Cellular correlates of enhanced anxiety caused by acute treatment with the selective serotonin reuptake inhibitor fluoxetine in rats
- National Centre for Biological Sciences, Tata Institute of Fundamental Research, Bangalore, India
Selective serotonin reuptake inhibitors (SSRIs) are used extensively in the treatment of depression and anxiety disorders. The therapeutic benefits of SSRIs typically require several weeks of continuous treatment. Intriguingly, according to clinical reports, symptoms of anxiety may actually increase during the early stages of treatment although more prolonged treatment alleviates affective symptoms. Consistent with earlier studies that have used animal models to capture this paradoxical effect of SSRIs, we find that rats exhibit enhanced anxiety-like behavior on the elevated plus-maze 1 h after a single injection of the SSRI fluoxetine. Next we investigated the potential neural substrates underlying the acute anxiogenic effects by analyzing the morphological and physiological impact of acute fluoxetine treatment on principal neurons of the basolateral amygdala (BLA), a brain area that plays a pivotal role in fear and anxiety. Although earlier studies have shown that behavioral or genetic perturbations that are anxiogenic for rodents also increase dendritic spine density in the BLA, we find that a single injection of fluoxetine does not cause spinogenesis on proximal apical dendritic segments on BLA principal neurons an hour later. However, at the same time point when a single dose of fluoxetine caused enhanced anxiety, it also enhanced action potential firing in BLA neurons in ex vivo slices. Consistent with this finding, in vitro bath application of fluoxetine caused higher spiking frequency and this increase in excitability was correlated with an increase in the input resistance of these neurons. Our results suggest that enhanced excitability of amygdala neurons may contribute to the increase in anxiety-like behavior observed following acute fluoxetine treatment.
Introduction
Selective serotonin reuptake inhibitors (SSRIs) are used routinely in the treatment of depression and a wide range of anxiety disorders (Sheehan et al., 1993; van der Kolk et al., 1994; Stokes and Holtz, 1997; Kent et al., 1998; Bezchlibnyk-Butler et al., 2000; Bondareff et al., 2000; Stahl, 2000; Gorman, 2003). The therapeutic benefits of SSRI treatment in patients requires several weeks of continuous treatment. Paradoxically, while such chronic treatment regimens decrease symptoms of anxiety and depression, acute effects of SSRI treatment may actually lead to an increase in anxiety and an increased risk for suicidal ideation (Gorman et al., 1987; Teicher et al., 1990; Mir and Taylor, 1997; Goldstein and Goodnick, 1998; Masand and Gupta, 1999; Spigset, 1999; Fergusson et al., 2005).
Unfortunately, little is known about the neural mechanisms underlying this acute anxiogenic effect of SSRIs. The choice of appropriate animal models poses a key challenge in gaining mechanistic insights into this effect. Nevertheless, the acute anxiogenic effect of SSRIs has been demonstrated in numerous rodent models of anxiety-like behavior (Bodnoff et al., 1989; Griebel et al., 1994; Sanchez and Meier, 1997; Dekeyne et al., 2000; Kurt et al., 2000; Silva and Brandao, 2000; Bagdy et al., 2001; Belzung et al., 2001; Koks et al., 2001; Salchner and Singewald, 2002). Another promising lead comes from LeDoux and colleagues who have shown that the same acute SSRI treatment that increases anxiety-like behavior also enhances the acquisition of fear memory in an auditory fear conditioning task (Burghardt et al., 2004, 2007). Considerable evidence points to a crucial role for the basolateral amygdala (BLA) in the acquisition and expression of conditioned fear, and the underlying cellular and molecular mechanisms have been studied extensively (McKernan and ShinnickGallagher, 1997; Rogan et al., 1997; LeDoux, 2000; Maren, 2000; Maren and Quirk, 2004). Therefore, findings on the enhanced acquisition of auditory fear conditioning caused by acute SSRI treatment provide a powerful framework for investigating the cellular basis of this effect in the BLA. Results from animal studies mentioned above gain further significance in light of neuroimaging studies that have revealed enhanced amygdala activity in patients of affective disorders (Anand and Shekhar, 2003; Hasler et al., 2004; Hasler and Northoff, 2011). Furthermore, acute SSRI administration has been shown to enhance amygdala activity in healthy humans (Del-Ben et al., 2005; McKie et al., 2005). However, despite the pivotal role played by the amygdala in fear, anxiety, and depression, little is known about how antidepressants such as SSRIs affect neurons in this brain area.
In light of the accumulating evidence pointing to a potential role for the BLA in the heightened fear and anxiety triggered by acute SSRI treatment, the current study is aimed at examining the cellular correlates of the anxiogenic effect of the SSRI fluoxetine. To this end, we focus on two potential structural and physiological mechanisms emerging from earlier studies. First, behavioral or genetic manipulations that trigger the formation of dendritic spines on excitatory principal neurons in the BLA also enhance anxiety in rodents (Roozendaal et al., 2009). For instance, both acute and chronic immobilization stress increase anxiety-like behavior in the elevated plus-maze (EPM). Both of these stress paradigms also increase the number of spines on BLA principal neurons (Mitra et al., 2005). Further, BLA spinogenesis caused by transgenic overexpression of brain-derived neurotrophic factor (BDNF) is accompanied by enhanced anxiety-like behavior in mice (Govindarajan et al., 2006). Dendritic spines are highly dynamic structures and stable changes in spine number have been observed as early as an hour after behavioral training (Hofer and Bonhoeffer, 2010). Thus, an increase in spine number in the BLA may serve as a potential cellular substrate for the acute anxiogenic effects of SSRIs. A second prediction comes from a number of electrophysiological studies that have suggested a link between enhanced fear and anxiety-like behavior and increased amygdala activity. Behavioral stress that enhances fear learning also enhances the excitability of BLA neurons (Manzanares et al., 2005). Bath application of stress levels of the corticosterone enhanced the excitability of BLA cells (Duvarci and Pare, 2007). Further, hyper-excitability of the BLA can also be induced by intra-BLA infusion of a corticotrophin releasing factor (CRF) receptor agonist, which is normally released during stress (Rainnie et al., 2004). Thus, anxiety induced by acute SSRI treatment may also be mediated by an increase in neuronal excitability in the BLA. Therefore, in the present study we test these two predictions by analyzing if the acute anxiogenic effects of a single dose of the SSRI fluoxetine is accompanied by increases in the density of dendritic spines and spiking activity in amygdalar principal neurons.
Materials and Methods
Experimental Animals
Male Wistar rats (50- to 60- days old, 250–300 g) were used for all experiments. All animals were housed in groups of two or three with ad libitum access to food and water. They were maintained in a temperature-controlled room, with a 14-h/10-h day/night cycle. All procedures related to animal maintenance and experiments were approved by the Institutional Animal Ethics Committee (National Centre for Biological Sciences).
