




Abstract
Predictive genetic testing for Huntington's disease (HD) has revealed early cognitive deficits in asymptomatic gene carriers, such as altered working memory, executive function and impaired recognition memory. The perirhinal cortex processes aspects of recognition memory and the underlying mechanism is believed to be long-term depression (LTD) of excitatory neurotransmission, the converse of long-term potentiation (LTP). We have used the R6/1 mouse model of HD to assess synaptic plasticity in the perirhinal cortex. We report here a progressive derailment of both LTD and short-term plasticity at perirhinal synapses. Layer II/III neurones gradually lose their ability to support LTD, show early nuclear localization of mutant huntingtin and display a progressive loss of membrane integrity (depolarization and loss of cell capacitance) accompanied by a reduction in the expression of D1 and D2 dopamine receptors visualized in layer I of the perirhinal cortex. Importantly, abnormalities in both short-term and long-term plasticity can be reversed by the introduction of a D2 dopamine receptor agonist (Quinpirole), suggesting that alterations in dopaminergic signalling may underlie early cognitive dysfunction in HD.
INTRODUCTION
Huntington's disease (HD) is one of a family of neurodegenerative disorders attributable to an unstable CAG trinucleotide repeat expansion. For HD, this occurs within the open reading frame of the gene that codes for huntingtin (1) and the age at onset of this disorder is determined by the length of the CAG repeat expansion (2). In humans, the symptoms which usually appear in the third to fifth decades of life, often include an impairment of cognitive function that can eventually lead to dementia (3). The primary sites of neurodegeneration are the striatum and cerebral cortex (4–7). Several studies have suggested that early cognitive impairment can appear in patients before the onset of the classical symptoms (8–12). Furthermore, post-mortem studies (7) suggest that the first symptoms (both motor and cognitive) appear in the absence of overt neuronal cell loss, suggesting that impaired cognition is likely to be caused by a synaptic dysfunction rather than a consequence of neuronal cell death. With the recent introduction of genetic testing for the HD gene, it is now possible to assess the cognitive performance of known gene carriers prior to the onset of overt motor symptoms. These studies reveal early alterations in working memory, executive function and recognition memory (13–16). Early cognitive decline has also been correlated with a loss of dopamine receptors in both the striatum and cortex of asymptomatic patients (17,18).
In primates, recognition memory is registered by a change in the activity of neural circuits in the perirhinal cortex (19). Neurones within this brain region respond to novel cues by an increase in action potential firing rate and responsiveness declines as the cue becomes familiar to the animal. The process believed to underlie the neural encoding of recognition memory, the transition of the firing rate from a ‘novel’ to a ‘familiar’ state, is synaptic long-term depression (LTD); an activity-dependent decrease in the efficacy of neurotransmission at glutamatergic synapses within the perirhinal cortex (20).
The identification of the HD gene mutation in 1993 (21) has led to the successful creation of several genetically modified mouse models that recapitulate aspects of this human disease [reviewed in (22)]. To date, the most widely studied models are the R6/1 and R6/2 lines developed by Gillian Bates et al. (23). Briefly, these mice express exon 1 of the human gene under the control of the human huntingtin promoter and carry ∼116 and ∼150 CAG repeats, respectively. Both lines exhibit a progressive phenotype but with very different patterns of severity. The R6/2 line has a greatly shortened life-span, living to approximately 13–18 weeks, whereas the R6/1 line can survive for 12–14 months. Consequently, the latter is considered to be a better model for assessing the subtle and early changes in neural processing and cognition (24) that may occur in human asymptomatic gene carriers.
We report here a series of experiments, using the R6/1 line, designed to test the hypothesis that impaired recognition memory in HD is caused by synaptic and cellular dysfunction in the perirhinal cortex. In particular, we also assessed the role of the dopaminergic signalling system as a possible mediator of synaptic dysfunction in this brain region.
RESULTS
At weaning, transgenic mice were indistinguishable from their littermate controls, but by 13 weeks of age some exhibited hind-limb clasping upon tail suspension and by 19 weeks of age more than 80% exhibited this neurological phenotype [for further details concerning the Open University colony see (24,25)].
Basal neurotransmission at perirhinal synapses
Field potentials were recorded in layer II/III of the perirhinal cortex in response to stimulation of adjacent layer I. At all ages studied, the potentials recorded in transgenic slices were notably different from those recorded in age-matched littermate controls in which they tended to exhibit a second and prolonged component with an onset latency of ∼9 ms (Fig. 1). This late component was not susceptible to receptor pharmacological manipulation (such as dopamine receptor blockade, NMDA and metabotropic glutamate receptor antagonism and GABAergic manipulation; data not shown) and is likely to reflect an alteration in an intrinsic property of layer II/III neurones.
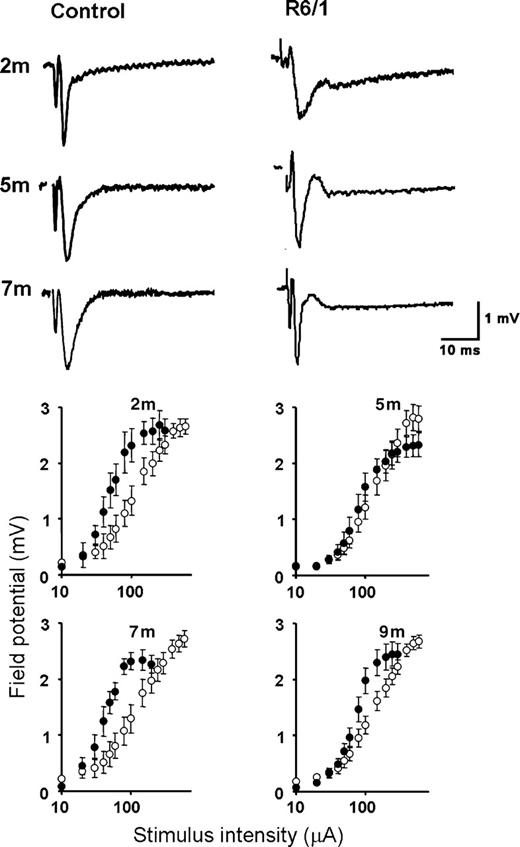
Basal synaptic transmission is altered in transgenic slices. Top: Typical field potentials recorded from non-transgenic littermates (left) and R6/1 transgenic slices (right) at the ages indicated. Note the prominent late component seen at all ages in transgenic slices. Bottom: Input/output relationships in transgenic and non-transgenic littermates at the ages indicated. Transgenic and non-transgenic data points are denoted by filled and open symbols, respectively. Each data point is expressed as pooled EPSP amplitude (mean±SEM) from 6 to 18 slices (three to six mice) per condition.
Input/output curves were constructed and are shown in Figure 1. Once again, there was a marked difference between transgenic and control littermates. At 2, 7 and 9 months of age, the transgenic input/output curves were shifted leftwards, suggesting an enhanced level of synaptic activation at transgenic synapses or a loss of inhibitory neuromodulation.
