




Abstract
Chronically isolated neocortex develops chronic hyperexcitability and focal epileptogenesis in a period of days to weeks. The mechanisms operating in this model of post-traumatic epileptogenesis are not well understood. We hypothesized that the spontaneous burst discharges recorded in chronically isolated neocortex result from homeostatic plasticity (a mechanism generally assumed to stabilize neuronal activity) induced by low neuronal activity after deafferentation. To test this hypothesis we constructed computer models of neocortex incorporating a biologically based homeostatic plasticity rule that operates to maintain firing rates. After deafferentation, homeostatic upregulation of excitatory synapses on pyramidal cells, either with or without concurrent downregulation of inhibitory synapses or upregulation of intrinsic excitability, initiated slowly repeating burst discharges that closely resembled the epileptiform burst discharges recorded in chronically isolated neocortex. These burst discharges lasted a few hundred ms, propagated at 1–3 cm/s and consisted of large (10–15 mV) intracellular depolarizations topped by a small number of action potentials. Our results support a role for homeostatic synaptic plasticity as a novel mechanism of post-traumatic epileptogenesis.
Introduction
Chronically isolated slabs of neocortex, produced by gray and white matter lesions, develop chronic hyperexcitability and focal epileptogenesis (Echlin and Battista, 1963; Sharpless, 1969; Prince, 1999). Spontaneous bursts of activity appear a few days after isolation; these bursts occur more frequently during subsequent days and weeks (Grafstein and Sastry, 1957; Sharpless and Halpern, 1962; Burns and Webb, 1979), while field potentials display slow waves at frequencies around 1 Hz (Echlin and Battista, 1963) (Fig. 1A). At the same time the isolated cortex develops an increased susceptibility to experimentally induced epileptiform activity (Grafstein and Sastry, 1957; Sharpless and Halpern, 1962; Echlin and Battista, 1963), similar to the phenomenon of disuse supersensitivity encountered in peripheral structures after deprivation of afferent inputs (Sharpless, 1969). In slices of chronically (>1–2 weeks) isolated cortex, electrical stimulation may evoke ‘epileptiform’ burst discharges (Prince and Tseng, 1993; Fig. 1B) that are initiated in layer 5 (Hoffman et al., 1994) and resemble in some ways the up-states of the slow (<1 Hz) oscillation in cortical slices (Sanchez-Vives and McCormick, 2000) and naturally sleeping cats (Steriade et al., 2001). Similar burst discharges are observed in lesioned organotypic hippocampal slice cultures (McKinney et al., 1997) and cell cultures subjected to chronic blockade of activity (Furshpan and Potter, 1989; Ramakers et al., 1990; van den Pol et al., 1996; Rutherford et al., 1997). The mechanisms underlying epileptogenesis in chronically isolated neocortex are unclear. Previous proposals involve the sprouting of new excitatory connections (Purpura and Housepian, 1961; Salin et al., 1995; McKinney et al., 1997), decreases in synaptic inhibition (Ribak and Reiffenstein, 1982), and increases of NMDA currents and pyramidal cell excitability (Bush et al., 1999).
![Spontaneous and evoked activities in chronically isolated cortex. (A) Field potentials and multi-unit recordings from the unanesthetized intact cortex and the chronically isolated slab on the fifth day after isolation during a period of REM sleep. The fragment indicated by the horizontal bar is expanded on the right as indicated by the arrow. The chronically isolated slab was characterized by slow waves and clustered neuronal firing. (B) Characteristics of epileptiform events recorded in slices of chronically isolated (‘undercut’) cortex [modified from Prince and Tseng (1993), used with permission]. Electrical stimulation evoked all-or-none polyphasic epileptiform field potentials in undercut cortex (left panel), but only short-latency responses in slices of contralateral control cortex. Burst discharges propagated across cortex at an average speed of 1–4 cm/s (middle panel). The distance between positions a–d was ∼5 mm. Intracellular recording of a layer 2 pyramidal cell during an evoked epileptiform event (right panel).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cercor/15/6/10.1093_cercor_bhh184/2/m_cercorbhh184f01_lw.jpeg?Expires=1716401464&Signature=ma3w-DTD9~cUModHrbCTjepxkn~27UYiKmwoxo1pwRwthMOE3nHkG~eBDuH-d~pwjjl8vZFw0Uxu27~Av~kkgT9xEiJ7gwQGZOyAmzM3JIvjuaZt3ZFxqJ~kwx6nDpzRGdSyyo4ZRD2WrS4LgvTLhiGfg1aiuvqvOAudJJrW~PbinqxQzIx3v7omciAUb~iVrwcQN8tep-T~fqyxgbG9mE2WoP1E8--G6KYJ2bBJsX6ZvkGzL-n6EzCF8BF-BuI8m04BmljJl-S0MtWNZLHhhk3Y849LUevT0aOan-Ludhfa~tIxrKmUfON4YZMCM~NfI3WeWRya20GEp9pcGtAETw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
{kind=link}
Spontaneous and evoked activities in chronically isolated cortex. (A) Field potentials and multi-unit recordings from the unanesthetized intact cortex and the chronically isolated slab on the fifth day after isolation during a period of REM sleep. The fragment indicated by the horizontal bar is expanded on the right as indicated by the arrow. The chronically isolated slab was characterized by slow waves and clustered neuronal firing. (B) Characteristics of epileptiform events recorded in slices of chronically isolated (‘undercut’) cortex [modified from Prince and Tseng (1993), used with permission]. Electrical stimulation evoked all-or-none polyphasic epileptiform field potentials in undercut cortex (left panel), but only short-latency responses in slices of contralateral control cortex. Burst discharges propagated across cortex at an average speed of 1–4 cm/s (middle panel). The distance between positions a–d was ∼5 mm. Intracellular recording of a layer 2 pyramidal cell during an evoked epileptiform event (right panel).