Drugs
Fluoxetine Hydrochloride (courtesy of Hikal Ltd.) was dissolved in 0.9% sterile saline and injected intraperitoneally (i.p.) at a dose of 10 mg/kg body weight. The 10 mg/kg, i.p. dose of fluoxetine was used based on a number of previously published studies that have already established the anxiogenic effect of this dose in various tests for anxiety-like behavior and fear conditioning (Bodnoff et al., 1989; Silva and Brandao, 2000; Bagdy et al., 2001; Burghardt et al., 2007). The drug solution was made fresh daily and animals were weighed before each injection to ensure the accuracy of drug dosage given. For the in vitro bath application of fluoxetine in brain slices, fluoxetine was dissolved in artificial cerebrospinal fluid (aCSF) at a concentration of 50 μM and the brain slices were perfused with this solution (Karson et al., 1993; Mukherjee et al., 1998).
Elevated Plus-Maze
The EPM, consisting of two opposite open arms (60 cm × 15 cm) and two enclosed arms (60 cm × 15 cm, surrounded by a 15-cm high opaque wall), was elevated 75 cm from the ground. The animals were tested on the maze 60 min after an injection of fluoxetine or saline. Individual trials of 5 min each were videotaped for subsequent off-line analysis. At the beginning of each trial, animals were placed at the center of the maze, facing an enclosed arm. All trials were conducted between 10 a.m. and 2 p.m., and the maze was cleaned with an ethanol solution after each trial.
Morphological Analysis
After testing for anxiety-like behavior on the EPM, all rats were sacrificed under deep anesthesia. The brains were dissected out and processed for Golgi staining (Shankaranarayana Rao and Raju, 2004; Govindarajan et al., 2006). Coronal sections (120 μm thick) were prepared and mounted on slides (Vyas et al., 2002). Slides were coded before quantitative analysis, and the code was broken only after the analysis was completed. Dendrites directly originating from cell soma were classified as main shafts, and those originating from the main shafts were called primary dendrites. Starting from the origin of the branch, and continuing away from the cell soma, spines were counted along the first 80 μm stretch of the primary dendrite. Spine density was analyzed using the NeuroLucida image analysis system with the Olympus BX61 microscope. Spines were identified at a final magnification of 1000× (10× eyepiece and 100× objective) in the microscope and their position was marked on an 85 μ/pixel image displayed on the computer screen. All protrusions, irrespective of their morphological characteristics, were counted as spines if they were in direct continuity with the dendritic shaft. Finally, it may be noted that our analysis, like all those involving Golgi staining, is likely to lead to a systematic underestimation of spine density because it is not possible to visualize spines pointing directly toward the surface or extending beneath the dendrite (Feldman and Peters, 1979; Trommald et al., 1995; Trommald and Hulleberg, 1997). In the present study, however, no attempt was made to correct for these hidden spines, because of previously reported validation (Horner and Arbuthnott, 1991) of the use of visible spine counts for comparison between different experimental conditions.
In vitro Slice Electrophysiology
Fluoxetine or saline injected rats were sacrificed under deep anesthesia. The brain was removed rapidly and 400 μm thick coronal brain slices containing the amygdala were prepared using a Vibratome 1000 Plus (Vibratome, St. Louis, MO, USA). Slices were transferred to a submerged chamber containing aCSF (126 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 10 mM Glucose, 1 mM MgCl2, and 2 mM CaCl2) equilibrated with 95%O2–5%CO2 at room temperature. Slices were incubated for at least 1 h before being transferred to a superfused recording chamber. Patch electrodes (3–6 MΩ) were pulled from borosilicate glass pipettes (O.D.: 1.5 mm; I.D.: 0.86 mm; Warner Instruments, Hamden, CT, USA) on a P-97 Flaming-Brown Micropipette Puller (Sutter Instruments, Novato, CA, USA) and filled with a solution containing 115 mM K-gluconate, 20 mM KCl, 10 mM HEPES, 0.5 mM EGTA, 3 mM MgATP, and 0.3 mM NaGTP (pH 7.4, 290 mOsm). The internal solution was filtered through a 0.2-μm filter before use. Excitatory principal neurons in the lateral amygdala (LA) were visually identified with infrared video-microscopy using an upright microscope equipped with a 60X objective (Olympus BX-50WI microscope, water immersion lens, 0.9 NA). Data was acquired with an HEKA EPC9 (HEKA Elektronik, Lambrecht, Germany) amplifier, filtered at 2.9 kHz, and digitized at 20 kHz. Whole-cell patch clamp recordings were performed at room temperature in the current-clamp mode (with the membrane potential kept manually at −70 mV). To obtain the frequency–current (f–I) relationship, action potential firing in response to a series of depolarizing current steps was recorded. The instantaneous firing rate for each current step was calculated from the inter-spike interval. Saturating current intensities were excluded from the analysis. Series resistance (Rs) was monitored throughout the experiment by applying hyperpolarizing current steps. Only cells with a Rs value less than 25 MΩ that did not change by more than 10% were taken for analysis.
Statistics
All data values are expressed as mean ± SEM. All statistical analyses were performed using SPSS 9 and OriginPro 8. The behavior and morphology data were analyzed using the Student’s t-test. For the electrophysiology experiments, statistical analysis of the f/I curve data was done using a two-way mixed-model ANOVA (within subject factor – current injected, between subject factor – treatment) when the experiment was done after in vivo fluoxetine or saline administration. Subsequent pairwise comparisons were made using independent sample t-tests. For the in vitro fluoxetine application experiment, statistical analysis of the f/I curve data was done using a two-way within subject ANOVA (within subject factors – treatment and current injection). Subsequent pairwise comparisons were made using paired t-tests. Statistical comparison of firing rates and input resistance before, during, and after fluoxetine application was done using paired t-tests.
Results
Acute Fluoxetine Treatment Increases Anxiety-Like Behavior in the Elevated Plus-Maze
In order to establish the anxiogenic effect of acute fluoxetine treatment (Griebel et al., 1994; Kurt et al., 2000; Silva and Brandao, 2000; Koks et al., 2001), we first examined how a single injection of fluoxetine (10 mg/kg, i.p.) affected anxiety-like behavior of male Wistar rats on the EPM 60 min later (Figure 1A). Fluoxetine-treated rats displayed elevated anxiety levels when compared to saline injected controls as indicated by the significant reduction in open-arm time (Figure 1B; Saline: 58.9 ± 3.7, N = 13; Fluoxetine: 35.0 ± 5.4, N = 12; p < 0.01) and open-arm entries (Figure 1B; Saline: 63.5 ± 13.6, N = 13; Fluoxetine: 45.9 ± 13.2, N = 12; p < 0.01). The total number of entries into both open and closed arms was not different between saline and fluoxetine-treated groups, suggesting that locomotor activity was not affected. Thus, consistent with previous reports, we find that acute treatment with fluoxetine increases anxiety-like behavior.
