





Abstract
Frontotemporal dementia with inclusion body myopathy and Paget disease of bone is a rare, autosomal-dominant disorder caused by mutations in the gene valosin-containing protein (VCP). The CNS pathology is characterized by a novel pattern of ubiquitin pathology distinct from sporadic and familial frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U) without VCP mutations. TAR DNA binding protein 43 (TDP-43) was recently identified as a major disease protein in the ubiquitin-positive inclusions of sporadic and familial FTLD-U. To determine whether the ubiquitin pathology associated with mutations in VCP is characterized by the accumulation of TDP-43, we analyzed TDP-43 in the CNS pathology of five patients with VCP gene mutations. Accumulations of TDP-43 colocalized with ubiquitin pathology in inclusion body myopathy and Paget disease of bone, including both intranuclear inclusions and dystrophic neurites. Similar to FTLD-U, phosphorylated TDP-43 was detected only in insoluble brain extracts from affected brain regions. Identification of TDP-43, but not VCP, within ubiquitin-positive inclusions supports the hypothesis that VCP gene mutations lead to a dominant negative loss or alteration of VCP function culminating in impaired degradation of TDP-43. TDP-43 is a common pathologic substrate linking a variety of distinct patterns of FTLD-U pathology caused by different genetic alterations.
Introduction
Frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U) is the most common pathologic change underlying the clinical syndrome of frontotemporal dementia (FTD) (1, 2). FTLD-U is characterized by ubiquitin-positive neuronal cytoplasmic and nuclear inclusions as well as by dystrophic neurites that are not detected with antibodies recognizing other cellular proteins including tau, α-synuclein, β-amyloid, neuronal intermediate filaments, and expanded polyglutamines (3-6). Recently, TAR DNA binding protein 43 (TDP-43) was identified as a major disease protein in the ubiquitinated inclusions characteristic of both sporadic and familial FTLD-U as well as sporadic amyotrophic lateral sclerosis (ALS) (7). Pathologic TDP-43 in FTLD-U and ALS is abnormally phosphorylated, ubiquitinated, and cleaved to generate C-terminal fragments and is recovered only from affected CNS regions including hippocampus, neocortex, and spinal cord (7).
Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD) is a rare autosomal dominant disorder characterized by variable penetrance of this unusual triad of clinical features (8). Mutations in the valosin-containing protein (VCP) gene, a member of the AAA-ATPase (i.e. ATPase associated with diverse cellular activities) gene superfamily have been identified in affected individuals (9). The expressed protein functions as a molecular chaperone in a plethora of distinct cellular activities including ubiquitin-dependent, endoplasmic reticulum-associated protein degradation (ERAD), stress responses, programmed cell death, membrane fusion, nuclear envelope reconstruction, and post-mitotic Golgi reassembly (10, 11). Many of these activities are directly or indirectly regulated by the ubiquitin-proteasome system. We recently performed a detailed, systematic analysis of the neuropathologic changes in eight patients with VCP mutations from five different families (12). A novel pattern of ubiquitin pathology was identified in IBMPFD that was distinct from sporadic and familial FTLD-U without VCP gene mutations. IBMPFD cases were characterized by ubiquitin-positive neuronal intranuclear inclusions and dystrophic neurites in neocortex with relatively few intracytoplasmic inclusions (12, 13). Only rare inclusions were labeled with antibodies to VCP, and there was no ubiquitination, aggregation, or alteration in solubility of VCP protein. These findings are consistent with the hypothesis that the pathology associated with VCP gene mutations is the result of impairment of ubiquitin-based degradation pathways such as ERAD (14).
In this study, we sought to determine whether the ubiquitinated inclusions in the CNS associated with mutations in VCP are characterized by the accumulation of TDP-43 that has been identified in both sporadic and familial FTLD-U (7). Toward this end, we analyzed the TDP-43 protein in affected CNS regions in five patients with FTD and VCP gene mutations. We demonstrate that whereas the pattern of ubiquitinated inclusions associated with VCP gene mutations is distinctive, the pathology is characterized by the accumulation of phosphorylated TDP-43 similar to that in sporadic and familial FTLD-U.
Materials and Methods
Patients
We studied postmortem material from five affected members from four different families with IBMPFD and a clinical diagnosis of FTD, as previously described (8, 9, 12). Consent for autopsy was obtained from the legal next-of-kin of all subjects in accordance with state law as well as local institutional review boards.