A striking feature of acutely isolated cortex is its strongly reduced neuronal activity (Burns, 1951; Sharpless and Halpern, 1962; Timofeev et al., 2000). In vitro evidence suggests that chronic blockade of activity may modify synaptic strength and intrinsic neuronal excitability. A few days of pharmacological activity blockade in cortical cell cultures increases the amplitude of miniature (m-) excitatory postsynaptic currents (EPSCs) and evoked EPSCs in pyramidal cells (Turrigiano et al., 1998; Watt et al., 2000). Conversely, prolonged enhanced activity reduces the amplitude of mEPSCs (Lissin et al., 1998; Turrigiano et al., 1998; Leslie et al., 2001). Similar bidirectional activity-dependent changes in mEPSC size have been observed in spinal cell cultures (O'Brien et al., 1998). Synaptic scaling occurs in part postsynaptically by changes in the number of glutamate receptors (Rao and Craig, 1997; Lissin et al., 1998; O'Brien et al., 1998; Turrigiano et al., 1998; Liao et al., 1999; Watt et al., 2000) but also presynaptically (Murthy et al., 2001). Interestingly, activity blockade scales down the amplitude of miniature inhibitory postsynaptic currents (mIPSCs), in the opposite direction of excitatory currents (Rutherford et al., 1997). This effect is accompanied by a reduction in the number of GABAA receptors clustered at synaptic sites (Kilman et al., 2002). Intrinsic excitability is also regulated by activity. Chronic activity blockade enhances Na+ currents and reduces K+ currents, resulting in an enhanced responsiveness of pyramidal cells to current injections (Desai et al., 1999). These observations suggest the existence of a fundamental mechanism, termed ‘homeostatic plasticity’ (Turrigiano, 1999), that controls the levels of neuronal activity, possibly firing rates (Turrigiano et al., 1998; Murthy et al., 2001). Several beneficial roles of homeostatic plasticity have been postulated (Miller, 1996; Turrigiano et al., 1998; Craig, 1998). For example, homeostatic synaptic plasticity may prevent the runaway of synaptic strengths during Hebbian modifications or maintain the balance between excitation and inhibition and facilitate the competition between synapses during development. In model neurons (and networks) with activity-dependent regulation of maximal conductances of ionic currents, conductances adjust to restore a given pattern of electrical activity when perturbed (LeMasson et al., 1993; Siegel et al., 1994; Liu et al., 1998; Golowasch et al., 1999). Thus, it is believed that homeostatic plasticity has a role in preserving a ‘normal’ level of activity.
Recent evidence suggests that some of these homeostatic processes also occur in vivo (Desai et al., 2002). In pyramidal cells of chronically isolated cortex, synaptic currents (Li and Prince, 2002) and intrinsic excitability (Prince and Tseng, 1993) are altered consistent with homeostatic plasticity. We hypothesized that the pathological development of bursting activity in chronically isolated cortex was the result of homeostatic plasticity of synaptic and/or intrinsic excitability induced by reduced neuronal activity. This hypothesis was investigated in computational models incorporating a biologically based homeostatic plasticity rule.
Materials and Methods
Animal preparation and recording procedure
Experiments were conducted on two adult cats. Surgical procedures for chronic implantation of recording electrodes were carried out in sterile conditions under deep pentobarbital (35 mg/kg) anesthesia, followed by three administrations, every 12 h, of buprenorphine (0.03 mg/kg, i.m.) to prevent pain. In addition, cats were injected with 500 000 units of penicillin (i.m.) for three consecutive days. Isolated neocortical slabs (6 × 10 mm) from area 5 were prepared as described in Timofeev et al. (2000). Briefly, a custom-made crescent knife was inserted along its curve into the cortex until the tip of the knife appeared ∼10 mm frontally under the pia. The knife was then turned 90° in both right and left directions. The pia was intact except where the knife entered. Tungsten electrodes (8–12 MΩ) were implanted in the slab and in intact cortical regions outside the slab at a depth of 1 mm to record focal field potentials and multi-unit activities. Additional pairs of electrodes were placed in the ocular cavities and neck muscles to monitor states of vigilance by recording the electro-oculogram (EOG) and electromyogram (EMG). A few bolts were cemented to the cranium to allow non-painful fixation of the cat's head to a stereotaxic frame. The opening in the cranium was covered with a thin layer of polyethylene and dental cement. After a recovery period (2–3 days), cats were adapted to be in the frame for 1–2 h/day. After a few days of training, cats started to sleep in the frame and they displayed clearly identifiable states of waking, slow-wave sleep (SWS) and rapid eye movement (REM) sleep. Electrophysiological recordings started 4–5 days after isolation of the slab. Each day three recording sessions were performed that lasted 2–3 h and usually contained periods of waking, SWS and REM sleep. Electrophysiological recordings were digitally filtered in two bands (0.1–300 Hz and 300–10 000 Hz) to obtain separate traces for local field potentials and multi-unit activities. All procedures were conducted according to institutional guidelines.
Cortex Model
The responses of neocortical pyramidal cells (PY) and interneurons (IN) were simulated using Hodgkin–Huxley-style reduced two-compartment models (Mainen and Sejnowski, 1996). The axosomatic compartment contained a leak current Ileak and fast Na+ and K+ currents for action potential generation, INa and IK. The dendritic compartment contained Ileak, INa, a high-voltage activated Ca2+ current ICa, a slow non-inactivating K+ current IM and a Ca2+-activated K+ current IKCa. A persistent Na+ current INaP (Golomb and Amitai, 1997) was added to the dendritic compartment of PY cells. Maximal conductance values (ḡ) and descriptions of currents were identical to those in Mainen and Sejnowski (1996), except for ḡNaP = 0.07 mS/cm2 and ḡCa = 0.01 mS/cm2. An entire spectrum of firing patterns could be produced when the ratio of axosomatic to dendritic membrane area (ρ) or the coupling resistance between axosomatic and dendritic compartments (κ) was varied. PY cells were taken to be regular-spiking (ρ = 140, κ = 10) and IN cells fast-spiking (ρ = 50, κ = 10). The leak reversal potential Eleak of each cell was drawn from a normal distribution with mean −70 mV and standard deviation σ = 4 mV. All simulations used NEURON (Hines and Carnevale, 1997).
The one-dimensional cortical model consisted of 5000 PY cells and 1250 IN cells. Synaptic connections between cells were both sparse and local, i.e. each cell connected with a probability of 20% to cells within a radius of 5% of the size of the network. We also considered networks with dense local connectivity (100% connection probability within the synaptic footprint) and networks with sparse all-to-all connectivity. On average, each PY cell connected to 100 (±9) PY cells with AMPA and NMDA synapses and to 25 (±4) IN cells with AMPA synapses, and each IN cell connected to 100 (±9) PY cells and 25 (±4) IN cells with GABAA synapses. All synapses contacted the dendritic compartments. The synaptic footprint shape was square, i.e. synaptic conductance values of a particular type were the same for different pairs of connected cells. NMDA current was included at PY–PY synapses with a ratio of NMDA:AMPA peak current of 0.15 (in zero Mg2+) in agreement with experimental values of 0.1–0.2 (Spruston et al., 1995; McAllister and Stevens, 2000). A small amount of short-term synaptic depression was incorporated at PY–PY synapses, with U = 0.05 and τD = 750 ms (see equation 5), which depressed EPSPs at firing rates of 5 Hz to ∼86% of the maximal size. In the intact cortex model each PY and IN cell received AMPAergic synaptic input from extrinsic afferents, modeled as an independent Poisson process with a rate of 1000 Hz and an average synaptic conductance of Gex–PY and Gex–IN respectively.