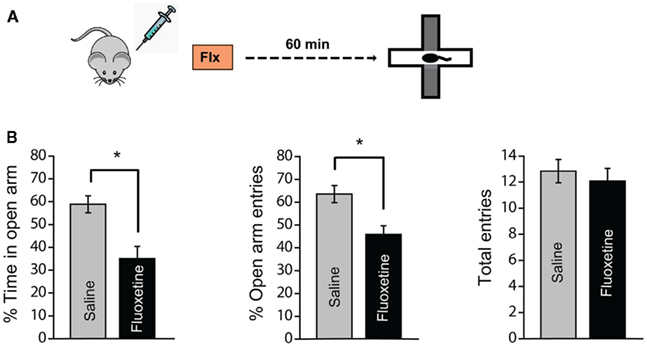
Figure 1. Acute fluoxetine treatment increases anxiety-like behavior in the elevated plus-maze. (A) Schematic representation of the experimental protocol. Male Wistar rats were injected with fluoxetine (10 mg/kg, i.p.) or saline and tested for anxiety-like behavior in the elevated plus-maze 60 min later. (B) Anxiety-like behavior was assessed by measuring the open-arm exploration, i.e., the percentage of time spent in open-arm (left) and the percentage of open-arm entries (middle). Fluoxetine injected rats spent less time in the open-arm and made fewer entries to the open-arm when compared to saline injected rats indicating high anxiety levels. The total entries (right) were not different suggesting that there was no difference in locomotor activity between the two groups. Saline: N = 13, Fluoxetine: N = 12. Error bars represent SEM; *p < 0.01.
Acute Fluoxetine Treatment Does Not Affect Spine Density on Primary Apical Dendrites of BLA Principal Neurons
Past studies have suggested a link between spinogenesis in the BLA and an increase in anxiety-like behavior in the EPM (Mitra et al., 2005; Govindarajan et al., 2006). These earlier studies showed that manipulations, as diverse as acute stress or transgenic up-regulation of BDNF, that increase anxiety-like behavior also enhance spine density on the first 80 μm of primary apical dendritic segments on BLA principal neurons. Since acute fluoxetine treatment had the same anxiogenic effect, we hypothesized that it could also have the same morphological effects on BLA principal neurons. In order to test this prediction, a subset of the rats tested for anxiety-like behavior were sacrificed immediately after behavioral testing and the brains were processed for Golgi staining. The density of dendritic spines was quantified on Golgi-impregnated spiny principal neurons in the BLA (see Materials and Methods; Figures 2A,B). We found no difference in the total number of spines in the first 80 μm of the primary apical dendrites of BLA neurons from fluoxetine and saline treated animals (Figures 2C,D; Saline: 115.4 ± 2.8, N = 5; Fluoxetine: 111 ± 3.7, N = 5). Further, a more detailed segmental analysis in steps of 10 μm did not show any effect of acute fluoxetine treatment on spine density along any segment of the dendrites analyzed (Figure 2E). These results show that the acute anxiogenic effects of fluoxetine are not accompanied by an increase in spine density of the primary apical dendrites on BLA principal neurons.
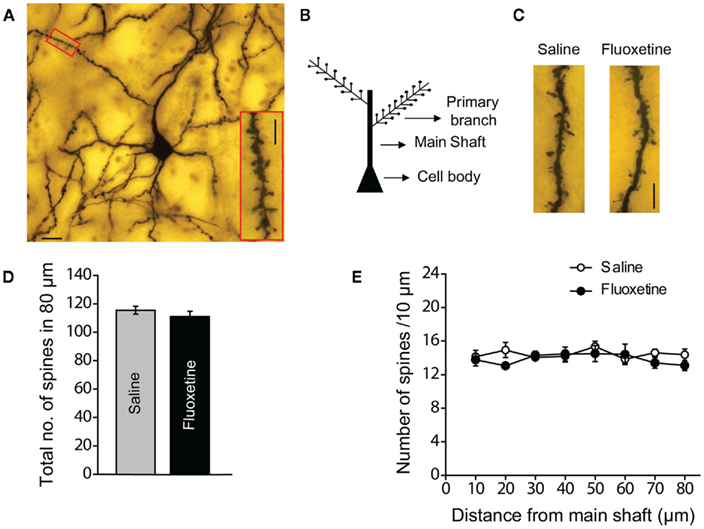
Figure 2. Acute fluoxetine treatment does not change spine densities in the basolateral amygdala (BLA). (A) Representative image of a Golgi-stained principal neuron in the BLA; scale bar: 20 μm. The inset is a high magnification image of the primary dendrite segment marked by the red box; scale bar: 5 μm. (B) Schematic of a BLA principal neuron; spines are counted along the first 80 μm stretch of the primary dendrite. (C) Representative dendritic segments from BLA spiny principal neurons of saline and fluoxetine injected animals; scale bar: 5 μm. (D) Total number of spines in the 80 μm stretch of the dendrite analyzed. (E) The distribution of spine density over 10 μm segments along the 80 μm stretch of the dendrite analyzed. Saline: N = 5, Fluoxetine: N = 5. Error bars represent SEM.
Acute in vivo Treatment with Fluoxetine Increases Excitability of Amygdala Neurons ex vivo
Next we explored the possibility that the short-term effects of fluoxetine may be mediated by physiological rather than structural changes in amygdalar neurons. An increase in amygdala activity is often predictive of fear and anxiety-like behavior in rodents (Maren and Quirk, 2004). Since acute fluoxetine treatment enhances these behavioral outputs of the amygdala (Figure 1; Burghardt et al., 2004, 2007), we hypothesized that the same acute in vivo administration of fluoxetine may also enhance the firing output of principal neurons in the lateral amygdala (LA). To this end, rats were subjected to a single injection of either fluoxetine (10 mg/kg, i.p.) or saline and 60 min later amygdala slices were prepared from these injected animals (Figure 3A). Whole-cell current-clamp recordings were carried out in LA principal neurons, which exhibit spike frequency adaptation upon depolarizing current injections (Figure 3B). Action potential firing in response to somatic injections of increasing steps of depolarizing currents was recorded and the average instantaneous firing frequency was calculated for each current value. A comparison of these frequency–current (f–I) relationship plots for LA neurons from saline and fluoxetine-treated animals is presented in Figure 3C. Cells from fluoxetine injected rats had significantly higher firing rates compared to saline injected rats [F (1, 23) = 4.41, p < 0.05]. There was also a significant increase in firing rates with current injection [F (5, 115) = 409.94, p < 0.001]. However, the interaction between treatment and current injection was not significant [F (5, 115) = 1.24]. Pairwise comparisons also revealed that LA neurons from the fluoxetine-treated rats exhibited a significant increase in spiking frequency (Figure 3C; for 350 pA current injection, Saline: 18.8 ± 0.7 Hz, n = 11 neurons; Fluoxetine: 21.2 ± 0.6 Hz, n = 14 neurons; p < 0.5). Thus, acute in vivo treatment with fluoxetine enhances spike firing of LA principal neurons at the same time point after acute fluoxetine administration when animals exhibited increased anxiety-like behavior.