Immunohistochemistry
Tissue blocks from frontal, temporal, parietal, and occipital neocortex were fixed in neutral-buffered formalin and paraffin wax-embedded. Immunohistochemistry was carried out as described previously with sections pretreated with formic acid (5 minutes) or microwaving in 10 mmol/L citrate buffer, pH 6.0 (3 minutes) to enhance anti-TDP-43 immunoreactivity (12, 15). Antibodies used in this study included 1) anti-ubiquitin antibodies: mouse monoclonal antibody 1510 (Chemicon, Temecula, CA) and rabbit polyclonal antibody (Dako, Carpinteria, CA); and 2) anti-TDP-43 antibodies: rabbit polyclonal antibody (ProteinTech Group, Chicago, IL) and mouse monoclonal antibody 2E2-D3(Abnova, Taipei, Taiwan). Double-labeling immunofluorescence was performed as previously described (7) using Alexa Fluor 488 and 594 conjugated secondary antibodies (Molecular Probes, Eugene, OR).
Biochemistry
Postmortem brain tissue was dissected, weighed, and sequentially extracted with buffers of increasing strength (7, 15).Briefly, gray and white matter was extracted at 5mL/g (v/w) with low-salt buffer (10 mmol/L Tris, pH 7.5, 5 mmol/L EDTA, 1 mmol/L dithiothreitol, 10% sucrose, and a cocktail of protease inhibitors), high-salt Triton X buffer (low-salt buffer + 1% Triton X-100 + 0.5 mmol/L NaCl), myelin flotation buffer (Triton X buffer containing 30% sucrose), and Sarkosyl buffer (low-salt buffer + 1% N-lauroyl sarcosine + 0.5 mol/L NaCl). The detergent-insoluble materials were extracted in 0.25 mL/g urea buffer (7 mol/L urea, 2 mol/L thiourea, 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, and 30 mmol/L Tris, pH 8.5). For Western blot analysis, protein extracts were resolved in Tris-glycine 5% to 20% gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA) and probed with antibodies to TDP-43. Primary antibodies were detected with alkaline phosphatase- conjugated anti-mouse or anti-rabbit IgG (Dako) and visualized by incubation with nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (Roche Molecular Biochemicals, Mannheim, Germany). Where indicated, TDP-43 was dephosphorylated by dialysis (50 mmol/L Tris and 0.2 mmol/L EDTA, pH 8.0) and treated with Escherichia coli alkaline phosphatase (Sigma-Aldrich, St. Louis, MO) for 2 hours at 56°C.
Results
TDP-43 in Inclusions of FTD Patients With VCP Gene Mutations; Colocalization With Ubiquitin
We performed immunohistochemistry on multiple neocortical brain regions from five FTD subjects with two different VCP gene mutations (12). All affected cortical regions showed abundant TDP-43-positive neuronal intranuclear inclusions and dystrophic neurites (Fig. 1), and the density of pathologic changes was similar to that observed with ubiquitin immunohistochemistry (Table) (12). TDP-43-immunoreactive cytoplasmic inclusions were only infrequently identified (Fig. 1D). The intranuclear inclusions had a characteristic lentiform or rod shape (Fig. 1E-H). Both the intranuclear inclusions and dystrophic neurites were most numerous in the upper cortical layers but were also present in neurons throughout the entire cortical thickness. In neurons and glia without ubiquitin pathology, both in unaffected and affected brains regions, physiologic TDP-43 was robustly detected in the nuclei of neurons and glia (Fig. 1C). In contrast, there was a dramatic reduction in the labeling intensity of nuclear TDP-43 in affected cortex. To determine whether the TDP-43 pathology is associated with the ubiquitin-positive inclusions, we performed double-labeling immunofluorescence. Antibodies to TDP-43 immunolabeled both the neuronal intranuclear inclusions and dystrophic neurites detected by antibodies to ubiquitin (Fig. 2).Only rare inclusions were detected with antibodies to either TDP-43 or ubiquitin alone. Collectively, similar to what has been described in both sporadic and familial FTLD-U (7), these studies show that TDP-43 is a novel disease-associated protein found in the distinctive pattern of ubiquitinated inclusions characteristic of IBMPFD.