Field potentials evoked in both transgenic and non-transgenic slices were abolished in the presence of an AMPA glutamate receptor antagonist (10 µm, 6-cyano-7-nitroquinoxaline-2,3-dione; data not shown) confirming that glutamatergic neurotransmission is largely intact at superficial perirhinal synapses.
Altered short-term synaptic plasticity
Short-term plasticity was investigated using a paired-pulse protocol. Stimuli were applied at an intensity that evoked a single response ∼80% of the maximum of peak (negative) amplitude. Paired-pulses were applied at intervals between 20 and 2000 ms. In control slices, the second response was smaller than the first at all intervals and all ages studied (Fig. 2). Responses recorded in transgenic slices differed in an age-dependent manner. At 2 and 5 months of age, the paired-pulse profiles were similar to those seen in control slices in which they too exhibited a paired-pulse depression (PPD). However, at 7 and 9 months, the outcome of paired-pulse stimulation diverged markedly from those seen in age-matched controls. Instead of exhibiting PPD, the paired-pulse profiles were shifted upwards, exhibiting a more facilitatory profile [paired-pulse facilitation (PPF)]. Both profiles were significantly different compared with controls (two-way ANOVAs: P<0.001, F11,263=4.285 at 7 months and P<0.008, F11,378=2.34 at 9 months).
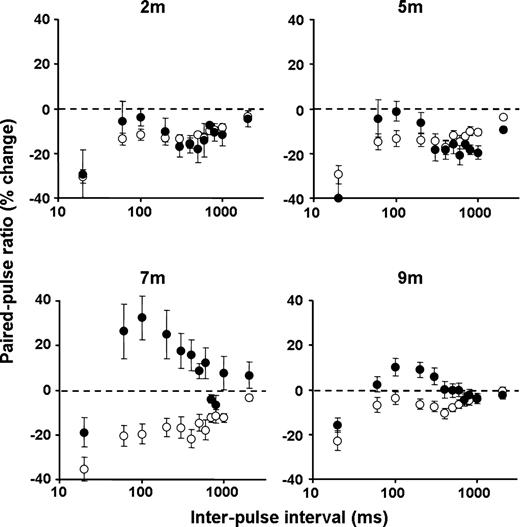
Age-dependent alteration in transgenic paired-pulse ratio. Non-transgenic slices consistently expressed PPD at all ages, whereas transgenic slices exhibited an age-dependent shift towards a more facilitatory profile. Transgenic and non-transgenic data points are denoted by closed and open symbols, respectively. Each data point is expressed as pooled paired-pulse ratio (mean±SEM) from 5 to 7 slices (three to six mice) per condition.
Loss of LTD
Long-term synaptic plasticity was also investigated in perirhinal cortical slices. LTD was readily induced in slices prepared from non-transgenic littermate controls (Fig. 3) at all ages studied. However, the pattern of LTD expression was strikingly different in slices prepared from transgenic mice (Fig. 3). At 2 months of age, the absolute magnitude of LTD expressed in transgenic slices was ∼120% greater than that seen in control slices (P<0.05). At 5 months, this position was reversed and there was now an evident trend towards a reduction in LTD expression. By 7 months of age, the majority of transgenic slices (86%) were no longer able to express LTD (P<0.0001). At 9 months, LTD was rarely observed in transgenic slices (<10%).
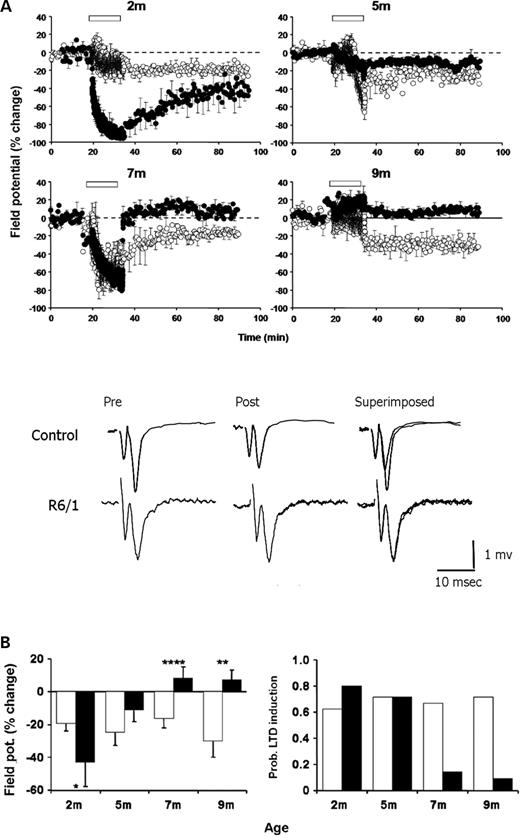
Progressive derailment of LTD in transgenic slices. (A) LTD was reliably induced in non-transgenic littermate slices at all ages examined. In contrast, LFS (open bar) induced an enhanced LTD at 2 months in transgenic slices followed by a progressive age-dependent decline in the ability to support LTD. Each data point is the pooled EPSP area (mean±SEM) normalized with respect to the 10-min period prior to LFS plotted against time. Transgenic and non-transgenic data indicated by closed and open symbols, respectively. Each pooled data set represents between 5 and 16 slices (three to six mice). Representative traces prior to and 1 h post-conditioning are shown from experiments recorded in slices prepared from 9-month-old animals (traces are averages of five successive responses). (B) Left: Summary histogram of mean EPSP change measured for the 50–60 min period post-LFS in non-transgenic (open histobars) and transgenic (filled histobars) slices. *P<0.05; **P<0.003; ****P<0.0001. Right: Probability of LTD induction. Non-transgenic tissue expressed LTD in >60% of experiments at all ages studied. In contrast, the probability of induction in transgenic slices was 80% at 2 months, but then declined until LTD was rarely induced at 7 and 9 months of age.
Progressive decline of intrinsic intracellular properties
The basal intracellular properties of layer II/III pyramidal neurones were assessed using standard sharp microelectrodes and are summarized in Figure 4 and Table 1. Recordings were made from more than 51 non-transgenic and 27 transgenic pyramidal neurones. The resting membrane potential of non-transgenic neurones varied from −66 to −90 mV with a mean resting membrane potential of −79.2±0.6 mV (n=51 cells; data from non-transgenic neurones have been pooled across all ages examined, as no age-dependent differences were detectable) and followed a Gaussian distribution (P>0.3). For 35 of these cells, a family of membrane voltage responses to a series of square (200 ms) hyperpolarising current pulses (from −0.2 to −0.8 nA) were recorded and the slope of steady-state voltage deflection against current injection used to calculate the membrane input resistance (Rin) for each neurone. In each case, the r2 value for each slope was greater than 0.98 and the mean input resistance was 39.2±2.4 MΩ. The membrane capacitance (Cm) was calculated from the membrane input resistance and the time constant (τm) using the following relationship: Cm=τm/Rin. Values for the time constant for each neurone were obtained using the protocol of Bindman et al. (26); (see Materials and Methods) and the mean time constant was found to be 9.1±0.7 ms. The membrane capacitance was then calculated for each cell and the mean value was found to be 0.24±0.02 nF. The specific membrane capacitance of biological membranes is ∼10 nF/mm2 (27); applying this constant to the data obtained from non-transgenic layer II/III pyramidal neurones suggests that these perirhinal cells had an approximate surface area of 0.02 mm2.