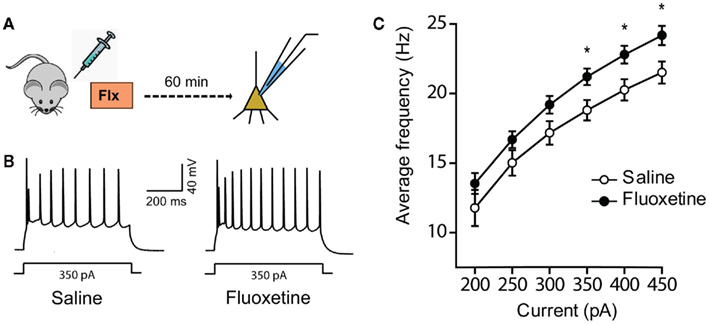
Figure 3. In vivo acute fluoxetine administration increases the intrinsic excitability of LA principal neurons measured ex vivo. (A) Schematic representation of the experimental protocol. Male Wistar rats were injected with fluoxetine (10 mg/kg, i.p.) or saline and sacrificed 60 min later. Whole-cell patch clamp recordings were then carried out on excitatory principal neurons in the LA in the current-clamp mode. (B) Representative spike trains evoked by a 350-pA somatic current injection. (C) Comparison of the average f–I curves (Instantaneous firing frequency versus current; Saline: n = 11 neurons, Fluoxetine: n = 14 neurons). The instantaneous firing frequency was calculated for a series of somatic current injections from the average inter-spike interval for each current. Error bars represent SEM; *p < 0.01.
In vitro Application of Fluoxetine Increases Excitability of Amygdala Neurons in Brain Slices
Our results from the ex vivo slice recordings raise the possibility that acute administration of fluoxetine enhances spike firings in LA neurons. To examine this possibility more directly we extended our analysis by examining if in vitro bath application of fluoxetine in brain slices enhances action potential firing in LA neurons. We used the same experimental design involving whole-cell current-clamp recordings in LA excitatory principal neurons that showed spike frequency accommodation in response to depolarizing current injections. The spiking response of each cell in response to a series of somatic depolarizing current injection steps was recorded before and 30 min after a 15-min bath application of 50 μM fluoxetine followed by a 30-min washout with normal aCSF (Figure 4A). We then compared the average firing frequency versus current (f–I) plots before the fluoxetine wash-in and at the end of the washout period (Figure 4C). Fluoxetine application significantly enhanced the firing rate of LA neurons [F (1, 7) = 10.73, p < 0.05]. There was also a significant effect of current injection on firing rates [F (4, 28) = 280.55, p < 0.001]. Further, the interaction between treatment and current injection was significant [F (4, 28) = 10.17, p < 0.001]. This was manifested as a lowering of the amplitude of the depolarizing current steps required to attain saturating firing frequencies in the presence of fluoxetine. Pairwise comparisons also show that bath application of fluoxetine increases pyramidal cell excitability (Figures 4B,C; for 150 pA current injection, Pre-fluoxetine: 12 ± 0.5 Hz, Post fluoxetine 16 ± 2 Hz, n = 8 neurons, p < 0.01).
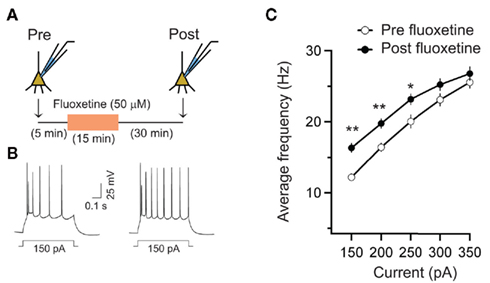
Figure 4. Acute fluoxetine application increased excitability of LA neurons. (A) Schematic representation of the experimental protocol. Excitability was measured using a range of supra-threshold current injections both during baseline and 30 min after the end of fluoxetine (50 μM, 15 min) application. (B) Action potential firing induced by depolarizing current injections from a representative neuron pre (left) and post (right) fluoxetine treatment. (C) Averaged firing frequency versus current relationship (f–I curve) shows the increase in excitability after fluoxetine application (n = 8 neurons) across a range of current injections. Error bars represent SEM; *p < 0.05, **p < 0.01.
In order to follow the time-course of this increase in spiking output caused by fluoxetine application, we next followed the spiking activity of individual cells in response to a 500-ms current step of fixed magnitude during the pre-fluoxetine baseline, fluoxetine application, and washout period. The current step was adjusted to elicit an average of four to five action potentials. After obtaining a steady baseline (Figure 5A, left, Baseline), bath application of fluoxetine (50 μM, 15 min) resulted in an increase in action potential firing (Figure 5A, middle, Fluoxetine). Mean values of firing frequency averaged over 5 min at the end of fluoxetine application increased significantly when compared to pre-drug baseline (Figure 5B; Baseline: 9.2 ± 0.2 Hz, Fluoxetine: 11 ± 0.2 Hz, n = 9 neurons, p < 0.01). This increase in firing rate observed after the end of fluoxetine application was persistent as is evident from the time-course plot (Figure 5A). The increase in firing rate remained significantly higher than pre-drug baseline even after the drug was washed out for 30 min (Figure 5B, Baseline: 9.2 ± 0.2 Hz, Washout: 12 ± 0.3 Hz, n = 9 neurons, p < 0.001). In control experiments, treatment with aCSF alone did not induce any significant changes in spiking (Figures 5A,B, open circles).
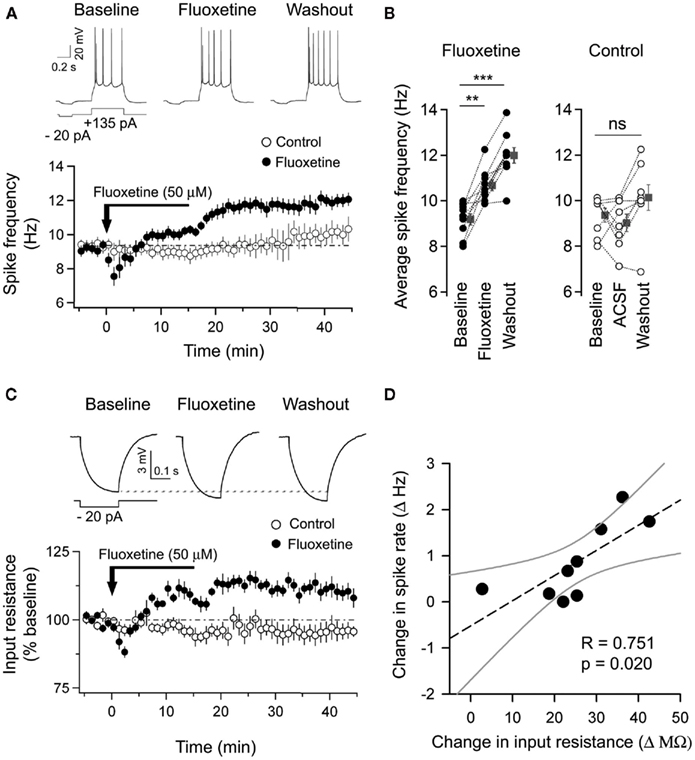
Figure 5. Fluoxetine application led to a correlated increase in both spike frequency and input resistance in LA neurons. (A) Top, Representative traces of action potentials elicited by supra-threshold current injections from an LA neuron during baseline, at the end of fluoxetine application and after washout. Bottom, time-course plot showing the persistent increase in spike frequency found only in fluoxetine-treated neurons and not in aCSF-treated control neurons. (B) Individual (fluoxetine – black circles, saline – white circles) and averaged (individual values averaged over 5 min, gray squares) values of spike frequency for fluoxetine (left, n = 9 neurons) and aCSF (right, n = 8 neurons) treated control neurons. (C) Top, the voltage response to hyperpolarizing current injections used to measure the input resistance is shown for a representative neuron before, during, and after fluoxetine application. Bottom, time-course of input resistance averaged (per minute) and normalized (%) to the baseline for fluoxetine and aCSF-treated control neurons. (D) Correlation plot indicating that the rise in spike frequency in the majority of fluoxetine-treated neurons was significantly correlated with a corresponding increase in input resistance. The dotted and gray lines indicate the linear-fit and 95% confidence bands respectively. Error bars represent SEM; **p < 0.01, ***p < 0.001.