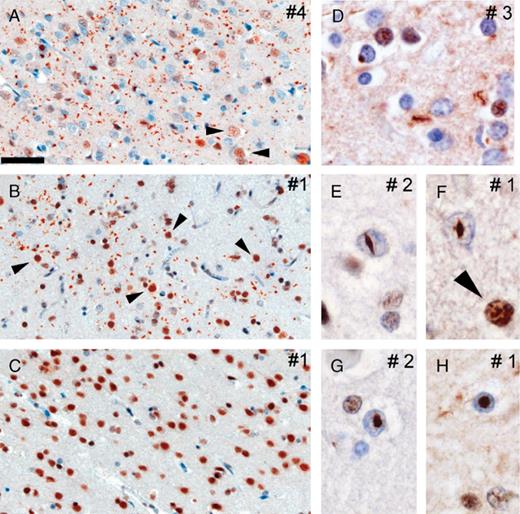
Spectrum of TAR DNA binding protein 43 (TDP-43)-positive pathology in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD). Immunohistochemistry with anti-TDP-43 reveals robust staining of dystrophic neurites and intranuclear inclusions in affected cortical regions in IBMPFD cases (A, parietal lobe; B, temporal lobe). Note the reduced nuclear TDP-43 staining compared to robust physiological nuclear TDP-43 immunoreactivity in adjacent nonaffected neurons (arrowheads in A, B, and F) or neurons in nonaffected cortical regions (C, occipital lobe). Higher magnification of dystrophic neurites and rare cytoplasmic inclusions (D) as well as lentiform (E, F) and round intranuclear inclusions (G, H). Numbering of panels refers to patients described in the Table. Scale bars = (A-C) 50 μm; (D-H) 15 μm.
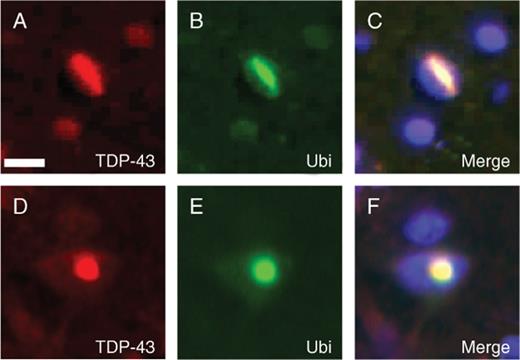
Double labeling of inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD) inclusions with TAR DNA binding protein 43 (TDP-43) and ubiquitin. (A-F) Double label immunofluorescence with anti-TDP-43 (red) and anti-ubiquitin (Ubi) (green) demonstrates colocalization of both proteins in the pathologic inclusions in IBMPFD brains. Scale bar = (A) 10 μm.
Demographic Information of IBMPFD Patients with and Semiquantitative Assessment of TDP-43 Immunoreactivity
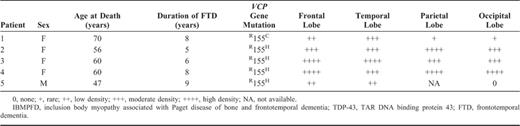
Demographic Information of IBMPFD Patients with and Semiquantitative Assessment of TDP-43 Immunoreactivity
Accumulation of Insoluble, Phosphorylated TDP-43 in FTD Patients With VCP Gene Mutations
A disease-specific biochemical signature of pathologically altered TDP-43 was reported in detergent-insoluble, urea-soluble extracts of sporadic and familial FTLD-U brains (7). To biochemically characterize TDP-43 protein in the CNS of IBMPFD, Western blot analysis was performed on samples of cortical gray matter from patients with FTD and VCP gene mutations that were sequentially extracted with buffers of increasing strength. Full-length TDP-43 protein of ~43 kDa was detected in all soluble and insoluble fractions of affected and unaffected brain regions from IBMPFD patients, as described for FTLD-U, Alzheimer disease, and control brains (data not shown) (7). Additional protein bands of ~25 and 45 kDa, as well as a high molecular smear, were detected in detergent-insoluble, urea-soluble fractions from affected brain regions from cases of IBMPFD and FTLD-U but not controls (Fig. 3A). The quantity of these modified TDP-43 species was variable but correlated with the amount of pathologic change detected by immunohistochemistry. Furthermore, the 45-kDa species was collapsed into the 43-kDa band upon dephosphorylation with alkaline phosphatase, indicating that TDP-43 is abnormally phosphorylated in the CNS in IBMPFD (Fig. 3B). Thus, these data suggest that, despite the distinct genetic alterations and pattern of ubiquitin pathology in IBMPFD, the molecular signature of the TDP-43 disease protein is similar to that of both sporadic and familial FTLD-U including those individuals with PGRN gene mutations (7, 15, 16).