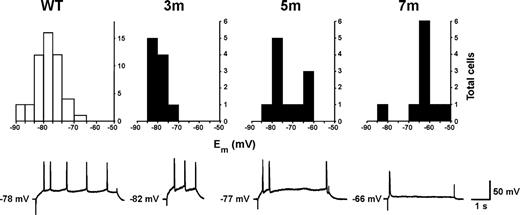
Basal intrinsic membrane properties are changed in an age-dependent manner. Top: Frequency distribution for resting membrane potential (Em). The resting membrane potential was found to be normal in cells from 3-month-old R6/1 slices. At 5 months, two populations of cells were discernable: those with normal resting membrane potentials and those that were depolarized. At 7 months, the majority of cells were found to be depolarized. Non-transgenic cells show a Gaussian distribution for resting membrane potentials (NB. Different ordinate axes for transgenic plots). Bottom: Typical intracellular recordings in response to a depolarising square pulse of current. Cells from non-transgenic and young R6/1 animals show regular action potential firing whilst cells from older R6/1 animals show a loss in the ability to sustain action potential firing.
Electrophysiological properties of neurones within layer II/III of the perirhinal cortex
| Cellular property | Mean±SEM | |||
|---|---|---|---|---|
| WT (n=35) | 3 months (n=6) | 5 months (n=7) | 7 months (n=5) | |
| Em | −79.2±0.6 mVa | −79.4±1.0 mVb | 69.5±2.5 mVc | −63.1±2.6 mVd |
| Rin | 39.2±2.4 MΩ | 42.5±2.5 MΩ | 47.5±2.5 MΩ | 57.8±6.7 MΩ† |
| τm | 9.1±0.7 ms | 10.0±0.3 ms | 9.8±0.5 ms | 6.6±0.4 ms |
| Cm | 0.24±0.02 nF | 0.23±0.02 nF | 0.21±0.02 nF | 0.12±0.01 nF* |
| APthreshold | −51.3±1.3 mV | −47.3±2.5 mV | −51.4±4.5 mV | −52.9±3.9 mV |
| APovershoot | +26.8±2.7 mV | +28.2±1.5 mV | +27.3±5.4 mV | +5.2±2.1 mV** |
| APamplitude | 78.2±1.8 mV | 75.7±1.6 mV | 78.7±6.9 mV | 60.0±2.1 mV* |
| APwidth | 1.83±0.09 ms | 1.73±0.10 ms | 2.75±0.48 ms | 4.05±0.21 ms* |
| Cellular property | Mean±SEM | |||
|---|---|---|---|---|
| WT (n=35) | 3 months (n=6) | 5 months (n=7) | 7 months (n=5) | |
| Em | −79.2±0.6 mVa | −79.4±1.0 mVb | 69.5±2.5 mVc | −63.1±2.6 mVd |
| Rin | 39.2±2.4 MΩ | 42.5±2.5 MΩ | 47.5±2.5 MΩ | 57.8±6.7 MΩ† |
| τm | 9.1±0.7 ms | 10.0±0.3 ms | 9.8±0.5 ms | 6.6±0.4 ms |
| Cm | 0.24±0.02 nF | 0.23±0.02 nF | 0.21±0.02 nF | 0.12±0.01 nF* |
| APthreshold | −51.3±1.3 mV | −47.3±2.5 mV | −51.4±4.5 mV | −52.9±3.9 mV |
| APovershoot | +26.8±2.7 mV | +28.2±1.5 mV | +27.3±5.4 mV | +5.2±2.1 mV** |
| APamplitude | 78.2±1.8 mV | 75.7±1.6 mV | 78.7±6.9 mV | 60.0±2.1 mV* |
| APwidth | 1.83±0.09 ms | 1.73±0.10 ms | 2.75±0.48 ms | 4.05±0.21 ms* |
Intracellular recordings were made from cells within layer II/III of the perirhinal cortex. The current–voltage relationship was used to calculate input resistance, membrane time constant, and from these values, the capacitance of the cell. Action potential (AP) amplitude was calculated from threshold to maximum overshoot. AP width was measured at half amplitude. Significant differences from the previous age group are indicated. n, number of cells.
an=51 cells.
bn=10 cells.
cOver all mean; but two sub-populations were discernable, one showing normal Em the other with depolarized Em (see main text and Fig. 4); n=11 cells.
dn=10 cells.
*P<0.05.
**P<0.01.
†P<0.001 two-tailed, unpaired t-test.
Electrophysiological properties of neurones within layer II/III of the perirhinal cortex
| Cellular property | Mean±SEM | |||
|---|---|---|---|---|
| WT (n=35) | 3 months (n=6) | 5 months (n=7) | 7 months (n=5) | |
| Em | −79.2±0.6 mVa | −79.4±1.0 mVb | 69.5±2.5 mVc | −63.1±2.6 mVd |
| Rin | 39.2±2.4 MΩ | 42.5±2.5 MΩ | 47.5±2.5 MΩ | 57.8±6.7 MΩ† |
| τm | 9.1±0.7 ms | 10.0±0.3 ms | 9.8±0.5 ms | 6.6±0.4 ms |
| Cm | 0.24±0.02 nF | 0.23±0.02 nF | 0.21±0.02 nF | 0.12±0.01 nF* |
| APthreshold | −51.3±1.3 mV | −47.3±2.5 mV | −51.4±4.5 mV | −52.9±3.9 mV |
| APovershoot | +26.8±2.7 mV | +28.2±1.5 mV | +27.3±5.4 mV | +5.2±2.1 mV** |
| APamplitude | 78.2±1.8 mV | 75.7±1.6 mV | 78.7±6.9 mV | 60.0±2.1 mV* |
| APwidth | 1.83±0.09 ms | 1.73±0.10 ms | 2.75±0.48 ms | 4.05±0.21 ms* |
| Cellular property | Mean±SEM | |||
|---|---|---|---|---|
| WT (n=35) | 3 months (n=6) | 5 months (n=7) | 7 months (n=5) | |
| Em | −79.2±0.6 mVa | −79.4±1.0 mVb | 69.5±2.5 mVc | −63.1±2.6 mVd |
| Rin | 39.2±2.4 MΩ | 42.5±2.5 MΩ | 47.5±2.5 MΩ | 57.8±6.7 MΩ† |
| τm | 9.1±0.7 ms | 10.0±0.3 ms | 9.8±0.5 ms | 6.6±0.4 ms |
| Cm | 0.24±0.02 nF | 0.23±0.02 nF | 0.21±0.02 nF | 0.12±0.01 nF* |
| APthreshold | −51.3±1.3 mV | −47.3±2.5 mV | −51.4±4.5 mV | −52.9±3.9 mV |
| APovershoot | +26.8±2.7 mV | +28.2±1.5 mV | +27.3±5.4 mV | +5.2±2.1 mV** |
| APamplitude | 78.2±1.8 mV | 75.7±1.6 mV | 78.7±6.9 mV | 60.0±2.1 mV* |
| APwidth | 1.83±0.09 ms | 1.73±0.10 ms | 2.75±0.48 ms | 4.05±0.21 ms* |
Intracellular recordings were made from cells within layer II/III of the perirhinal cortex. The current–voltage relationship was used to calculate input resistance, membrane time constant, and from these values, the capacitance of the cell. Action potential (AP) amplitude was calculated from threshold to maximum overshoot. AP width was measured at half amplitude. Significant differences from the previous age group are indicated. n, number of cells.