Fluoxetine Induced Increase in Spiking is Accompanied by an Increase in Input Resistance
In addition to recording the spiking response of cells, we also measured the input resistance for all the cells by measuring their response to a brief hyperpolarizing current step preceding each depolarizing current step. Bath application of fluoxetine (50 μM, 15 min) caused a marked increase (10%) in input resistance which was significantly different from the baseline (242 ± 13 MΩ) both immediately after fluoxetine application (Figure 5C, Fluoxetine: 264 ± 14 MΩ, n = 9 neurons, p < 0.001) and at the end of the washout period (Figure 5C, Washout: 267 ± 14 MΩ, n = 9 neurons, p < 0.01). In contrast, control cells treated with aCSF alone, showed no significant change in input resistance (Figure 5C, Open circles). Importantly, the increase in input resistance measured at the end of the washout period for each fluoxetine-treated cell was positively correlated with the increase in firing rate (Figure 5D; R = 0.75, p < 0.05). This suggests that the increase in input resistance may contribute to the enhanced spiking output of LA neurons induced by fluoxetine application, thereby providing a possible mechanism underlying the firing changes observed in vitro.
Discussion
Despite growing evidence pointing to a pivotal role for the amygdala in the debilitating emotional symptoms of depression and anxiety, few studies have addressed the role of cells and circuits in the amygdala in mediating the behavioral effects of antidepressants. While studies on the hippocampus and cortex have helped elucidate some of the cellular and molecular mechanisms that may underlie the cognitive deficits observed in affective disorders, relatively little is known about the cellular basis of the emotional and mood related symptoms. In this study, we have attempted to address this gap in knowledge by investigating if changes in morphological and physiological properties of BLA neurons may provide a cellular correlate for the acute effects of SSRI treatment. Consistent with previous studies, we find that a single injection of the SSRI fluoxetine leads to a significant increase in anxiety-like behavior in the plus-maze an hour later. Strikingly, this increase in anxiety is not paralleled by an increase in the density of dendritic spines in the first 80 μm of the primary dendrites of BLA principal neurons. Previous studies have reported that an increase in the density of spines, on the same class of dendrites in the same type of BLA neurons, may in itself be adequate to increase behavioral anxiety in rodents (Mitra et al., 2005; Govindarajan et al., 2006). However, our results show that anxiety-like behavior can be enhanced even in the absence of such modifications in the structural connectivity of BLA neurons. Although we could not detect any stable changes in spine density 1 h after SSRI administration, this does not rule out the possibility that SSRI treatment affects spine dynamics that only result in stable changes in spine number over longer periods of time or after longer durations of treatment. Indeed, earlier observations on enhanced anxiety-like behavior being correlated with BLA spinogenesis were manifested on a longer time-scale. For example, a robust increase in BLA spine density was observed after 10 days of chronic stress. Even when a single 2-h episode of acute stress was able to elicit spinogenesis in the BLA, the effect was manifested 10 days, but not 1 day, after acute stress (Mitra et al., 2005). Further, our morphological analysis was restricted to the quantification of a specific parameter, i.e., spine density in the first 80-μm segment of primary apical dendrites of BLA principal neurons, and does not rule out the possibility that spine density changes may have occurred in other dendritic compartments (e.g., in higher-order branches or in basal dendrites) or changes in other morphological characteristics such as spine morphology (size or shape of spines) or dendritic morphology of BLA principal neurons. Future studies would need to address these possibilities.
The absence of an obvious spine density change led us to shift our attention to physiological changes in amygdala neurons. Interestingly, we found that the acute anxiogenic effect of fluoxetine treatment was accompanied by an increase in the excitability of principal neurons in amygdala slices. Whole-cell recordings in amygdala slices prepared 1 h after in vivo fluoxetine administration, as well as in vitro fluoxetine application directly onto amygdala slices, enhanced action potential firing of LA principal neurons. Moreover, this increase in excitability of LA cells in response to fluoxetine application was accompanied by an increase in the input resistance of these neurons. Importantly, the change in input resistance exhibits a significant positive correlation with the increase in firing frequency in individual cells, suggesting that this increase in input resistance is likely to contribute to enhanced neuronal excitability triggered by fluoxetine.
Many lines of evidence suggest that the enhanced serotonin availability due to SSRIs may mediate the increase in BLA excitability due to acute fluoxetine treatment. Firstly, in addition to fluoxetine, the anxiogenic effects of acute SSRI treatment have also been demonstrated using other SSRIs such as citalopram, paroxetine, sertraline etc. (Griebel et al., 1994; Sanchez and Meier, 1997; Dekeyne et al., 2000; Kurt et al., 2000; Bagdy et al., 2001; Koks et al., 2001; Burghardt et al., 2004). Although various SSRIs are known to differ in their specificities and binding affinities, all of them bind to the serotonin transporter with maximum affinity supporting a role for some serotonin based mechanism in the observed behavioral effects. Secondly, acute SSRI administration has been shown to increase extracellular serotonin (Bosker et al., 2001), and glutamate levels in the amygdala (Reznikov et al., 2007). Many studies have previously shown that serotonin is capable of modulating excitatory and inhibitory currents in the amygdala (Stutzmann et al., 1998; Stutzmann and Ledoux 1999; Rainnie, 1999) and hence can tip the balance of excitation–inhibition in favor of increased excitability of BLA neurons. Finally, numerous serotonin receptor subtypes have also been implicated in the modulation of anxiety-like behavior and a growing body of data suggests that anxiety induced by serotonin via the 5HT2c receptor may underlie the acute effects of SSRIs (Dekeyne et al., 2000; Salchner and Singewald, 2006). Thus, many lines of evidence suggest a serotonin dependent mechanism for the acute effects of SSRIs and future studies will be needed to further explore a specific role for serotonin in the BLA neuronal excitability changes reported here.
On the other hand, anxiety can be induced by a wide range of pharmacological agents that affect glutamatergic receptors, inhibitory GABA receptors, and neuromodulatory systems such as serotonin, norepinephrine, and various neuropeptides. Thus, SSRI induced anxiety could be brought about by a range of different mechanisms. Fluoxetine, in addition to increasing serotonin levels, also increases the extracellular levels of other catecholamines such as norepinephrine and dopamine. The anxiogenic effects of fluoxetine may thus be mechanistically similar to other anxiogenic agents impinging on these neurotransmitter systems (Singewald et al., 2003). Further, although originally identified as a selective serotonin reuptake inhibitor, numerous studies have shown that fluoxetine can bind to a wide range of other targets at physiologically relevant concentrations (Bianchi, 2008). These alternate binding targets of fluoxetine include certain classes of potassium channels whose activation could lead to the increase in input resistance in fluoxetine-treated cells (Choi et al., 1999, 2001, 2004; Hahn et al., 1999; Kobayashi et al., 2004; Kennard et al., 2005; Norman et al., 2005). These mechanisms await further investigation.