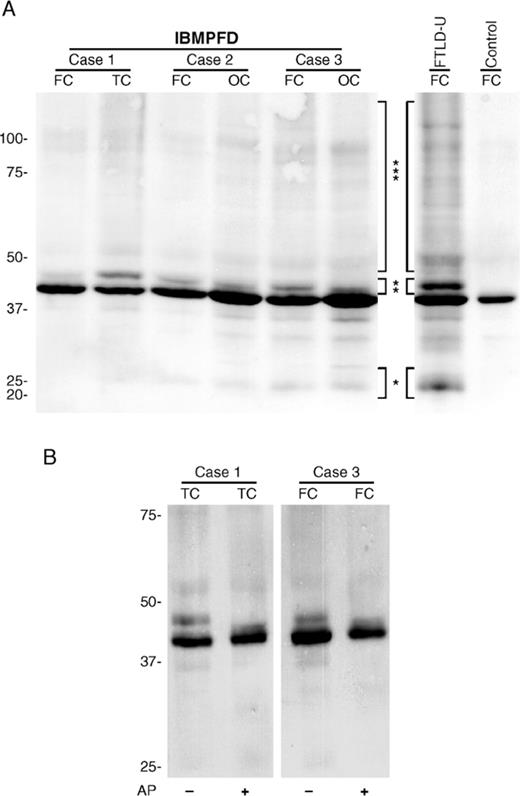
Biochemical analysis of TAR DNA binding protein 43 (TDP-43) in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD). (A) Immunoblot analysis of urea fractions from gray matter of frontal (FC), temporal (TC), and occipital (OC) cortex of IBMPFD brains probed with anti-TDP-43 antibody demonstrates variable presence of pathologic ~25-kDa bands (*), 45-kDa bands (**), and high molecular weight smears (***) similar to the distinct pathologic profile of TDP-43 obtained in frontotemporal lobar degeneration with ubiquitin-positive inclusions brains. Pathologic TDP-43 bands were not detected in control brains. (B) Dephosphorylation of IBMPFD urea extracts with alkaline phosphatase (AP) followed by immunoblotting with anti-TDP-43 shows a collapse of the 45-kDa band into the 43-kDa band, indicating that pathologic, detergent-insoluble TDP-43 is abnormally phosphorylated.
Discussion
FTLD-U, the most common pathology underlying the clinical syndrome of FTD, is characterized by ubiquitin-positive neuronal cytoplasmic and nuclear inclusions as well as dystrophic neurites (3-6). Up to 4 patterns of FTLD-U have been delineated on the basis of morphology and distribution of the ubiquitin-positive inclusions (7, 12, 15). A family history of a similar neurodegenerative disease is reported in up to 40% of FTD patients, strongly implicating a genetic etiology in FTLD-U pathogenesis (17). Causative mutations have been identified in 3 genes: VCP (9), PGRN (16, 18, 19), and CHMP2B (20). In addition, 2 genome-wide screens demonstrated linkage of ALS with FTLD-U to 2 additional distinct loci on chromosome 9 (21, 22). Although it remains unclear how these distinct genes all lead to FTD and ubiquitin-positive inclusions, the pattern of pathologic changes may correlate with specific genetic alterations (7, 12).
The pathologic characteristics of FTLD-U may be the result of impairment of ubiquitin-based degradation pathways (23). Alternatively, there may be accumulation and ubiquitination of a specific "abnormal" protein, similar to that seen in other neurodegenerative diseases including Alzheimer disease, the tauopathies, and synucleinopathies (24). Insights into this question were recently provided when TDP-43 was identified as a major component of the ubiquitin pathology in FTLD-U, including patients with PGRN gene mutations (7). Furthermore, in affected brain regions, phosphorylated and ubiquitinated TDP-43 was detected in detergent-insoluble, urea-soluble extracts. We have extended these findings to the neuropathology associated with IBMPFD caused by VCP gene mutations. These cases are characterized by a unique pattern of FTLD-U pathology consisting predominantly of neuronal intranuclear inclusions and dystrophic neurites (12, 13). In IBMPFD, only rare intracytoplasmic inclusions are present, and the dentate gyrus is specifically spared (12). In the current study, we demonstrated that both the ubiquitin-positive intranuclear inclusions and dystrophic neurites in IBMPFD are immunolabeled with antibodies to TDP-43. Biochemical analysis of affected brain regions also demonstrate the disease-specific signature consisting of phosphorylated and ubiquitinated TDP-43 in detergent-insoluble, urea-soluble extracts identical to that seen in other cases of FTLD-U and ALS (7).
