antagonist, relieves pathophysiological pain associated with inflammation and tissue injury in rats. J. Pharmacol. Exp. Ther. 314, 410–421 (2005).
Quiding, H. et al. TRPV1 antagonistic analgesic effect: a randomized study of AZD1386 in pain after third molar extraction. Pain 154, 808–812 (2013).
Gavva, N. R. et al. The vanilloid receptor TRPV1 is tonically activated in vivo and involved in body temperature regulation. J. Neurosci. 27, 3366–3374 (2007). This study establishes the role of TRPV1 in core body temperature regulation and shows that peripheral inhibition of TRPV1 using selective antagonists causes hyperthermia.
Steiner, A. A. et al. Nonthermal activation of transient receptor potential vanilloid-1 channels in abdominal viscera tonically inhibits autonomic cold-defense effectors. J. Neurosci. 27, 7459–7468 (2007).
Gavva, N. R. Body-temperature maintenance as the predominant function of the vanilloid receptor TRPV1. Trends Pharmacol. Sci. 29, 550–557 (2008).
Gavva, N. R. et al. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 136, 202–210 (2008).
Garami, A. et al. Contributions of different modes of TRPV1 activation to TRPV1 antagonist-induced hyperthermia. J. Neurosci. 30, 1435–1440 (2010).
Fischer, M. J., Btesh, J. & McNaughton, P. A. Disrupting sensitization of transient receptor potential vanilloid subtype 1 inhibits inflammatory hyperalgesia. J. Neurosci. 33, 7407–7414 (2013).
Fosgerau, K. et al. Drug-induced mild therapeutic hypothermia obtained by administration of a transient receptor potential vanilloid type 1 agonist. BMC Cardiovasc. Disord. 10, 51 (2010).
Cao, Z., Balasubramanian, A. & Marrelli, S. P. Pharmacologically induced hypothermia via TRPV1 channel agonism provides neuroprotection following ischemic stroke when initiated 90 min after reperfusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 306, R149–R156 (2014).
Liu, B. et al. TRPM8 is the principal mediator of menthol-induced analgesia of acute and inflammatory pain. Pain 154, 2169–2177 (2013).
Almeida, M. C. et al. Pharmacological blockade of the cold receptor TRPM8 attenuates autonomic and behavioral cold defenses and decreases deep body temperature. J. Neurosci. 32, 2086–2099 (2012). This study establishes the role of TRPM8 in core body temperature regulation and shows that peripheral blockade of TRPM8 inhibits involuntary and voluntary cold defence responses.
Tajino, K. et al. Application of menthol to the skin of whole trunk in mice induces autonomic and behavioral heat-gain responses. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R2128–R2135 (2007).
Chen, J. et al. Selective blockade of TRPA1 channel attenuates pathological pain without altering noxious cold sensation or body temperature regulation. Pain 152, 1165–1172 (2011).
Coste, B. et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330, 55–60 (2010).
Yang, Y. D. et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455, 1210–1215 (2008).
Caputo, A. et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322, 590–594 (2008).
Schroeder, B. C., Cheng, T., Jan, Y. N. & Jan, L. Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134, 1019–1029 (2008).
Vig, M. et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223 (2006).
Feske, S. et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185 (2006).
Kiyonaka, S. et al. Genetically encoded fluorescent thermosensors visualize subcellular thermoregulation in living cells. Nature Methods 10, 1232–1238 (2013).
Clapham, D. E. & Miller, C. A thermodynamic framework for understanding temperature sensing by transient receptor potential (TRP) channels. Proc. Natl Acad. Sci. USA 108, 19492–19497 (2011).
Schmidt, M., Dubin, A. E., Petrus, M. J., Earley, T. J. & Patapoutian, A. Nociceptive signals induce trafficking of TRPA1 to the plasma membrane. Neuron 64, 498–509 (2009).
Jordt, S. E. & Julius, D. Molecular basis for species-specific sensitivity to “hot” chili peppers. Cell 108, 421–430 (2002).
Bandell, M. et al. High-throughput random mutagenesis screen reveals TRPM8 residues specifically required for activation by menthol. Nature Neurosci. 9, 493–500 (2006).
Voets, T., Owsianik, G., Janssens, A., Talavera, K. & Nilius, B. TRPM8 voltage sensor mutants reveal a mechanism for integrating thermal and chemical stimuli. Nature Chem. Biol. 3, 174–182 (2007).
Janssens, A. & Voets, T. Ligand stoichiometry of the cold- and menthol-activated channel TRPM8. J. Physiol. 589, 4827–4835 (2011).
Macpherson, L. J. et al. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445, 541–545 (2007).
Hinman, A., Chuang, H. H., Bautista, D. M. & Julius, D. TRP channel activation by reversible covalent modification. Proc. Natl Acad. Sci. USA 103, 19564–19568 (2006).
Salazar, H. et al. A single N-terminal cysteine in TRPV1 determines activation by pungent compounds from onion and garlic. Nature Neurosci. 11, 255–261 (2008).