The values of the synaptic parameters were constrained using the following assumptions: (i) average firing rates are 5 and 10 Hz for PY and IN cells, respectively; (ii) synaptic activity increases input conductances by 150% in agreement with observations in barbiturate-anesthetized animals where spontaneous synaptic activity increases input conductances of isolated cells by (at least) 33–250% (Paré et al., 1998); (iii) cells in intact cortex fire asynchronously. An asynchronous state with these properties was obtained only when the average conductance G of recurrent synapses was smaller than that of extrinsic synapses: GPY–PY< Gex–PY and GPY–IN< Gex–IN (not shown). We took GPY–PY/(GPY–PY + Gex–PY) = GPY–IN/(GPY–IN + Gex–IN) = 0.25, which resulted in asynchronous firing at rates close to 5 Hz (PY cells) and 10 Hz (IN cells) and input resistances of about 40% of those of isolated cells. The corresponding synaptic conductance values were: Gex–PY = 0.975 nS, Gex–IN = 0.45 nS, ḡPY–PY,AMPA = 12.6 nS, ḡPY–PY,NMDA = 2.1 nS, ḡPY–IN = 6 nS, ḡIN–PY = 128 nS, ḡIN–IN = 40 nS. Values for individual connections were obtained by dividing ḡ by the average number of connections for each type.
For some networks the local network potential (LNP), a measure related to the local field potential, was calculated by convolving the spike trains of individual PY cells with a synaptic (conductance) response function, a decaying exponential with time constant 5 ms, and summing these traces over all PY cells within a synaptic footprint.
Simplified Firing Rate Model
Results
Electrical Activity of Chronically Isolated Neocortex
Acutely isolated neocortical slabs (6 × 10 mm) are typically silent (Timofeev et al., 2000). In the chronically (4–21 days) isolated slab, however, field potentials and multi-unit recordings displayed a slow oscillatory pattern consisting of silent periods and periods of burst firing (Fig. 1A, slab), regardless of the state of vigilance, as previously reported (Grafstein and Sastry, 1957; Sharpless and Halpern, 1962; Echlin and Battista, 1963; Burns and Webb, 1979). Field potential recordings from the intact area 4 during REM sleep revealed a typical activated pattern (Fig. 1A, area 4) and multi-unit recordings suggested that neurons fired in a tonic mode (not shown). Slow periodic activities were recorded from chronically isolated slabs for the duration of our observations (4–21 days). We never recorded clear-cut spontaneous electrographic seizures, e.g. consisting of spike-wave complexes and fast runs (Topolnik et al., 2003), in chronically isolated slabs.
In slices of chronically (1–21 weeks) isolated cortex, electrical stimulation evokes all-or-none ‘epileptiform’ field potentials that last a few hundred milliseconds (Fig. 1B, left panel) and occur spontaneously in a small number of slices (Prince and Tseng, 1993; Hoffman et al., 1994). These events propagate along the long axis of the slice at speeds of 1–5 cm/s (Fig. 1B, middle panel), with intracellular activities consisting of large (10–25 mV) prolonged (<400–500 ms) depolarizations topped by a small number of action potentials (Fig. 1B, right panel).
We hypothesized that the periodic bursting activity in chronically isolated cortex was the result of homeostatic plasticity of synaptic and/or intrinsic excitability. In the following this hypothesis was explored using computational models.
Acute Effects of Deafferentation in the Cortical Model
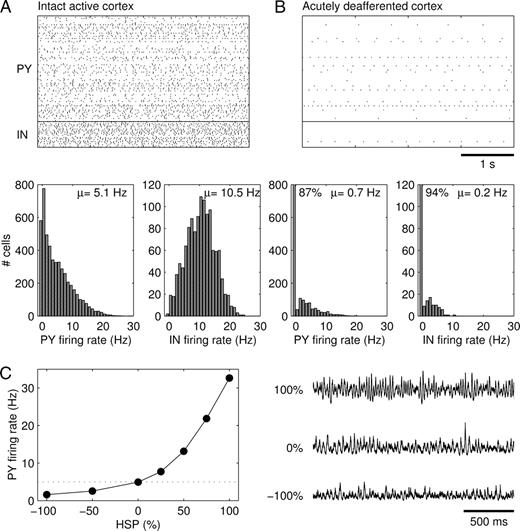
Our model of intact cortex consisted of 5000 PY and 1250 IN cells, with local (within 10% of the size of the network) but sparse (20% connection probability) synaptic connections between all cell types. Extrinsic afferent synapses (e.g. from the thalamus and other cortical areas) provided glutamatergic inputs to both PY and IN cells. Synaptic conductance values were naturally constrained using the following assumptions (see Materials and Methods): (i) average firing rates are about 5 and 10 Hz for PY and IN cells respectively (Fig. 2A); (ii) synaptic activity provides a large contribution to input conductances (Paré et al., 1998); (iii) cells in intact cortex fire asynchronously.
{kind=link}
Intact and acutely deafferented cortex models. Spike rasterplots and firing rate histograms of (A) intact cortex and (B) acutely deafferented cortex. In the rasterplots, spike trains of every 50th PY and IN cell are represented. (C) Synaptic scaling of ḡPY−PY and ḡIN−PY (in opposite directions) in intact cortex modulated the average firing rate of PY cells while maintaining a low-amplitude irregular LNP. The LNPs are scaled by the reverse of the square root of their means for comparison purposes. The simulations in (C) were performed in a network with 500 PY and 125 IN cells and sparse all-to-all synaptic connectivity.
Deafferentation consisted of removing the extrinsic excitatory inputs from all PY and IN cells. The average membrane potential of PY cells dropped from −62.2 ± 1.6 mV to −69.2 ± 4.3 mV, which closely resembled measured values in awake (Steriade et al., 2001) and deafferented cortex (Timofeev et al., 2000). Deafferentation also dramatically reduced the firing rates of PY and IN cells to on average 0.7 and 0.2 Hz respectively (Fig. 2B). A small fraction of PY (13%) and IN (6%) cells was spontaneously active due to a variability in leak current reversal potential (Eleak) values. The size of individual EPSPs in PY cells was ∼0.3 mV.