an=51 cells.
bn=10 cells.
cOver all mean; but two sub-populations were discernable, one showing normal Em the other with depolarized Em (see main text and Fig. 4); n=11 cells.
dn=10 cells.
*P<0.05.
**P<0.01.
†P<0.001 two-tailed, unpaired t-test.
In each cell, action potentials were evoked by the injection of depolarising current (0.2–2 nA) and the firing threshold, overshoot potential, action potential amplitude and duration recorded (summarized in Table 1). Cells typically fired within the first 500 ms of a depolarising current step and exhibited rapid accommodation.
Recordings made in transgenic neurones in slices prepared from 3-month-old mice were similar to those obtained in non-transgenic slices (Table 1). However, at 5 months of age it was possible to discern two populations of neurones, the first had a normal resting membrane potential (−77.5±0.9 mV, n=6; P>0.3) and the second displayed a depolarized resting membrane potential (−64.8±2.5 mV, n=5; P<0.001), however, the overall mean value was −69.5±2.5 mV. By 7 months of age the vast majority of cells exhibited a highly depolarized membrane potential (−63.1±2.6 mV, n=10; P<0.0001) and this was evident further at 9 months of age (−61.1±2.1 mV, n=8; P<0.0001). Input resistances in transgenic cells at 3 months of age were similar to those seen in control cells (Table 1). At 5 months, there was a trend towards an increased input resistance and at 7 months there was a highly significant increase (Table 1). The membrane time constant was found to be normal at both 3 and 5 months of age. At 7 months, there was a modest reduction in the mean time constant but this did not achieve significance. Likewise, the mean membrane capacitance, at 3 and 5 months, was similar to the control value. However, at 7 months there was a 50% reduction in the membrane capacitance corresponding to an approximation of cell surface area of 0.01 mm2. The properties of action potentials evoked in transgenic slices from 3, 5 and 7 month mice are summarized in Table 1. The action potential firing threshold was not altered at any age but overshoot and duration was significantly altered at 7 months. The pattern of action potential generation was also affected in a progressive manner. At 3 months of age, depolarization gave rise to a pattern of regular cell firing at a frequency of ∼1 Hz. At 5 months, however, the cells fired immediately following depolarization, but failed to maintain regular action potential firing. A similar deficit was seen for cells recorded in slices prepared from 7-month-old transgenic animals.
Localization of mutant huntingtin and aggregate formation
The localization of mutant huntingtin and aggregates were investigated within the perirhinal cortex of R6/1 mice using a polyclonal antibody (S830) raised against exon 1 of human huntingtin containing 52 glutamines. At all ages, slices prepared from non-transgenic animals exhibited no immunoreactivity. At 1 month, diffuse staining was discernible in transgenic material in a small number of cell nuclei located in layer II/III. At 3 months of age, diffuse nuclear staining was observed in a highly cortical layer-specific manner (Fig. 5). Nuclei of layer II/III pyramids were immunoreactive for mutant huntingtin while the deeper layers of the cortex showed little if any staining. At 7 months, diffuse nuclear staining for mutant huntingtin was seen in all cortical layers and frank nuclear aggregates were evident in most neurones. Cytoplasmic aggregates were also present, but were largely distributed to the deeper layers of the cortex. The pattern of progression and distribution for both nuclear localization and aggregate formation revealed by mutant huntingtin immunoreactivity are summarized in Table 2.
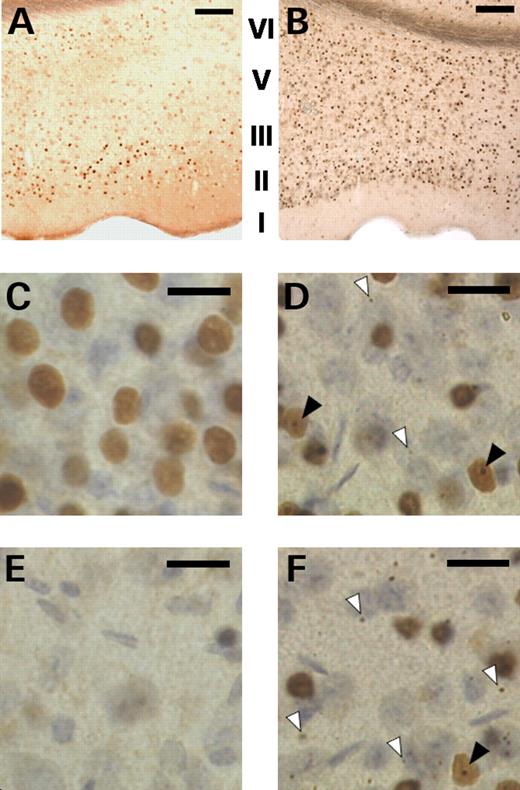
Mutant huntingtin expression in the perirhinal cortex is age- and layer-specific. (A and B) slices from 3 and 7 month-old transgenic mice respectively showing differential and regional-specific diffuse nuclear staining in the perirhinal cortex. (C and D) higher powered images of layers II/III taken at 3 and 7 months respectively; note the distinct pattern of immunostaining in the nuclei of the principal cells. (E and F) higher powered images of layers V/VI taken at 3 and 7 months respectively; note the near absence of nuclear staining at 3 months. At 7 months of age distinct nuclear inclusions (filled arrowheads) and micro-aggregates within the neuropil (open arrowhead) were observed in layers II/III (D) and layers V/VI (F). Scale bar: Images A and B (100× magnification) bar represents 250 µm; images C–F (400× magnification) bar represents 20 µm. Cortical layers I–VI indicated in images A and B; note the absence of layer IV in perirhinal cortex.