Karashima, Y. et al. Bimodal action of menthol on the transient receptor potential channel TRPA1. J. Neurosci. 27, 9874–9884 (2007).
McNamara, F. N., Randall, A. & Gunthorpe, M. J. Effects of piperine, the pungent component of black pepper, at the human vanilloid receptor (TRPV1). Br. J. Pharmacol. 144, 781–790 (2005).
Everaerts, W. et al. The capsaicin receptor TRPV1 is a crucial mediator of the noxious effects of mustard oil. Curr. Biol. 21, 316–321 (2011).
Macpherson, L. J. et al. The pungency of garlic: activation of TRPA1 and TRPV1 in response to allicin. Curr. Biol. 15, 929–934 (2005).
Yao, J., Liu, B. & Qin, F. Modular thermal sensors in temperature-gated transient receptor potential (TRP) channels. Proc. Natl Acad. Sci. USA 108, 11109–11114 (2011).
Yang, F., Cui, Y., Wang, K. & Zheng, J. Thermosensitive TRP channel pore turret is part of the temperature activation pathway. Proc. Natl Acad. Sci. USA 107, 7083–7088 (2010).
Grandl, J. et al. Temperature-induced opening of TRPV1 ion channel is stabilized by the pore domain. Nature Neurosci. 13, 708–714 (2010).
Valente, P. et al. Identification of molecular determinants of channel gating in the transient receptor potential box of vanilloid receptor I. FASEB J. 22, 3298–3309 (2008).
Brauchi, S. et al. Dissection of the components for PIP2 activation and thermosensation in TRP channels. Proc. Natl Acad. Sci. USA 104, 10246–10251 (2007).
Vlachova, V. et al. Functional role of C-terminal cytoplasmic tail of rat vanilloid receptor 1. J. Neurosci. 23, 1340–1350 (2003).
Wang, H., Schupp, M., Zurborg, S. & Heppenstall, P. A. Residues in the pore region of Drosophila transient receptor potential A1 dictate sensitivity to thermal stimuli. J. Physiol. 591, 185–201 (2013).
Acknowledgements
The authors thank all members of the Laboratory of Ion Channel Research for helpful discussions. This work was supported by grants from the Belgian Science Policy (IUAP P7/13 to T.V.), the Research Foundation - Flanders (G.0825.11 to T.V. and J.V.), the Research Council of the KU Leuven (PF-TRPLe to T.V.) and the Planckaert-De Waele fund (to J.V.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
DATABASES
Glossary
- Transient receptor potential channels
-
(TRP channels). A family of cation channels homologous to the product of the Drosophila melanogaster trp gene. Several mammalian members of this family function as molecular thermosensors.
- Aδ fibres
-
Sensory neurons with medium-diameter, thinly myelinated axons and a medium conduction velocity of between 2 and 30 metres per second.
- C fibres
-
Sensory neurons with small-diameter, non-myelinated axons and a low conduction velocity of between 0.5 and 2 metres per second.
- Adaptation
-
The reduction over time of the response of a neuron to a constant sensory stimulus.
- Q10 value
-
A dimensionless value to quantify the temperature dependence of a process. It is defined as the ratio between reaction rates or current amplitudes measured at two temperatures 10 degrees apart.
- Hyperalgesia
-
Increased sensitivity to a painful stimulus.
- Tetrodotoxin
-
(TTX). A toxin from puffer fish that potently blocks several types of voltage-gated Na+ channels.
- Enthalpy
-
A thermodynamic measure of the internal energy of a system. In a protein, formation of stabilizing intramolecular bonds causes a reduction in enthalpy.
- Entropy
-
A thermodynamic measure of the disorder of a system. It is determined by the number of different configurations (microstates) that correspond to a specific macrostate.
- Allodynia
-
Pain in response to a stimulus that does not normally evoke pain.
- Complete Freund's adjuvant
-
(CFA). An emulsion of inactivated and dried mycobacteria. When injected subcutaneously, it induces local inflammation and hypersensitivity to thermal and mechanical stimuli.
- Carrageenan
-
A sulphated polysaccharide extracted from red edible seaweeds. When injected subcutaneously, it induces local inflammation and hypersensitivity to thermal and mechanical stimuli.
Rights and permissions
About this article

Cite this article
Vriens, J., Nilius, B. & Voets, T. Peripheral thermosensation in mammals. Nat Rev Neurosci 15, 573–589 (2014). https://doi.org/10.1038/nrn3784
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrn3784
This article is cited by
-
Whole genome sequencing analysis of alpaca suggests TRPV3 as a candidate gene for the suri phenotype
BMC Genomics (2024)
-
The cellular coding of temperature in the mammalian cortex
Nature (2023)
-
Classic and exertional heatstroke
Nature Reviews Disease Primers (2022)
-
TRP channels: a journey towards a molecular understanding of pain
Nature Reviews Neuroscience (2022)
-
Real time monitoring of cold Ca2+ dependent transcription and its modulation by NCX inhibitors
Scientific Reports (2022)