Homeostatic Synaptic Plasticity in Deafferented Cortex
We hypothesized that the strongly reduced levels of neuronal activity in deafferented cortex activated homeostatic synaptic plasticity (HSP). The plasticity rule (equation 7) involved the upregulation of ḡPY–PY and downregulation of ḡIN–PY as long as the average firing rate of PY cells F was below a target firing rate T, which was set at the original average firing rate of PY cells before deafferentation (5 Hz). The rule states that a k% increase in PY–PY peak conductance occurs concurrently with a 0.5k% decrease in IN–PY conductance. Thus, in close agreement with observations in cell cultures after 2 days of activity blockade (Rutherford et al., 1997, 1998; Turrigiano et al., 1998; Watt et al., 2000; Kilman et al., 2002), a 100% upregulation of ḡPY–PY is accompanied by a 50% downregulation of ḡIN–PY, which is designated an HSP value of 100%.
To get some intuition on the possible effects of homeostatic synaptic plasticity we scaled PY–PY and IN–PY synaptic strengths (in opposite directions) in the intact cortex model. Synaptic scaling modulated firing rates while maintaining network activity characterized by a low-amplitude irregular LNP (local network potential, see Materials and Methods) (Fig. 2C).
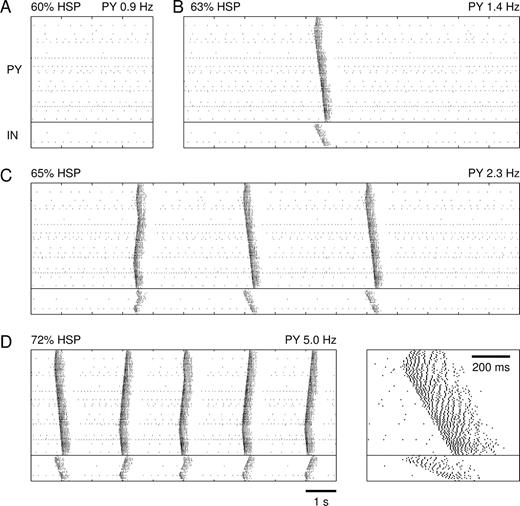
In the deafferented cortex, homeostatic synaptic plasticity had initially little effect on network activity. Up to 60% HSP, firing rates were similar to those in the acutely deafferented network (Fig. 3A). PY cells fired at an average rate of 0.9 Hz and IN cells at 0.3 Hz. After 63% HSP, the spontaneous activity of the network changed in a qualitative manner (Fig. 3B). Occasionally, locally generated spike bursts propagated through the network. These burst discharges were 200–400 ms in duration and involved multiple spikes in PY and IN cells (see Fig. 5B). Bursts were generated by network interactions because none of the cells possessed intrinsic bursting mechanisms (all PY cells were of the regular-spiking type). As the average number of spikes per PY cell measured over long periods of time (1.4 Hz) was still below the homeostasis target frequency of 5 Hz, further HSP increased the occurrence of network bursts (Fig. 3C, 65% HSP). Eventually a steady state was reached where bursts repeated at frequencies of ∼0.5 Hz and the average PY cell firing rate (5.0 Hz) equaled the homeostasis target firing rate (Fig. 3D, 72% HSP). The amounts of HSP needed to reach steady state in four different simulations (with different seeds of the random number generator) were 65%, 69%, 70% and 71% (mean 69.4%). Note that our approach did not provide the timecourse of homeostasis, which requires an exact description of how the rates of homeostatic plasticity depend on average firing rate F, target firing rate T and conductance values ḡj (functions hj(F, T, ḡj) in equation 6).
{kind=link}
Emergence of propagating burst discharges in deafferented cortex after homeostatic synaptic plasticity. Spike rasterplots of network activity after (A) 60% HSP, (B) 63% HSP, (C) 65% HSP and (D) 72% HSP. After 72% HSP a steady state was reached for which PY cells fired on average 5.0 Hz. The inset shows an expanded spontaneous burst at 72% HSP.
Upregulation of ḡPY–PY and downregulation of ḡIN–PY did not necessarily lead to bursting. For example, excess synaptic scaling (beyond the amount needed to restore firing rates) could result in sustained activity at high firing rates (∼50 Hz at 160% HSP) or in PY cells being locked into a strongly depolarized state with Na+ channel inactivation (not shown).
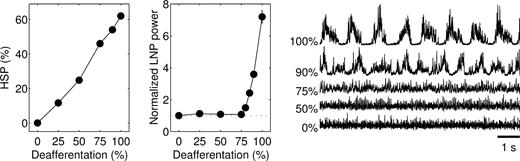
Because cortical trauma may not necessarily lead to complete (100%) deafferentation, we systematically varied the degree of deafferentation and studied the effects of homeostatic synaptic plasticity. Surprisingly, only nearly complete deafferentation resulted in slow oscillatory network activity (Fig. 4). When the model was deprived of only a fraction (<80%) of extrinsic inputs, homeostatic synaptic plasticity restored a low-amplitude irregular LNP similar to that of intact cortex. Homeostatic upregulation of the remaining extrinsic synapses on PY cells (not modeled here) may also contribute to restoring an asynchronous network state. These results indicate that only after severe deafferentation is homeostatic synaptic plasticity unable to restore an asynchronous state and periodic bursting occurs.
{kind=link}
Only nearly complete deafferentation leads to network bursting activity. Amount of HSP that was required to restore the average PY cell firing rate to 5 Hz in networks that were partially deprived of afferents (left). The middle panel plots the average power of the LNPs computed for 10 contiguous blocks of 100 PY cells, normalized with respect to that of intact cortex. LNPs computed over all PY cells are shown on the right. Homeostatic synaptic plasticity restored a low-amplitude irregular LNP after partial (<80%) deafferentation. The simulations in partially deafferented cortex were performed in networks with 1000 PY and 250 IN cells and dense local synaptic connectivity.
Properties of Burst Discharges
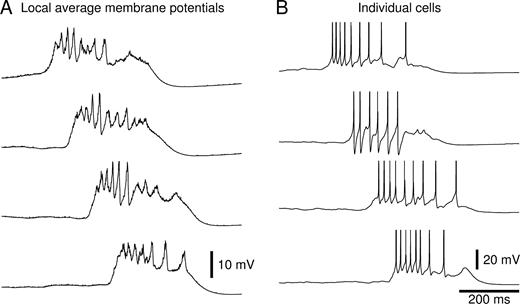
Inspection of the local average membrane potentials (of 500 PY cells, corresponding to one synaptic footprint) during a spontaneous network burst revealed the presence of large-amplitude multiphasic activity on top of a strong (10–15 mV) prolonged depolarization (Fig. 5A). The appearance of these local average membrane potentials was similar to the epileptiform field potentials recorded in slices of chronically isolated cortex (Prince and Tseng, 1993; Hoffman et al., 1994) (Fig. 1B). Membrane potentials of individual PY and IN cells showed a similar depolarization, lasting 200–400 ms and carrying a variable number of action potentials (Fig. 5B). There was a close resemblance between these membrane potentials and intracellular recordings from slices of chronically isolated cortex during epileptiform events (Prince and Tseng, 1993) (Fig. 1B). The observation that IN cells generated action potentials during bursts is in agreement with recordings of pyramidal cells in slices of chronically isolated cortex, which reveal large amplitude IPSCs in voltage-clamp (Salin et al., 1995; Li and Prince, 2002) and IPSP-like hyperpolarizing potentials in current-clamp (Prince and Tseng, 1993).