Layer-specific and cellular localisation of mutant huntingtin
| 1 month | 3 month | 7 month | |||||||
|---|---|---|---|---|---|---|---|---|---|
| NS | NII | NPI | NS | NII | NPI | NS | NII | NPI | |
| Layer I | − | − | − | − | − | − | − | − | − |
| Layer II/III | + | − | − | +++ | − | − | ++++ | ++ | + |
| Layer V/VI | − | − | − | + | − | − | +++ | ++ | ++ |
| 1 month | 3 month | 7 month | |||||||
|---|---|---|---|---|---|---|---|---|---|
| NS | NII | NPI | NS | NII | NPI | NS | NII | NPI | |
| Layer I | − | − | − | − | − | − | − | − | − |
| Layer II/III | + | − | − | +++ | − | − | ++++ | ++ | + |
| Layer V/VI | − | − | − | + | − | − | +++ | ++ | ++ |
–, none detected; +, low levels detected; ++, moderate detection; +++, high detection; NS, nuclear staining; NII: neuronal intranuclear inclusion; NPI: neuropil inclusion.
Layer-specific and cellular localisation of mutant huntingtin
| 1 month | 3 month | 7 month | |||||||
|---|---|---|---|---|---|---|---|---|---|
| NS | NII | NPI | NS | NII | NPI | NS | NII | NPI | |
| Layer I | − | − | − | − | − | − | − | − | − |
| Layer II/III | + | − | − | +++ | − | − | ++++ | ++ | + |
| Layer V/VI | − | − | − | + | − | − | +++ | ++ | ++ |
| 1 month | 3 month | 7 month | |||||||
|---|---|---|---|---|---|---|---|---|---|
| NS | NII | NPI | NS | NII | NPI | NS | NII | NPI | |
| Layer I | − | − | − | − | − | − | − | − | − |
| Layer II/III | + | − | − | +++ | − | − | ++++ | ++ | + |
| Layer V/VI | − | − | − | + | − | − | +++ | ++ | ++ |
–, none detected; +, low levels detected; ++, moderate detection; +++, high detection; NS, nuclear staining; NII: neuronal intranuclear inclusion; NPI: neuropil inclusion.
Progressive loss of dopamine receptors in the perirhinal cortex
The shift in the paired pulse profile from depression to facilitation is indicative of a change in a neuromodulatory process. The neurotransmitter dopamine is known to be a powerful neuromodulator at cortical synapses and importantly, several studies have highlighted a loss of dopamine receptors within the striatum of R6/2 mice (28–31). We therefore investigated the hypothesis that loss of dopamine receptor expression in the cortex might contribute to the synaptic phenotype observed in the perirhinal cortex of R6/1 mice.
At the end of recording, slices were retrieved from the recording chamber, fixed and treated with antibodies specific for either D1A or D2 dopamine receptors and then labelled with a fluorescent marker. In order to determine the specificity of the D2 dopamine receptor antibody, slices prepared from an adult D2 knock-out mouse (32) were processed together with transgenic and non-transgenic slices. Crucially, very little fluorescence was detected in the D2 knock-out slices, indicating the high specificity of the D2 antibody used in this study (Fig. 6). While a D1 dopamine receptor knock-out mouse was not available at the time this study was conducted, we were nonetheless able to verify the specificity of the D1 antibody used as application of only the primary or the secondary antibody revealed very little non-specific staining.
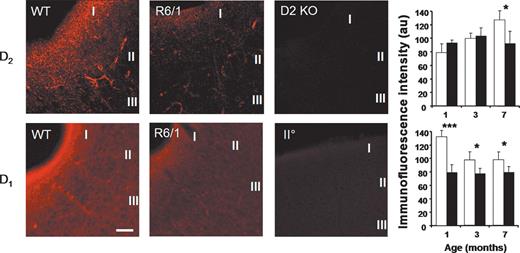
Altered dopamine receptor expression in the R6/1 cortex. Top: D2 dopamine receptor levels, measured using immunofluoresence, were found to be reduced in layer I of the perirhinal cortex in R6/1 mice. Representative fluorescent (confocal) images prepared from 7-month-old non-transgenic (WT) and transgenic (R6/1) mice are shown together with material processed from a D2 dopamine receptor knock-out (D2KO) mouse. There is a clear reduction in immunofluoresence observed in the R6/1 section. No staining was seen in the perirhinal cortex of the D2KO mouse demonstrating specificity of the antibody used. Histograms show the detection levels of D2 dopamine receptors in layer I of the perirhinal cortex at 1, 3 and 7 months of age. There is a clear age-dependent increase in immunofluoresence in non-transgenic tissue, this increase is not maintained in R6/1. Bottom: D1 dopamine receptors also demonstrate an age-dependent reduction in expression levels. Representative images prepared from 7-month-old non-transgenic (WT) and transgenic (R6/1) mice are shown together with control material processed using only the secondary (IIO) antibody (indicating the relative specificity of the D1 antibody used). The histogram shows receptor detection at 1, 3 and 7 months. In non-transgenic perirhinal cortex, an age-dependent decrease in D1 dopamine receptor expression is seen. From the earliest age examined, D1 receptor levels in R6/1 cortex were reduced compared with controls. (*P<0.05; ***P<0.0001; Fisher LSD test). Images taken with a 40× objective; scale bar: 100 µm (D1 WT image). Cortical layers I–III as indicated (three animals used for each condition).
In all sections labelled for D1 and D2 dopamine receptors, the majority of staining was within layer I of the perirhinal cortex and therefore only layer I is considered here for analysis. In sections prepared from non-transgenic mice, an age-dependent increase in D2 dopamine receptors was evident (Fig. 6). At 3 months of age, receptor detection increased to 126% of that seen at 1 month. While this did not reach significance at this age, a further increase, which was significant, was apparent by 7 months (161% increase in fluorescence with respect to that seen at 1 month; P<0.05). Slices from R6/1 animals showed a similar pattern of increasing levels of fluorescence at 3 months, but at 7 months there was, instead, a significant decrease compared with age-matched controls (72%; P<0.05).
In contrast, D1 receptors showed a developmental down-regulation in expression levels in slices prepared from non-transgenic animals (Fig. 6). At 3 months of age, D1 receptor detection was only 73% of 1 month levels (P<0.05) and remained at this level at 7 months. From the earliest age examined (1 month), there was a significant decrease in fluorescence in R6/1 slices compared with age-matched littermates. At 1 month, fluorescence was 60% of control levels (P<0.0001), 78% at 3 months (P<0.05) and 80% at 7 months (P<0.05).
Recovery of normal plasticity in R6/1 perirhinal cortex
A reduction in dopamine release or/and reduced levels of dopamine receptor expression reported here, might be sufficient to account for the altered plasticity seen in R6/1 perirhinal cortex. To assess the role of D2 dopamine receptors in the modulation of synaptic plasticity at perirhinal synapses, the D2 receptor agonist Quinpirole was applied to transgenic slices. Unexpectedly, exposure to Quinpirole (10 µm) had a dramatic effect on both short-term and long-term plasticity (Fig. 7).