A number of studies have previously suggested a relationship between increased amygdala activity and enhanced fear and anxiety-like behavior (Rainnie et al., 2004; Manzanares et al., 2005; Duvarci and Pare, 2007; Karst et al., 2010). How may the increase in BLA neuronal excitability observed in our study lead to heightened fear and anxiety caused by acute fluoxetine treatment? First, such increase in excitability could contribute to a non-specific increase in spontaneous activity. This, in turn, would lead to generalized, cue non-specific fear that is manifested as enhanced anxiety. Second, fear conditioning has been shown to induce increases in conditional stimulus (CS)-evoked spike firing in LA neurons (Quirk et al., 1995; Collins and Pare, 2000; Maren, 2000; Maren and Quirk, 2004). Thus, increase in excitability in these LA neurons following fluoxetine treatment would make them more susceptible to greater CS-evoked firing compared to LA neurons in fear-conditioned animals that have not received fluoxetine injection, and this too would be manifested as a facilitation of auditory fear conditioning as reported earlier (Burghardt et al., 2004, 2007).
Our findings from this study may also provide a useful experimental framework for investigating another interesting aspect of the effects of SSRI treatment on anxiety and fear memory formation. Clinical reports indicate that while anxiety is often increased during early stages of SSRI treatment, it is reduced after several weeks of treatment. In other words, acute versus chronic treatment with the same SSRI can elicit opposite effects on anxiety and earlier studies have captured this dichotomy in rodent models of auditory fear conditioning (Burghardt et al., 2004). Thus, future studies will be needed to examine if more prolonged SSRI treatment leads to differential effects on the same physiological and morphological measures used in the present study. It is also important to note that the amygdala is a heterogeneous collection of numerous nuclei and sub-nuclei, and our present analysis has focused only on the input interface of the amygdala – the lateral and basolateral divisions. Although the BLA is most well studied for its role in fear memory acquisition and expression, more recent studies suggest important roles for output nuclei of the amygdala (Pare et al., 2004; Ehrlich et al., 2009; Amano et al., 2010) as well as extended amygdala structures such as the bed nucleus of stria terminalis (BNST) in the regulation of fear and anxiety (Grillon, 2008; Hammack et al., 2009; Davis et al., 2010). The cellular effects of SSRI treatment in these regions may differ considerably to those observed in the BLA. For example, pharmacologically diverse anxiogenic agents induce Fos expression in the CeA (Singewald et al., 2003). Interestingly, repeated restraint stress leads to an up-regulation of the extracellular matrix protease tissue plasminogen activator (tPA) in the medial (MeA) and central nucleus of the amygdala (CeA), but not BLA. Further, the same repeated stress that elicits enhanced anxiety and spinogenesis in the BLA actually lowers spine density in the MeA. Importantly, stress-induced spine loss in the MeA is tPA-dependent, but stress-induced spinogenesis in the BLA is not (Bennur et al., 2007; Roozendaal et al., 2009). It will, therefore, be important to assess the potential contributions of these output nuclei of the amygdala to the anxiogenic effects of acute SSRI treatment. Together, a more rigorous analysis of the impact of both acute and chronic treatment with SSRIs on cells and microcircuits of the amygdala will be critically important for improving our understanding of both the beneficial, as well as the undesirable side-effects, of antidepressant treatments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funds from National Centre for Biological Sciences. Fluoxetine Hydrochloride was provided by Hikal Ltd. The authors would like to thank Ms. Shobha Anilkumar for technical assistance and advice with Golgi staining.
References
Amano, T., Unal, C. T., and Pare, D. (2010). Synaptic correlates of fear extinction in the amygdala. Nat. Neurosci. 13, 489–494.
Anand, A., and Shekhar, A. (2003). Brain imaging studies in mood and anxiety disorders: special emphasis on the amygdala. Ann. N. Y. Acad. Sci. 985, 370–388.
Bagdy, G., Graf, M., Anheuer, Z. E., Modos, E. A., and Kantor, S. (2001). Anxiety-like effects induced by acute fluoxetine, sertraline or m-CPP treatment are reversed by pretreatment with the 5-HT2C receptor antagonist SB-242084 but not the 5-HT1A receptor antagonist WAY-100635. Int. J. Neuropsychopharmacol. 4, 399–408.
Belzung, C., Le Guisquet, A. M., Barreau, S., and Calatayud, F. (2001). An investigation of the mechanisms responsible for acute fluoxetine-induced anxiogenic-like effects in mice. Behav. Pharmacol. 12, 151–162.
Bennur, S., Shankaranarayana Rao, B. S., Pawlak, R., Strickland, S., Mcewen, B. S., and Chattarji, S. (2007). Stress-induced spine loss in the medial amygdala is mediated by tissue-plasminogen activator. Neuroscience 144, 8–16.
Bezchlibnyk-Butler, K., Aleksic, I., and Kennedy, S. H. (2000). Citalopram – a review of pharmacological and clinical effects. J. Psychiatry Neurosci. 25, 241–254.
Bianchi, M. T. (2008). Non-serotonin anti-depressant actions: direct ion channel modulation by SSRIs and the concept of single agent poly-pharmacy. Med. Hypotheses 70, 951–956.
Bodnoff, S. R., Suranyicadotte, B., Quirion, R., and Meaney, M. J. (1989). A comparison of the effects of diazepam versus several typical and atypical anti-depressant drugs in an animal model of anxiety Psychopharmacology (Berl.) 97, 277–279.
Bondareff, W., Alpert, M., Friedhoff, A. J., Richter, E. M., Clary, C. M., and Batzar, E. (2000). Comparison of sertraline and nortriptyline in the treatment of major depressive disorder in late life. Am. J. Psychiatry 157, 729–736.
Bosker, F. J., Cremers, T. I., Jongsma, M. E., Westerink, B. H., Wikstrom, H. V., and Den Boer, J. A. (2001). Acute and chronic effects of citalopram on postsynaptic 5-hydroxytryptamine(1A) receptor-mediated feedback: a microdialysis study in the amygdala. J. Neurochem. 76, 1645–1653.
Burghardt, N. S., Bush, D. E. A., Mcewen, B. S., and Ledoux, J. E. (2007). Acute selective serotonin reuptake inhibitors increase conditioned fear expression: blockade with a 5-HT2C receptor antagonist. Biol. Psychiatry 62, 1111–1118.
Burghardt, N. S., Sullivan, G. M., Mcewen, B. S., Gorman, J. M., and Ledoux, J. E. (2004). The selective serotonin reuptake inhibitor citalopram increases fear after acute treatment but reduces fear with chronic treatment: a comparison with tianeptine. Biol. Psychiatry 55, 1171–1178.
Choi, B. H., Choi, J. S., Yoon, S. H., Rhie, D. J., Min, D. S., Jo, Y. H., Kim, M. S., and Hahn, S. J. (2001). Effects of norfluoxetine, the major metabolite of fluoxetine, on the cloned neuronal potassium channel Kv3.1. Neuropharmacology 41, 443–453.