{kind=link}
Membrane potentials during burst discharges. (A) Average PY cell membrane potentials at four locations (20–40–60–80% along the length of the network) during a spontaneous network burst that occurred after 70% HSP. (B) Membrane potentials of three PY cells and one IN cell (second from top) at each of the four locations.
Bursts were also evoked by ‘electrical’ stimulation, which consisted of activating AMPA synapses on a set of neighboring PY cells to evoke a single action potential. At small HSP values (≤41%), stimulation of any number of PY cells evoked spikes in the stimulated PY cells (and in some of the neighboring PY and IN cells) but discharges never propagated. At HSP values of 42–46%, stimulation evoked local bursts of spikes in PY cells that propagated for variable distances from the site of stimulation. Whether a burst was generated, and over what distance it propagated, depended randomly on the number and locations of cells that were stimulated. At HSP values of 47% and higher, stimulation of ∼60 or more PY cells triggered a burst of spikes that propagated through the entire network resembling spontaneous bursts. The response to stimulation at around ‘threshold’ HSP values was sensitive to the number of cells that were stimulated, with stimulation of large numbers of cells (>200–300) failing to generate network bursts. Epileptiform events evoked in slices of chronically deafferented cortex display a similar dependence on stimulus intensity (Prince and Tseng, 1993).
The frequency at which bursts repeated, the number of spikes in bursts and the speed of burst propagation all increased in the course to steady state (Fig. 6A). The average number of spikes per cell during a burst more than doubled from PY 4.5 ± 1.8 (SD) and IN 2.5 ± 1.4 at 47% HSP to PY 10.9 ± 2.4 and IN 6.5 ± 2.3 at 72% HSP. To obtain a measure for burst duration, we determined for each PY cell the time interval between the first and the last spike during a burst. Average burst duration increased from 146 ± 51 ms at 47% HSP to 251 ± 40 ms at 72% HSP (not shown). The velocity of burst propagation, measured in synaptic footprints (SF) per second, increased from 8.9 SF/s at 47% HSP to 30.1 SF/s at 72% HSP. If one synaptic footprint is taken to be 1 mm, these velocities become 0.89 and 3.01 cm/s respectively. Epileptiform events in slices of chronically isolated cortex propagate at a similar range of speeds of 1–5 cm/s (Prince and Tseng, 1993; Hoffman et al., 1994) (Fig. 1B).
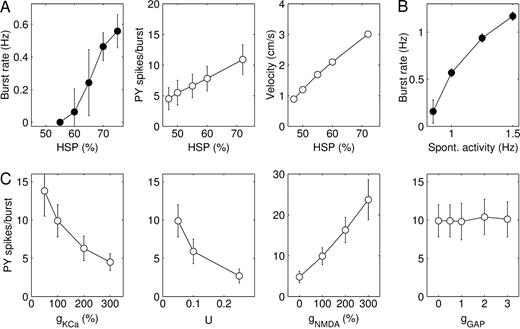
{kind=link}
Parameter dependence of burst discharges. (A) Rate at which bursts repeated, average number of PY cell spikes per burst and speed of burst propagation as a function of the amount of HSP. Number of spikes per burst and burst velocity were measured from evoked burst discharges in a model with reduced spontaneous activity. (B) Rate at which bursts repeated as a function of the spontaneous firing rate of PY cells. (C) Dependence of the average number of PY spikes per burst on ḡKCa, parameter U of short-term synaptic depression, ḡPY–PY,NMDA and the summated electrical coupling conductance gGAP. The values of ḡKCa and ḡPY–PY,NMDA are expressed as a percentage of their default values and that of gGAP as a fraction of the leak conductance of individual IN cells. Burst discharges were evoked after 70% HSP.
Analysis of Mechanisms Underlying Burst Generation
Increasing ḡKCa or the parameter U of short-term synaptic depression decreased burst duration and the number of spikes per burst (Fig. 6C). A complete block of IKCa (ḡKCa = 0) or short-term synaptic depression (U = 0) caused burst discharges to sustain indefinitely. NMDA current at PY–PY synapses modulated burst duration and number of spikes in a similar manner (Fig. 6C). Interestingly, blocking NMDA current approximately halved the number of spikes per burst in networks at 70% HSP, but completely blocked burst discharges at low HSP values (≤56%). Addition of electrical synapses between IN cells had no effect on our burst measures, even for very high coupling conductances (Fig. 6C). In these simulations each IN cell was electrically coupled to on average 10 neighboring IN cells (randomly chosen within the synaptic footprint) and the average summated coupling conductance (gGAP) that each cell received was of the same order of magnitude as the leak conductance of individual IN cells (Amitai et al., 2002).
The frequency at which bursts were initiated depended not only on the amount of HSP but also on the spontaneous firing rates of PY cells. In the acutely deafferented cortex a small fraction of cells (13%) was spontaneously active due to a variability in leak current reversal potential (Eleak) values (see Fig. 2B). To assess the role of spontaneous activity in triggering network bursts the variability in Eleak values was reduced and instead each cell in the network received glutamatergic inputs from a set of ‘virtual’ PY cells modeled as independent Poisson processes. The firing rate of the virtual PY cells influenced the rate at which bursts were generated (Fig. 6B). In a network that had been subjected to 70% HSP, bursts appeared when the spontaneous firing rate was larger than ∼0.8 Hz. Because of the random spontaneous activity, bursts were initiated at multiple locations in the network. When the spontaneous firing rate was 1.25 Hz, bursts repeated at regular intervals of ∼1 s. These results suggest that the spontaneous firing rates after deafferentation affect the amount of HSP needed to restore firing rates and possibly the nature of the resulting network state (e.g. see Fig. 4).