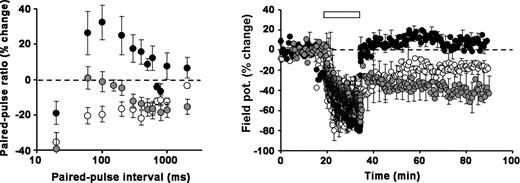
D2 dopamine receptor activation restores normal synaptic plasticity. Left: Paired-pulse ratio. In tissue prepared from 7-month-old animals application of the D2 receptor agonist, Quinpirole (10 µm), to transgenic slices shifted the paired-pulse ratio downwards, towards a less facilitatory profile (grey symbols, n=9) that resembled age-matched controls. Non-transgenic (open symbols, n=6) and non-drug treated transgenics (filled symbols, n=7) are shown for comparison. Right: LTD. Activation of D2 receptors enables R6/1 perirhinal synapses to support LTD. In one set of slices prepared from 7-month-old transgenic animals LFS was applied prior to (filled symbols) and after the addition of 10 µm Quinpirole to the bathing medium (grey symbols, n=9). The addition of the D2 receptor agonist reversed the apparent loss of LTD induction in R6/1 slices. LTD in non-transgenic 7-month-old animals shown for comparison (open symbols, n=6). LFS indicated by bar.
Exposure to Quinpirole reduced the degree of facilitation seen in the transgenic paired-pulse profile at both 5 and 7 months (9 months not tested). Indeed, the profiles were comparable with those seen in aged-matched controls (in the absence of Quinpirole) (two-way ANOVA: P>0.3, F11,527=1.107). The action of Quinpirole was restricted to transgenic slices as it had no effect on paired-pulse profiles in slices prepared from non-transgenic animals.
Remarkably, exposure to Quinpirole restored the ability of transgenic slices to support LTD at perirhinal cortical synapses. At 5 months, Quinpirole-treated slices readily expressed LTD (−31.3±4.1%, n=6; P<0.02 compared with untreated transgenic slices) that was similar in magnitude to that expressed in untreated control slices (P>0.3). In a second series of experiments, LFS was applied to a set of slices prepared from 7-month-old transgenic mice prior to and then after the bath application of Quinpirole. Consistent with earlier observations, LFS failed to induce LTD in the absence of Quinpirole, however, once Quinpirole had been added to the bathing medium, LFS successfully induced LTD (−42.1±13.7%, n=9; P<0.01, paired t-test). While Quinpirole had a dramatic effect on the induction of LTD in transgenic slices, it had no observable effect on the induction of LTD in age-matched control slices (P>0.5).
DISCUSSION
This is the first electrophysiological study to examine long-term synaptic plasticity within the neocortex of a mouse model of HD. Synaptic abnormalities at striatal synapses and alterations in long-term plasticity at hippocampal synapses have been reported previously in several mouse models of HD (33–42). The hippocampal studies reported a decrease in the ability of hippocampal synapses to express long-term potentiation (LTP), the converse of LTD. One of these studies (41), using R6/2 mice, noted that a conspicuous feature of transgenic CA1 synapses was the expression of an NMDA receptor-dependent form of LTD, a form of plasticity that was absent in age-matched controls (43). A similar augmentation of LTD has also been recently reported in R6/1 mice (24), demonstrating a progressive derailment of plasticity at CA1 synapses which was manifest prior to the development of a motor phenotype. We report here that augmentation of LTD is not a generic characteristic of HD synapses. We demonstrate an age-dependent, but biphasic change in the ability of perirhinal synapses to support LTD together with a marked alteration in short-term plasticity revealed by paired-pulse stimulation. Importantly, our observations are consistent with the view that altered plasticity in the perirhinal cortex of human HD gene carriers might contribute to deficits in cognition and recognition memory (13,19). Deficits in cortex-dependent cognition and neocortical plasticity have been reported in presymptomatic R6/1 mice. Neuroplasticity within the barrel-cortex of these mice, measured using the indirect technique of 2-deoxyglucose brain mapping, is severely suppressed and is associated with a failure in a whisker-dependent associative discriminatory task (44,45). These observations, together with the findings presented here, strongly suggest that plasticity within the neural circuits of the R6/1 cortex is severely impaired and is likely to be mediated by deficits in the plastic properties of glutamatergic synapses.
The electrical membrane properties of R6/1 perirhinal neurones also exhibit progressive deterioration. At 1 month of age transgenic neurones are indistinguishable from non-transgenic neurones, however, by 3 months of age some transgenic neurones show signs of abnormality. At 7 months, the majority of transgenic perirhinal neurones are abnormal. A similar pattern of neuronal dysfunction has also been reported for CA1 neurones in the R6/1 hippocampus (24). The pathogenic machinery underlying the alterations in membrane potential, input resistance and cell capacitance reported here is not known, however, R6/1 cortical neurones do recapitulate many of the changes previously reported in striatal medium spiny neurones in the R6/2 mouse model (37–39). Alterations in membrane potential and input resistance are consistent with a global change in a membrane property such as altered potassium and/or sodium conductance. Recently, changes in potassium channel number have been reported in both CAG100 knock-in and symptomatic R6/2 mice (46). Although alterations in sodium channel activity have not been described in HD models, mRNA analysis has suggested a reduction in sodium channel expression in symptomatic R6/2 mice (30,31). It is also possible that changes in the extracellular concentration of the excitatory amino acid, glutamate, might contribute to membrane depolarization. Extracellular glutamate levels have been found to be increased in both the cortex and striatum of R6/2 and R6/1 mice; changes associated with a corresponding reduction in mRNA and protein expression for the Glu1 glutamate transporter (47). Interestingly, it has recently been reported that glutamate currents are reduced in isolated R6/2 cortical neurones at an early age (48), possibly representing a compensatory mechanism that is sensitive to extracellular glutamate levels.
While neuronal cell death does not appear to be a major feature of most mouse models of HD, the majority do exhibit a profound loss of brain mass (49); a process believed to be related to a progressive retraction and malformation of neuritic processes (50,51). The reduction in cell capacitance reported here is indicative of a progressive loss of membrane surface area. Consistent with this view, it has recently been shown that there is a reduction in both dendritic spine density and spine length in neurones in the anterior cingulate cortex and striatum of 8 month old R6/1 mice (51). Similar changes in dendritic morphology have been reported in the late-stages of human HD (52) and spine loss has also been observed in R6/2 (38) and full-length huntingtin cDNA mice (53).
Aggregate deposition is believed to be a hallmark of HD (22,50,54) and neuronal intranuclear inclusions (NII) were seen in layer II/III perirhinal neurones at 7 months. The benign or malign nature of such inclusions is currently the source of much debate (55). While a consensus has yet to be reached, there is a growing body of evidence that cell death and/or dysfunction does not correlate with the presence of aggregates but rather with the accumulation of soluble mutant huntingtin within the nucleus (56). In agreement with this, we found that the development of aberrant cortical plasticity at perirhinal synapses was associated more closely with diffuse nuclear staining for mutant huntingtin than with the frank expression of NIIs.