Choi, J. S., Choi, B. H., Ahn, H. S., Kim, M. J., Han, T. H., Rhie, D. J., Yoon, S. H., Jo, Y. H., Kim, M. S., and Hahn, S. J. (2004). Fluoxetine inhibits A-type potassium currents in primary cultured rat hippocampal neurons. Brain Res. 1018, 201–207.
Choi, J. S., Hahn, S. J., Rhie, D. J., Yoon, S. H., Jo, Y. H., and Kim, M. S. (1999). Mechanism of fluoxetine block of cloned voltage-activated potassium channel Kv1.3. J. Pharmacol. Exp. Ther. 291, 1–6.
Collins, D. R., and Pare, D. (2000). Differential fear conditioning induces reciprocal changes in the sensory responses of lateral amygdala neurons to the CS(+) and CS(−). Learn. Mem. 7, 97–103.
Davis, M., Walker, D. L., Miles, L., and Grillon, C. (2010). Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology 35, 105–135.
Dekeyne, A., Denorme, B., Monneyron, S., and Millan, M. J. (2000). Citalopram reduces social interaction in rats by activation of serotonin (5-HT)(2C) receptors. Neuropharmacology 39, 1114–1117.
Del-Ben, C. M., Deakin, J. F. W., Mckie, S., Delvai, N. A., Williams, S. R., Elliott, R., Dolan, M., and Anderson, I. M. (2005). The effect of citalopram pretreatment on neuronal responses to neuropsychological tasks in normal volunteers: an fMRI study. Neuropsychopharmacology 30, 1724–1734.
Duvarci, S., and Pare, D. (2007). Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J. Neurosci. 27, 4482–4491.
Ehrlich, I., Humeau, Y., Grenier, F., Ciocchi, S., Herry, C., and Luthi, A. (2009). Amygdala inhibitory circuits and the control of fear memory. Neuron 62, 757–771.
Feldman, M. L., and Peters, A. (1979). A technique for estimating total spine numbers on Golgi-impregnated dendrites. J. Comp. Neurol. 188, 527–542.
Fergusson, D., Doucette, S., Cranley, K., Glass, K. C., Shapiro, S., Healy, D., Hebert, P., and Hutton, B. (2005). Association between suicide attempts and selective serotonin reuptake inhibitors: systematic review of randomised controlled trials. Br. Med. J. 330, 396–399.
Goldstein, B. J., and Goodnick, P. J. (1998). Selective serotonin reuptake inhibitors in the treatment of affective disorders – III. Tolerability, safety and pharmacoeconomics. J. Psychopharmacol. 12, S55–S87.
Gorman, J. M., Liebowitz, M. R., Fyer, A. J., Goetz, D., Campeas, R. B., Fyer, M. R., Davies, S. O., and Klein, D. F. (1987). An open trial of fluoxetine in the treatment of panic attacks. J. Clin. Psychopharmacol. 7, 329–332.
Govindarajan, A., Rao, B. S. S., Nair, D., Trinh, M., Mawjee, N., Tonegawa, S., and Chattarji, S. (2006). Transgenic brain-derived neurotrophic factor expression causes both anxiogenic and antidepressant effects. Proc. Natl. Acad. Sci. U.S.A. 103, 13208–13213.
Griebel, G., Moreau, J. L., Jenck, F., Misslin, R., and Martin, J. R. (1994). Acute and chronic treatment with 5-HT reuptake inhibitors differentially modulate emotional responses in anxiety models in rodents. Psychopharmacology (Berl.) 113, 463–470.
Grillon, C. (2008). Models and mechanisms of anxiety: evidence from startle studies. Psychopharmacology (Berl.) 199, 421–437.
Hahn, S. J., Choi, J. S., Rhie, D. J., Oh, C. S., Jo, Y. H., and Kim, M. S. (1999). Inhibition by fluoxetine of voltage-activated ion channels in rat PC12 cells. Eur. J. Pharmacol. 367, 113–118.
Hammack, S. E., Guo, J. D., Hazra, R., Dabrowska, J., Myers, K. M., and Rainnie, D. G. (2009). The response of neurons in the bed nucleus of the stria terminalis to serotonin: implications for anxiety. Prog. Neuropsychopharmacol. Biol. Psychiatry 33, 1309–1320.
Hasler, G., Drevets, W. C., Manji, H. K., and Charney, D. S. (2004). Discovering endophenotypes for major depression. Neuropsychopharmacology 29, 1765–1781.
Hasler, G., and Northoff, G. (2011). Discovering imaging endophenotypes for major depression. Mol. Psychiatry 16, 604–619.
Hofer, S. B., and Bonhoeffer, T. (2010). Dendritic spines: the stuff that memories are made of? Curr. Biol. 20, R157–R159.
Horner, C. H., and Arbuthnott, E. (1991). Methods of estimation of spine density – are spines evenly distributed throughout the dendritic field? J. Anat. 177, 179–184.
Karson, C. N., Newton, J. E., Livingston, R., Jolly, J. B., Cooper, T. B., Sprigg, J., and Komoroski, R. A. (1993). Human brain fluoxetine concentrations. J. Neuropsychiatry Clin. Neurosci. 5, 322–329.
Karst, H., Berger, S., Erdmann, G., Schutz, G., and Joels, M. (2010). Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc. Natl. Acad. Sci. U.S.A. 107, 14449–14454.
Kennard, L. E., Chumbley, J. R., Ranatunga, K. M., Armstrong, S. J., Veale, E. L., and Mathie, A. (2005). Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br. J. Pharmacol. 144, 821–829.
Kent, J. M., Coplan, J. D., and Gorman, J. M. (1998). Clinical utility of the selective serotonin reuptake inhibitors in the spectrum of anxiety. Biol. Psychiatry 44, 812–824.
Kobayashi, T., Washiyama, K., and Ikeda, K. (2004). Inhibition of G protein-activated inwardly rectifying K+ channels by various antidepressant drugs. Neuropsychopharmacology 29, 1841–1851.
Koks, S., Beljajev, S., Koovit, I., Abramov, U., Bourin, M., and Vasar, E. (2001). 8-OH-DPAT, but not deramciclane, antagonizes the anxiogenic-like action of paroxetine in an elevated plus-maze. Psychopharmacology (Berl.) 153, 365–372.
Kurt, M., Arik, A. C., and Celik, S. (2000). The effects of sertraline and fluoxetine on anxiety in the elevated plus-maze test in mice. J. Basic. Clin. Physiol. Pharmacol. 11, 173–180.
Manzanares, P. A. R., Isoardi, N. A., Carrer, H. F., and Molina, V. A. (2005). Previous stress facilitates fear memory, attenuates GABAergic inhibition, and increases synaptic plasticity in the rat basolateral amygdala. J. Neurosci. 25, 8725–8734.
Maren, S. (2000). Auditory fear conditioning increases CS-elicited spike firing in lateral amygdala neurons even after extensive overtraining. Eur. J. Neurosci. 12, 4047–4054.
Maren, S., and Quirk, G. J. (2004). Neuronal signalling of fear memory. Nat. Rev. Neurosci. 5, 844–852.