Different Strategies of Homeostatic Regulation
Next, we investigated how the individual homeostatic processes contributed to burst generation. In the default model the conductances of PY–PY synapses (both AMPA and NMDA components) and IN–PY synapses were subject to homeostatic regulation. Regulation of PY–PY synaptic conductance alone (of the AMPA component alone, or of both AMPA and NMDA components) restored PY cell firing rates (to 5 Hz) in the fully deafferented cortex after conductance increases of ∼100% (Fig. 7A). These increases are comparable in size to those of mEPSCs and evoked EPSCs in cell cultures after 2 days of activity blockade. Homeostatic regulation of IN–PY synaptic conductance alone required a 65% or larger downregulation to restore firing rates in partially (75–90%) deafferented cortex (Fig. 7B). Homeostatic regulation of synaptic conductances always restored bursting states in 90–100% deafferented networks (Fig. 7D).
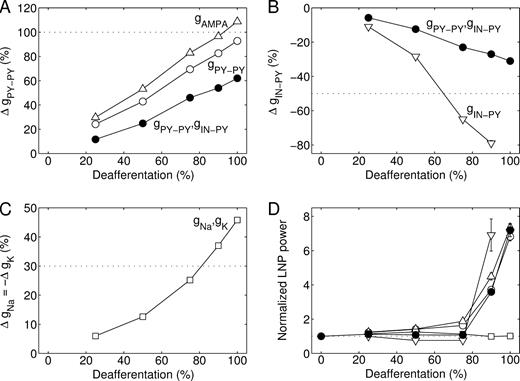
{kind=link}
Different strategies of homeostatic regulation. The conductance changes that were required to restore the average PY cell firing rate to 5 Hz after (A) upregulation of PY–PY synapses alone, with AMPA component only (open triangle) or both AMPA and NMDA components (open circle), (B) downregulation of IN–PY synapses alone (open inverted triangle), or (C) upregulation of intrinsic excitability alone (open square). The conductance changes required in the default model incorporating all homeostatic synaptic processes are shown for comparison (filled circle). Note that in fully deafferented cortex a 100% downregulation of IN–PY synapses alone was not sufficient to restore firing rates. (D) Average power of LNPs computed for 10 contiguous blocks of 100 PY cells, normalized with respect to that of intact cortex. These simulations were performed in networks with 1000 PY and 250 IN cells and dense local synaptic connections.
Thus far we did not consider homeostatic regulation of intrinsic excitability. This was modeled as an upregulation of fast and persistent Na+ conductances (ḡNa, ḡNaP) and a downregulation of K+ conductances (ḡM, ḡKCa) in PY cells, which made the relationship between injected current and spike firing frequency steeper. In the absence of an exact experimental description of how the rates of plasticity depend on F, T and maximal conductances our plasticity rule was again based on observations in cortical cell cultures after two days of activity blockade: fast Na+ conductances in pyramidal cells are upregulated by ∼30% and K+ conductances (of the delayed rectifier and a persistent outward current) are downregulated by ∼30–60% (Desai et al., 1999). Thus, we assumed that a 0.3k% increase in Na+ conductances occurs concurrently with a 0.3k% decrease in K+ conductances (and a k% increase in ḡPY–PY and a 0.5k% decrease in ḡIN–PY in case of additional HSP, see equation 7). Homeostatic regulation of intrinsic excitability alone always restored asynchronous states (Fig. 7D), although this required large (>35%) conductance changes in networks that were 90–100% deafferented (Fig. 7C). Homeostatic regulation of both intrinsic and synaptic excitability reduced the amount of HSP needed to restore firing rates without affecting the development of bursting in the network (not shown).
Homeostatic regulation of synaptic release probability (Murthy et al., 2001) affects ḡ in our deterministic synapse model (because ḡ represents the product of maximal conductance and release probability summed over all synapses between two cells) and U, the proportion of available synaptic resources used to generate an EPSP. A 135% upregulation of both ḡ and U at PY–PY synapses restored firing rates in the form of repetitive burst discharges (not shown). Regulation of ḡPY−PY alone required smaller increases to reach the homeostasis target firing rate (see Fig. 7A), because increased U values shortened burst discharges by increasing short-term synaptic depression (see Fig. 6C).
Simplified Firing Rate Model
Homeostatic synaptic plasticity robustly initiated bursting activity after deafferentation in a variety of networks with different physiological properties (see Supplementary Material). We also investigated whether the development of bursting activity could be explained in generic terms of dynamical systems behavior. The simplest system that we examined was a reduced rate model that consisted of a population of PY cells described by the average membrane potential X (equation 8) which received extrinsic input and recurrent excitation subject to depletion of its available synaptic resources R (equation 9) (Carpenter and Grossberg, 1983). With intact extrinsic input (Iext = 0.124) the population fired at 10 Hz (Fig. 8A, lower panel). In the (R,X)-plane (Fig. 8A, upper panel) this mode was reflected as a stable fixed point given by the intersection of the nullclines of the system, i.e. the steady-state solutions of the individual equations (8) and (9) (dX/dt = 0, dR/dt = 0). After acute deafferentation (Iext = 0) the firing rate dropped to 0.7 Hz due to a downward shift of the X-nullcline (Fig. 8B). The synaptic coupling W was then increased to model the effects of homeostatic synaptic plasticity. Initially, this led to slightly increased firing rates without altering the stability of the equilibrium point (Fig. 8C). At a critical value of W, however, the equilibrium point lost its stability due to a Hopf bifurcation and X started to oscillate (Fig. 8D). The oscillation consisted of a short burst with a firing rate up to ∼160 Hz which was terminated due to partial depletion of the synaptic resources, followed by a longer-lasting low-activity state during which synaptic resources slowly recovered until a level was reached that supported a new burst. On the (R,X)-plane the oscillation corresponded to a slow motion on the lower stable branch of the X-nullcline (low-activity state) interrupted by fast jumps into the vicinity of the upper branch (bursts). Further increases of W reduced the duration of the silent state until the time-averaged firing rate reached the homeostatic setpoint of 10 Hz (Fig. 8E). The oscillations are caused by the presence of an S-shaped (hysteresis) loop in the X-nullcline (dX/dt = 0). Many firing rate functions other than the parabolic f(X) obtained from our conductance-based PY cell model give rise to S-shaped loops (van Ooyen and van Pelt, 1994).