Using confocal microscopy, we also show that deficits in synaptic plasticity reported here correlate with reduced levels of dopamine receptor expression in layer I of the perirhinal cortex. This is the first demonstration of a layer-specific reduction in the levels of dopamine receptor expression in the neocortex of a HD mouse model. Reductions in striatal dopamine receptors, using immunofluoresence, in situ hybridization and ligand-binding techniques, have also been shown in R6/2 mice (28,29,57). Similar reductions in dopamine receptor levels have been recorded, using the non-invasive technique of positron emission tomography, in both asymptomatic and symptomatic human gene carriers (17,18). Importantly, the dopamine receptor reductions reported here underscore the value of the R6 mouse models in studying dopaminergic deficits in HD.
The loss of dopamine receptors in the perirhinal cortex of R6/1 mice was largely restricted to layer I. Interestingly, this was temporally associated with a co-incident reduction in the cell capacitance of layer II/III neurones, suggesting that alterations in, or retraction of, the apical dendrites of these neurones underlie the loss of dopamine receptor immunofluoresence.
The most significant finding reported here is the apparent reversal of perirhinal synaptic plasticity deficits by a D2 dopamine receptor agonist. In rodents, D2 receptor activation has been shown to enhance LTD (58,59) and modulate LTP (60–62). In humans, the role of dopamine receptors as mediators of synaptic plasticity and cognition has recently become an area of clinical interest as dopaminergic agents may have efficacy in rehabilitating memory and motor function after brain trauma (63). In healthy participants, antagonism of D2 receptors induces cognitive deficits in learning (64) while verbal learning is improved by administration of levodopa (65). Moreover, it has recently been demonstrated that levodopa can improve the formation of motor memory in both healthy subjects and chronic stroke patients (63,66). However, the most convincing demonstration that D2 receptors mediate aspects of human cortical plasticity comes from the recent work of Nitsche et al. (67). They used transcranial direct current stimulation to induce changes in cortical excitability, in human participants, that lasted for many tens of minutes. These changes were abolished in the presence of a D2 receptor antagonist and enhanced when D2 receptors were pharmacologically activated. Indeed, it is possible to recreate the key features of the R6/1 perirhinal cortical phenotype in brain slices prepared from wild-type animals by addition of the D2 receptor antagonist, remoxipride, to the bathing medium (68).
The observation that normal cortical plasticity can be restored in slices from symptomatic R6/1 mice by the application of a D2 dopamine agonist suggests that (i) the remaining D2 dopamine receptors are functional and (ii) the deficit is likely to be related to the release or bioavailability of dopamine. In a parallel series of experiments we have also discovered that LTP in the medial prefrontal cortex of R6/1 mice is impaired and that this impairment can be fully reversed by the addition of a D1 dopamine agonist (69); once again, suggesting that the deficit is related to a lack of dopamine. In support of this view we have recently concluded a series of experiments using amperometry to measure the real-time release of dopamine in the R6/1 striatum. Crucially, by 9 months of age, a time when LTD is entirely absent at R6/1 perirhinal synapses, the nigral-striatal pathway in R6/1 striatal slices is no longer capable of sustaining electrically evoked dopamine release. A similar reduction in striatal dopamine release has also been reported in R6/2 mice (70). [While these studies indicate a failure in the release of dopamine, it should be noted that the source of perirhinal cortical dopamine is the ventral tegmental area and not the substantia nigra]. Behavioural and neurochemical studies have also revealed dopaminergic deficits in HD mouse models. R6/2 mice are less responsive to the administration of cocaine and methamphetamine (71) and basal level of striatal dopamine, measured using microdialysis, is reported to be reduced in R6/1 (72).
In summary, we demonstrate an age-dependent derailment of both short-term and long-term plasticity at perirhinal synapses in an exon-1 model of HD and provide evidence that this is related to a specific deficit in signalling at D2 dopamine receptors. These data strongly suggest that therapy targeted to D2 dopamine receptors may provide a means to restore cognitive function in HD.
MATERIALS AND METHODS
Slice preparation and electrophysiology
Coronal brain slices containing the perirhinal cortex were prepared from transgenic and non-transgenic littermates. The mice were obtained from an in-house R6/1 mouse colony, maintained by breeding hemizygotic R6/1 males with CBA×C57BL/6 females and housed in an enriched environment (73). All the mice used in this study carried a R6/1 transgene containing ∼116 CAG repeats (23,25). Briefly, mice were killed by cervical dislocation and decapitation in accordance with UK legislation (Animal Scientific Procedures Act 1986). The brain was then rapidly removed and placed in chilled (1–4°C) dissection artificial cerebrospinal fluid [ACSF; comprising (in mm) 124 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 10 d-glucose, 10 MgSO4 and zero calcium] aerated with 95% O2 and 5% CO2 (Carbogen; British Oxygen Company). A block containing the perirhinal cortex was then fixed to a silicone stage with cyanoacrylate glue (Radio Spares, Corby, UK) and submerged in ice-cool dissection ACSF continuously bubbled with Carbogen. Coronal sections (400 µm in thickness) were cut with a manual vibrotome (Campden Instruments Ltd., Sileby, UK). Slices were then trimmed to leave the perirhinal and adjacent temporal (dorsal side) and entorhinal/ectorhinal (ventral side) cortices and then transferred to an interface recording chamber (Scientific Systems Design Inc., Mercerville, NJ, USA). Standard ACSF (modified dissection ACSF containing 2 mm CaCl2 and 1 mm MgSO4), bubbled with Carbogen, was continuously perfused into the chamber at a flow rate of ∼125 µl/min (Watson-Marlow Ltd., UK) and maintained at a temperature of 27±0.1°C (Proportional Temperature Controller; Scientific Systems Design Inc.). Once placed in the recording chamber, slices were allowed to recover for 90 min prior to experimentation.
Extracellular recording electrodes were pulled from borosilicate glass capillaries (Harvard Apparatus Limited, Edenbridge, UK) using a horizontal micropipette puller (P87, Sutter Instruments Corp., USA) and filled with 1 m sodium chloride and 2% pontamine sky-blue 5BX (electrode impedance: 4–8 MΩ). Recording electrodes were connected to an Axoclamp 2B DC-preamp amplifier (Axon Instruments Inc., Foster City, CA, USA), via an Axoclamp head-stage (gain: 0.1×). Electrodes were positioned in layer II/III of the perirhinal cortex directly below the rhinal sulcus.
Stainless steel, monopolar stimulating electrodes (A-M Systems, Carlborg, WA, USA) were used to elicit (1–600 µA, 40 µs) field potentials (field EPSPs) and were positioned ∼150–350 µm from the recording electrode in layer I. Stimulating electrodes were connected via Neurolog isolated stimulators (NL800, Digitimer Ltd., UK), to an IT-16 computer interface digital-analogue converter (Instrutech Corp., NY, USA) and evoked responses recorded on a Macintosh PPC computer (A/Dvance 3.6 Software; Robert McKellar Douglas, Vancouver, Canada).