TDP-43 is a ubiquitously expressed, highly conserved nuclear protein (25) that may be a transcription repressor, an activator of exon skipping (26-28), and/or a scaffold for nuclear bodies through interactions with survival motor neuron protein (29). Physiologically, TDP-43 is localized primarily to the nucleus. It is unclear whether TDP-43 is normally present at significant levels in the cytoplasm, axons, and dendrites of neurons. However, in both sporadic and familial FTLD-U including IBMPFD, the immunodetection of TDP-43 in the nucleus is reduced in affected cortex, and there is a significant burden of pathologic changes within cellular processes (i.e. dystrophic neurites). Thus, the current evidence links TDP-43 to the pathogenesis of FTLD-U, although the pathologic significance of these alterations in the cellular distribution and post-translational modification of TDP-43 remain unknown. This reduced immunodetection of TDP-43 in the nucleus may be due to reduced protein expression, redistribution of TDP-43 from the nucleus, and/or sequestration of TDP-43 within ubiquitinated aggregates. One possible consequence of this altered pattern of expression could be loss of TDP-43 nuclear function. Future studies will need to address these and other mechanistic aspects of the aggregation of pathologic TDP-43 in cytoplasmic, neuritic, and nuclear inclusions and their role in the pathogenesis of neurodegeneration.
In addition to the recent identification of disease-causing mutations in IBMPFD (9), VCP has been implicated in the pathogenesis of other neurodegenerative diseases (30-36). VCP consists of 2 ATPase domains and an N-terminal domain that provides substrate specificity (10, 11). VCP functions as a molecular chaperone in a plethora of distinct cellular activities, including ERAD, and a majority of these activities are directly or indirectly regulated by the ubiquitin-proteasome system. Loss of VCP function leads to accumulation of polyubiquitinated proteins (30, 31) or vacuole and inclusion body formation (32-34), whereas two of the VCP gene mutations directly impair ERAD in a cell culture model (14). Furthermore, both VCP and TER94, the Drosophila homolog, have been shown to modulate polyglutamine-induced neurodegeneration (35, 36). Our data showing accumulation of ubiquitin-positive inclusions that are composed of TDP-43, but not VCP, support the hypothesis that VCP gene mutations in IBMPFD lead to a dominant negative loss or alteration of VCP function that might impair ubiquitin-proteasome system activity. However, the molecular link between VCP and TDP-43 is unknown.
The identification of TDP-43 as a major component of the ubiquitinated inclusions specific to both sporadic and familial FTLD-U resolves a long standing enigma concerning the nature of the ubiquitinated disease protein (7). However, an understanding of the pathogenesis of FTLD-U including the role of TDP-43 will have to integrate the biology of multiple distinct genetic elements in addition to VCP. For example, FTD-causing mutations in PGRN, a secreted growth factor involved in the regulation of multiple cellular processes including wound repair and inflammation, lead to premature termination codons that are subject to nonsense-mediated decay resulting in haploinsufficiency (18, 19). A mechanism linking VCP and PGRN to TDP-43 and neurodegeneration remains elusive and will surely be the focus of future studies into the pathogenesis of FTD.
Acknowledgments
We are indebted to the patients and their caregivers whose altruism and support have facilitated the research described in this study. We thank John Q. Trojanowski and Virginia M.-Y. Lee of the Center for Neurodegenerative Disease Research at the University of Pennsylvania for generously providing resources and technical advice. We also thank Dr. John C. Morris of the Alzheimer's Disease Research Center at Washington University, St. Louis for his clinical expertise in the characterization of one of the patients.
References
Author notes
This work was supported by grants from the National Institutes of Health (P30 AG10124 [MSF], K08 AG20073 [MSF], P30 AG028383 [WRM], P50 AG005144 [WRM and CDS], P30 AG021300 [PJB], and P50 AG005681 [NJC]), the Canadian Institute of Health Research (74580 [IRM]), and the Winspear Family Special Center for Research on the Neuropathology of Alzheimer's Disease (PJB).

{kind=link}
{kind=link}
{kind=link}