![Homeostatic synaptic plasticity restores burst firing after deafferentation in a simplified firing rate model. The reduced model consisted of a single PY unit, representing a population of PY cells with average membrane potential X ∈ [0,1] , that received extrinsic and recurrent synaptic inputs. The recurrent excitation was subject to short-term depression with R denoting the available synaptic resources. The upper panels show the nullclines of the system with intact extrinsic input (A) and of the deafferented system at different values of synaptic coupling W = 5.69w, where w = 1 in (B), w = 2.50 in (C), w = 3.85 in (D) and w = 4.01 in (E). The lower panels show the respective time evolutions of firing rate f(X). See Results for a detailed explanation. Parameter values were U = 0.05, τR = 750 ms, τX = 10 ms.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cercor/15/6/10.1093_cercor_bhh184/2/m_cercorbhh184f08_lw.jpeg?Expires=1716401464&Signature=3Hx9MCp7JqvO0XIWxABas5mWtccQ4JYcarV3GbHh84O9KdOg9G~XALBfHGuL~g2v3b9DpFNl94370TJinXIF-zlVxE7ND5HxSEbikqKRH5887o6kJekJljvqcPpkdWpuMTZZ8Oijs-qdCh8IQ98uV6yDzuuy6WCR~WUyGkT5mhbZg4V1~M0Pm5AxJ2u2X-Di4cAMMQ~s3o-0lKZNOL6o-UlDz~L5AfQhZlatxEf8AQl1fyxTlWYEfNVIyN7gL5qZeM18TtGh0Os3ndlGeUwzczOIhqpwMRl2gn9kwMFyKCRMN7Hit2ElVtzgUkYwKEVoxt1ikcmtKe-Khxjv3g7org__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
{kind=link}
Homeostatic synaptic plasticity restores burst firing after deafferentation in a simplified firing rate model. The reduced model consisted of a single PY unit, representing a population of PY cells with average membrane potential X ∈ [0,1] , that received extrinsic and recurrent synaptic inputs. The recurrent excitation was subject to short-term depression with R denoting the available synaptic resources. The upper panels show the nullclines of the system with intact extrinsic input (A) and of the deafferented system at different values of synaptic coupling W = 5.69w, where w = 1 in (B), w = 2.50 in (C), w = 3.85 in (D) and w = 4.01 in (E). The lower panels show the respective time evolutions of firing rate f(X). See Results for a detailed explanation. Parameter values were U = 0.05, τR = 750 ms, τX = 10 ms.
Discussion
It is generally believed that homeostatic synaptic plasticity works to restore a stable pattern of activity in networks when perturbed. We showed, however, that this process has limits and that after complete cortical deafferentation (such as may occur after severe head trauma) homeostatic synaptic plasticity may increase network excitability to a level where only synchronized bursting activity becomes possible. Interestingly, the same mechanisms may recover a ‘normal’ asynchronous state after partial deafferentation. Burst discharges in the model were similar in many aspects to the epileptiform events observed in slices of chronically isolated neocortex, including burst duration, speed of propagation and intracellular activities. This study supports the novel hypothesis that homeostatic synaptic plasticity is a primary mechanism of post-traumatic epileptogenesis and may have important implications for how trauma-induced epilepsy could be treated.
Network bursting activity resulted from the upregulation of excitatory synapses between pyramidal cells, either with or without a concurrent downregulation of inhibitory synapses and/or upregulation of intrinsic excitability. In contrast, homeostatic upregulation of pyramidal cell intrinsic excitability alone restored ‘normal’ asynchronous activity similar to that of intact cortex. Analysis of a simplified firing rate model showed that the development of bursting activity can be explained in generic terms of dynamical systems behavior. It is important to note that homeostatic synaptic plasticity does not necessarily lead to bursting. In fact, activity states other than bursting were observed (i) in partially (<80%) deafferented networks, (ii) in networks where PY cells lacked both IKCa and short-term synaptic depression (see Supplementary Material), (iii) after excess amounts of homeostatic synaptic plasticity and (iv) after synaptic scaling in intact cortex.
Experimental observations in slices of chronically isolated sensorimotor cortex are consistent with homeostatic plasticity. Frequencies of mEPSCs and spontaneous EPSCs (sEPSCs) are increased in layer 5 pyramidal cells, frequencies of mIPSCs and sIPSCs are reduced, and amplitudes of mEPSCs and sEPSCs are increased (Li and Prince, 2002). The amplitudes of EPSCs evoked by extracellular stimulation are enhanced for supra-threshold stimuli. In layer 5 pyramidal neurons of chronically deafferented cortex the relationship between applied current and adapted spike firing frequency is steeper than that in controls (Prince and Tseng, 1993), similar to the homeostatic effects on intrinsic excitability observed in cell cultures (Desai et al., 1999). It remains to be explored whether homeostatic plasticity operates in vivo in a similar way as in cell cultures. Recent evidence suggests that excitatory synapses in intact rodent visual cortex are upregulated after sensory deprivation. Two days of monocular deprivation scales up mEPSC amplitudes by 15–30% in a layer- and age-dependent manner (Desai et al., 2002). These changes are smaller than those seen in culture, probably because sensory deprivation only partially reduces activity compared to the complete activity block in cultures.
Surprisingly, chronic exposure to tetrodotoxin (TTX) reduces epileptogenesis in chronically (10–15 days) isolated cortex (Graber and Prince, 1999). It is possible that TTX blocked all residual neuronal activity after deafferentation and homeostatic plasticity somehow requires a minimal amount of spontaneous firing. Another likely possibility is that additional factors contribute to the epileptogenesis in chronically injured neocortex, such as increased input resistances (Prince and Tseng, 1993) and axonal sprouting (Salin et al., 1995) of layer 5 pyramidal cells with the formation of new synapses. It is not known, however, whether new synapses are formed predominantly on pyramidal cells. In our model, new synapses between pyramidal cells promoted bursting, but an additional equal number of new synapses from pyramidal cells to interneurons did not. It is possible that axonal sprouting and the formation of new synapses is a secondary effect induced by paroxysmal activity (McNamara, 1999). Indeed, the axonal sprouting of corticostriatal neurons after ischemic cortical lesions depends on synchronous neuronal activity in perilesion neocortex (Carmichael and Chesselet, 2002). A previous modeling study concluded that increased pyramidal cell excitability, i.e. increased input resistance (+150%) and decreased capacitance (−50%), and increased NMDA conductance (+100%) were both necessary to reproduce the experimental epileptiform data in slices of chronically isolated cortex (Bush et al., 1999). In our model these changes were sufficient, but not necessary, to evoke propagating burst discharges in acutely deafferented cortex. At variance with the Bush et al. (1999) model, bursts were also evoked when only pyramidal cell excitability was increased but not NMDA conductances. Also, upregulation of AMPA synapses, either alone or with concurrent downregulation of inhibitory synapses, was sufficient to generate network bursts (see Fig. 7D).