For each slice, prior to the induction of LTD, the input–output and paired-pulse profiles were assessed. For the LTD and paired-pulse experiments, the stimulus intensity was set to evoke field responses at 80% of the maximum amplitude. Test shocks were applied every 30 s (0.033 Hz) and synaptic potentials recorded for 15–20 minutes to establish a baseline. Low-frequency stimulation (LFS) was then applied to induce LTD. LFS consisted of 900 pulses at 1 Hz applied at test shock intensity. Following LFS, responses were monitored every 30 s for at least 1 h. EPSP responses are expressed as percent change from the last 10 min of the baseline period (mean±SEM). In some experiments, the D2 dopamine receptor agonist, Quinpirole (10 µm; Sigma, Poole, UK), was added to the bathing medium.
Intracellular recording electrodes (impedance 60–150 MΩ filled with 3 m potassium acetate, connected to an Axoclamp-2B amplifier in bridge mode) were used to impale layer II/III neurones. Passive membrane properties were recorded by square pulse (200–500 ms) current injection (−0.8 to ∼+0.6 nA) to generate a current–voltage (I/V) relationship, including positive pulses to assess threshold and action potential characteristics. Membrane resistance (Rin) was calculated by the slope of a line-of-best-fit through the linear portion of the I/V plot (r2 always >0.98), and the membrane time constant (τm) from the 0.4 nA deflection, by determining the time (ms) taken to achieve (1−1/e×100)% of the maximal and steady state potential. Membrane capacitance (Cm) was calculated from Rin and τm using the relationship Cm=τm/Rin (26).
Immunohistochemistry to detect mutant huntingtin
R6/1 and age-matched WT mice (three animals of each genotype at three age points, total 18) were sacrificed by cervical dislocation and immediate decapitation in accordance with UK legislation (Animal Scientific Procedures Act 1986). Brains were rapidly removed and 400 µm coronal slices were prepared on a vibratome (Campden Instruments Inc., USA). Slices were fixed in 4% paraformaldehyde (PFA, Sigma–Aldrich) then 2% PFA overnight before transfer to 0.1 m phosphate-buffered saline (PBS pH 7.4) and stored at 4°C. Slices were temporarily mounted in 5% agar and re-sectioned at 50 µm thickness before re-immersion in PBS, in preparation for immunohistochemistry. Free-floating sections were blocked/permeabilized (2% Fish gelatine, 0.01% sodium azide, 0.1% TritonX-100 in PBS), and endogenous peroxidase activity quenched (3% H2O2 30 min) prior to further rinsing. Sections were then treated with primary antibody (1:2000 dilution of S830 sheep polyclonal antibody) raised against the product of the N-terminal region to 52 glutamine residues of exon 1 of the human gene (a gift from Professor Gillian Bates, Department of Medical and Molecular Genetics, GKT School of Medicine, King's College London, UK), and left to incubate overnight under constant agitation at 4°C. Slices were again washed then exposed to secondary antibody (1:500 dilution of biotinylated anti-sheep IgG, Vector Laboratories, UK) and left to incubate for 1 h at room temperature. The immunoperoxidase reaction was performed using Avidin–biotin complex (Vectastrain Elite kit, Vector Laboratories Ltd.) and diaminobenzidene (0.05% in TBS +10 µl 30% H2O2) as chromogen. Nuclei were counterstained with methyl green or Harris' haematoxylin. Sections were dehydrated in a graded series of alcohols, cleared in xylene and coverslipped using DPX as mountant. Negative control sections were included, where the primary antibody was omitted.
Dopamine receptor immunohistochemistry
Coronal slices from R6/1 and age-matched littermate control mice were prepared as indicated, fixed and resectioned as described earlier. These were washed in PBS, blocked/permeabilized (2% Fish gelatine; 0.01% sodium azide; 0.1% TritonX-100 in PBS) for 2 h, and endogenous peroxidase activity quenched (3% H2O2 30 min). Subsequently, sections were incubated with the relevant primary antibody (AB1765P, rabbit polyclonal anti-dopamine D1A receptor or AB5840P rabbit polyclonal anti-dopamine D2 receptor; 1:1600 dilution of 1 mg/ml stock, Chemicon International Inc., UK) made up in 2% blocking solution for 48 h. Next, the sections were rinsed with PBS prior to incubation overnight with peroxidase-conjugated anti-rabbit antibody (tyramide signal amplification kit, Molecular Probes Inc., USA), according to manufacturer's instructions. This peroxidase-based amplification system is based on the deposition of fluorochrome-labeled tyramide molecules, and has been used to generate high-density labelling of target proteins. Sections were incubated in a 1:50 dilution of the amplification reagent and 0.0015% H2O2 for 5 h, rinsed in PBS (3×15 min), and coverslipped using minimal amount of fluorescence mounting medium, and left to dry for 48–62 h. Consecutive slices were visualized on an inverted confocal microscope (Leica DM IBRE scanning confocal microscope, Leica Microsystems, Heidelberg, Germany) under 568 nm excitation (PMT 907) with the TRIT-C channel optimized for emission at 576 nm. Image stacks (6 µm) of 12 sequential scans (0.5 µm) were collated for each section using Leica Confocal Software (Version 2.5, Leica). The relative intensity of fluorescence was quantified by generating a mean fluorescence value (in arbitrary units) from three manually placed non-overlapping sampling boxes in each region of interest (ROI) through the laminae of the perirhinal cortex (avoiding capillaries). A minimum of three consecutive sections was used per animal (WT, R6/1) and age (1, 3 and 7 months; three animals per genotype per time point). Negative control sections were included where the primary antibody was omitted. Antibody specificity was further confirmed on sections of the brain from mice deficient in D2 dopamine receptors (32) that were a gift from Professor Michael Levine (Mental Retardation Research Center, UCLA, USA). Sections prepared from D2 knock-out brains were processed for D2 immunoreactivity together with control and R6/1 tissue. No immunoreactivity was observed in the D2 knock-out material.
Analysis
Statistical analyses were performed as indicated. Data for each condition were pooled irrespective of experimental outcome and are expressed as mean±SEM. Student's t-test, Welch-corrected t-tests and one-way ANOVA were carried out using InStat 3.05 (GraphPad Software Inc.) and two-way ANOVA using Statistica 6.1 (StatSoft Inc.).
ACKNOWLEDGEMENTS
We would like to thank Mr Steve Walters, Mrs Dawn Sadler, Mrs Karen Evans and Mr Chris Hall at the Open University for their excellent technical assistance and Drs Tony Hannan and Anton van Dellen of Oxford University for their help in establishing our R6/1 colony. We would also like to thank Professor Gillan Bates of Kings' College London for the kind gift of the S830 antibody and Professor Michael Levine and Mr Ehud Gruen for providing D2 knock-out mouse brains. This work was funded by the Open University Research Development Committee and the Royal Society.
Conflict of Interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}