Masand, P. S., and Gupta, S. (1999). Selective serotonin-reuptake inhibitors: an update. Harv. Rev. Psychiatry 7, 69–84.
McKernan, M. G., and ShinnickGallagher, P. (1997). Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390, 607–611.
McKie, S., Del-Ben, C., Elliott, R., Williams, S., Del Vai, N., Anderson, I., and Deakin, J. F. W. (2005). Neuronal effects of acute citalopram detected by pharmacoMRI. Psychopharmacology (Berl.) 180, 680–686.
Mir, S., and Taylor, D. (1997). The adverse effects of antidepressants. Curr. Opin. Psychitary 10, 88–94.
Mitra, R., Jadhav, S., Mcewen, B. S., Vyas, A., and Chattarji, S. (2005). Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc. Natl. Acad. Sci. U.S.A. 102, 9371–9376.
Mukherjee, J., Das, M. K., Yang, Z. Y., and Lew, R. (1998). Evaluation of the binding of the radiolabeled antidepressant drug, 18F-fluoxetine in the rodent brain: an in vitro and in vivo study. Nucl. Med. Biol. 25, 605–610.
Norman, E. D., Egli, R. E., Colbran, R. J., and Winder, D. G. (2005). A potassium channel blocker induces a long-lasting enhancement of corticostriatal responses. Neuropharmacology 48, 311–321.
Pare, D., Quirk, G. J., and Ledoux, J. E. (2004). New vistas on amygdala networks in conditioned fear. J. Neurophysiol. 92, 1–9.
Quirk, G. J., Repa, C., and Ledoux, J. E. (1995). Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: parallel recordings in the freely behaving rat. Neuron 15, 1029–1039.
Rainnie, D. G. (1999). Serotonergic modulation of neurotransmission in the rat basolateral amygdala. J. Neurophysiol. 82, 69–85.
Rainnie, D. G., Bergeron, R., Sajdyk, T. J., Patil, M., Gehlert, D. R., and Shekhar, A. (2004). Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J. Neurosci. 24, 3471–3479.
Reznikov, L. R., Grillo, C. A., Piroli, G. G., Pasumarthi, R. K., Reagan, L. P., and Fadel, J. (2007). Acute stress-mediated increases in extracellular glutamate levels in the rat amygdala: differential effects of antidepressant treatment. Eur. J. Neurosci. 25, 3109–3114.
Rogan, M. T., Staubli, U. V., and Ledoux, J. E. (1997). Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390, 604–607.
Roozendaal, B., Mcewen, B. S., and Chattarji, S. (2009). Stress, memory and the amygdala. Nat. Rev. Neurosci. 10, 423–433.
Salchner, P., and Singewald, N. (2002). Neuroanatomical substrates involved in the anxiogenic-like effect of acute fluoxetine treatment. Neuropharmacology 43, 1238–1248.
Salchner, P., and Singewald, N. (2006). 5-HT receptor subtypes involved in the anxiogenic-like action and associated Fos response of acute fluoxetine treatment in rats. Psychopharmacology (Berl.) 185, 282–288.
Sanchez, C., and Meier, E. (1997). Behavioral profiles of SSRIs in animal models of depression, anxiety and aggression – are they all alike? Psychopharmacology (Berl.) 129, 197–205.
Shankaranarayana Rao, B. S., and Raju, T. R. (2004). “The golgi techniques for staining neurons,” in Brain and Behavior, eds T. R. Raju, B. M. Kutty, T. N. Sathyaprabha, and B. S. Shankaranarayana Rao (Bangalore: National Institute of Mental Health and Neurosciences), 108–111.
Sheehan, D. V., Raj, B. A., Trehan, R. R., and Knapp, E. L. (1993). Serotonin in panic disorder and social phobia International Clinical. Psychopharmacology (Berl.) 8, 63–77.
Silva, R. C. B., and Brandao, M. L. (2000). Acute and chronic effects of gepirone and fluoxetine in rats tested in the elevated plus-maze: an ethological analysis. Pharmacol. Biochem. Behav. 65, 209–216.
Singewald, N., Salchner, P., and Sharp, T. (2003). Induction of c-Fos expression in specific areas of the fear circuitry in rat forebrain by anxiogenic drugs. Biol. Psychiatry 53, 275–283.
Spigset, O. (1999). Adverse reactions of selective serotonin reuptake inhibitors – reports from a spontaneous reporting system. Drug Saf. 20, 277–287.
Stahl, S. M. (2000). Placebo-controlled comparison of the selective serotonin reuptake inhibitors citalopram and sertraline. Biol. Psychiatry 48, 894–901.
Stokes, P. E., and Holtz, A. (1997). Fluoxetine tenth anniversary update: the progress continues. Clin. Ther. 19, 1135–1250.
Stutzmann, G. E., and Ledoux, J. E. (1999). GABAergic antagonists block the inhibitory effects of serotonin in the lateral amygdala: a mechanism for modulation of sensory inputs related to fear conditioning. J. Neurosci. 19, RC8.
Stutzmann, G. E., Mcewen, B. S., and Ledoux, J. E. (1998). Serotonin modulation of sensory inputs to the lateral amygdala: dependency on corticosterone. J. Neurosci. 18, 9529–9538.
Teicher, M. H., Glod, C., and Cole, J. O. (1990). Emergence of intense suicidal preoccupation during fluoxetine treatment. Am. J. Psychiatry 147, 207–210.
Trommald, M., and Hulleberg, G. (1997). Dimensions and density of dendritic spines from rat dentate granule cells based on reconstructions from serial electron micrographs. J. Comp. Neurol. 377, 15–28.
Trommald, M., Jensen, V., and Andersen, P. (1995). Analysis of dendritic spines in rat CA1 pyramidal cells intracellularly filled with a fluorescent dye. J. Comp. Neurol. 353, 260–274.
van der Kolk, B. A., Dreyfuss, D., Michaels, M., Shera, D., Berkowitz, R., Fisler, R., and Saxe, G. (1994). Fluoxetine in posttraumatic stress disorder. J. Clin. Psychiatry 55, 517–522.
Keywords: amygdala, anxiety, neuronal excitability, SSRI, fluoxetine, dendritic spines
Citation: Ravinder S, Pillai AG and Chattarji S (2011) Cellular correlates of enhanced anxiety caused by acute treatment with the selective serotonin reuptake inhibitor fluoxetine in rats. Front. Behav. Neurosci. 5:88. doi: 10.3389/fnbeh.2011.00088
Received: 08 September 2011; Accepted: 12 December 2011;
Published online: 28 December 2011.
Edited by:
Andrew Holmes, National Institute on Alcohol Abuse and Alcoholism, USAReviewed by:
John F. Cryan, University College Cork, IrelandCara Lynn Wellman, Indiana University, USA
Copyright: © 2011 Ravinder, Pillai and Chattarji. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Sumantra Chattarji, National Centre for Biological Sciences, GKVK Campus, Bellary Road, Bangalore 560065, India. e-mail: shona@ncbs.res.in
† Present address: Anup G. Pillai , Rudolf Magnus Institute of Neuroscience, Stratenum building, Room STR5.203, Universiteitsweg 100, 3584 CG Utrecht, Netherlands.