If homeostatic synaptic plasticity has a role in post-traumatic epileptogenesis, then interfering with its processes immediately after cortical injury in patients at risk for epilepsy may reduce the risk of developing epilepsy. For example, brain-derived neurotrophic factor (BDNF) prevents the homeostatic plasticity of excitatory (Rutherford et al., 1998) and inhibitory synapses (Rutherford et al., 1997) after chronic activity blockade in cell cultures. Alternatively, pharmacologically induced increased levels of spontaneous activity or low-intensity electrical stimulation of the deafferented area with arrays of input electrodes may eliminate the drive for homeostatic regulation.
Homeostatic plasticity may contribute to epileptogenesis in other conditions with prolonged periods of reduced activity, such as in the GABA withdrawal syndrome (GWS) (Brailowsky et al., 1988). In GWS, chronic perfusion of GABA in the sensorimotor cortex results upon cessation in the appearance of continuous focal epileptic discharges. One difference between GWS and chronically isolated cortex is that extrinsic glutamatergic and cholinergic afferents are left intact. Thus, in addition to intracortical synapses, afferent synapses may be subject to homeostatic plasticity in GWS.
A better understanding of the cellular mechanisms of homeostatic plasticity is needed to further explore its effects in computational models. Which features of neuronal activity are subject to homeostatic regulation? Does homeostatic plasticity regulate activity at the level of individual neurons or at the level of networks? Recent studies have started to address these questions in cell cultures (Burrone et al., 2002). A mechanism similar to homeostatic synaptic plasticity at the single cell level has been explored in models of activity-dependent neurite outgrowth during development (van Ooyen and van Pelt, 1994, 1996). Interesting properties may emerge in these models when networks of excitatory neurons have bistable activity states for some range of excitatory connection strengths, including complex periodic behavior in electrical activity and synaptic connectivity of individual cells (at the slow time scale of neurite outgrowth).
During slow-wave sleep the neocortex is functionally deafferented due to the hyperpolarization of thalamocortical cells (Hirsch et al., 1983) and the reduction in cholinergic inputs, while cortical activity is characterized by synchronized slow (<1 Hz) and delta (1–4 Hz) oscillations (Steriade et al., 1993). The burst discharges in our model and those in slices of chronically deafferented cortex resemble the up-states of the slow oscillation in naturally sleeping cats (Steriade et al., 2001) and cortical slices (Sanchez-Vives and McCormick, 2000), with similar durations, levels of depolarization and numbers of action potentials. Thus, the slow oscillation and the spontaneous burst discharges in chronically deafferented cortex may share a fundamental underlying mechanism that operates in deafferented cortical networks. This mechanism may critically depend on the strength of recurrent excitatory synaptic connections, such that large intact cortical networks during sleep or acutely isolated cortical gyri or hemispheres may generate slow oscillations (Kellaway et al., 1966; Gloor et al., 1977; Timofeev et al., 2000), but small cortical slabs, which lack long-range corticocortical excitatory connections, may only support slow oscillatory activity after upregulation of recurrent excitation.
This research was supported by National Institute of Health (NS40522), Human Frontier Science Program, Howard Hughes Medical Institute and Canadian Institutes for Health Research (MT-3689, MOP-36545, MOP-37862).
References
Abbott LF, Varela JA, Sen K, Nelson SB (
Amitai Y, Gibson JR, Beierlein M, Patrick SL, Ho AM, Connors BW, Golomb D (
Brailowsky S, Kunimoto M, Menini C, Silva-Barrat C, Riche D, Naquet R (
Burns B (
Burns BD, Webb AC (
Burrone J, O'Byrne M, Murthy VN (
Bush PC, Prince DA, Miller KD (
Carmichael ST, Chesselet MF (
Carpenter GA, Grossberg S (
Desai NS, Rutherford LC, Turrigiano GG (
Desai NS, Cudmore RH, Nelson SB, Turrigiano GG (
Echlin F, Battista A (
Furshpan EJ, Potter DD (
Gloor P, Ball G, Schaul N (
Golomb D, Amitai Y (
Golowasch J, Casey M, Abbott LF, Marder E (
Graber KD, Prince DA (
Grafstein B, Sastry P (
Hirsch JC, Fourment A, Marc ME (
Hoffman SN, Salin PA, Prince DA (
Jahr CE, Stevens CF (
Kellaway P, Gol A, Proler M (
Kilman V, van Rossum MC, Turrigiano GG (
LeMasson G, Marder E, Abbott LF (
Leslie KR, Nelson SB, Turrigiano GG (
Li H, Prince DA (
Liao D, Zhang X, O'Brien R, Ehlers MD, Huganir RL (
Lissin DV, Gomperts SN, Carroll RC, Christine CW, Kalman D, Kitamura M, Hardy S, Nicoll RA, Malenka RC, von Zastrow M (
Liu Z, Golowasch J, Marder E, Abbott LF (
Mainen ZF, Sejnowski TJ (
McAllister AK, Stevens CF (
McKinney RA, Debanne D, Gahwiler BH, Thompson SM (
Miller KD (
Murthy VN, Schikorski T, Stevens CF, Zhu Y (
O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL (
Paré D, Shink E, Gaudreau H, Destexhe A, Lang EJ (
Prince DA, Tseng GF (
Purpura D, Housepian E (
Ramakers GJ, Corner MA, Habets AM (
Rao A, Craig AM (
Ribak CE, Reiffenstein RJ (
Rutherford LC, DeWan A, Lauer HM, Turrigiano GG (
Rutherford LC, Nelson SB, Turrigiano GG (
Salin P, Tseng GF, Hoffman S, Parada I, Prince DA (
Sanchez-Vives MV, McCormick DA (
Sharpless S (
Sharpless S, Halpern L (
Siegel M, Marder E, Abbott LF (
Spruston N, Jonas P, Sakmann B (
Steriade M, McCormick DA, Sejnowski TJ (
Steriade M, Timofeev I, Grenier F (
Timofeev I, Grenier F, Bazhenov M, Sejnowski TJ, Steriade M (
Topolnik L, Steriade M, Timofeev I (
Tsodyks MV, Markram H (
Turrigiano GG (
Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (
van den Pol AN, Obrietan K, Belousov A (
van Ooyen A, van Pelt J (
van Ooyen A, van Pelt J (
Wang XJ (
Author notes
1The Salk Institute, Computational Neurobiology Laboratory, La Jolla, CA 92037, USA, 2Program in Neurosciences, University of California San Diego, La Jolla, CA 92093, USA, 3Division of Biological Sciences, University of California San Diego, La Jolla, CA 92093, USA and 4Laboratoire de Neurophysiologie, Université Laval, Québec, Canada G1K 7P4