Article Text

Abstract
OBJECTIVE A pilot study of the density of dendritic spines on pyramidal neurons in layer III of human temporal and frontal cerebral neocortex in schizophrenia.
METHODS Postmortem material from a group of eight prospectively diagnosed schizophrenic patients, five archive schizophrenic patients, 11 non-schizophrenic controls, and one patient with schizophrenia-like psychosis, thought to be due to substance misuse, was impregnated with a rapid Golgi method. Spines were counted on the dendrites of pyramidal neurons in temporal and frontal association areas, of which the soma was in layer III (which take part in corticocortical connectivity) and which met strict criteria for impregnation quality. Altogether 25 blocks were studied in the schizophrenic group and 21 in the controls. If more than one block was examined from a single area, the counts for that area were averaged. All measurements were made blind: diagnoses were only disclosed by a third party after measurements were completed. Possible confounding affects of coexisiting Alzheimer’s disease were taken into account, as were the effects of age at death and postmortem interval.
RESULTS There was a significant (p<0.001) reduction in the numerical density of spines in schizophrenia (276/mm in control temporal cortex and 112/mm in schizophrenic patients, and 299 and 101 respectively in the frontal cortex). An analysis of variance, taking out effects of age at death and postmortem interval, which might have explained the low spine density for some of the schizophrenic patients, did not affect the significance of the results.
CONCLUSION The results support the concept of there being a defect in the fine structure of dendrites of pyramidal neurons, involving loss of spines, in schizophrenia and may help to explain the loss of cortical volume without loss of neurons in this condition, although the effect of neuroleptic drugs cannot be ruled out.
- cerebral cortex
- pyramidal neurons
- dendritic spines
- glutamate
- schizophrenia
Statistics from Altmetric.com
A consistent finding in studies of schizophrenia using neuroimaging or at postmortem1-7 is enlargement of the cerebral ventricles. Some studies have shown a decrease in volume of the cerebral cortex but no change in that of subcortical white matter,7-9 suggesting that there is no loss of axons, and probably no loss of cell bodies of neurons in the cortex.10 This could be explained by a differential loss of neuronal processes, such as axon terminals, dendritic processes, or dendritic spines, in the cortical neuropil, or their failure to develop. This was suggested by our recent finding of a loss of immunoreactivity for calcium binding proteins in the neuropil without loss of neuronal somata in schizophrenia,11 and in early reports of this current study,12 13 which first showed that dendritic spines in schizophrenic patients were abnormally sparse.
We have set out to investigate dendritic spines of pyramidal cells as a possible site for changes in the neuropil and to extend our preliminary report that, in area 38 of the temporal cortex, there is a decrease in spine density, defined as the number of spines/unit length of pyramidal neuron dendrite.12 Extending this finding to other cortical areas13 we briefly reported that spine density in schizophrenic patients was less than half that in non-schizophrenic controls. Glantz and Lewis14 15 have also given preliminary reports of 35% reduction of pyramidal dendritic spines in schizophrenic prefrontal cortex. Although our study must be regarded as a pilot study, based on tissue obtained from various sources with an attempt to rule out other pathology in a limited subsample, we think that our findings are novel and of sufficient interest to justify a more ambitious and rigorous investigation. This report expands the number of areas studied and gives greater detail on the cohort reported previously by abstract.12 13
A loss of dendritic spines, particularly of the pyramidal neurons in the cortex, could have implications for the glutamate hypothesis of schizophrenia,16 as the N-methyl-D-aspartate (NMDA) subtype of glutamate receptor is present on their dendrites (and probably dendritic spines).17 Our own findings also confirm immunoreactivity for GluR1 and GluR4 receptors in dendrites and spines in human cortex.18 19 Pyramidal cells in layer III are glutamatergic20 and are involved in projections between various cortical areas,21 22 and might therefore be expected to be important in higher cognitive function that is disturbed in schizophrenia.23
Materials and methods
SPECIMENS AND FIXATION
This study was carried out in conjunction with the Charing Cross Prospective Schizophrenia Project in which authorisation is obtained in life for neuropsychological evaluation of long stay schizophrenic patients and subsequent neuropathological study of their brains postmortem. Eight of the brains from schizophrenic patients were from an early phase of the Prospective Project, for which there had been limited assessment premortem. They are referred to as the “CX” group. Another five schizophrenic brains were not part of the Prospective Project but were obtained from other sources. They are referred to as the “archive” group. In all, the brains of 13 schizophrenic patients (three women, average age 67 years, range 54–78; 10 men, average age 65 years, range 44–83) and 11 non-schizophrenic controls (two women, average age 72 years, range 66–78; nine men, average age 62 years, range 50–77 years) were available (table 1). One further brain was obtained from an 87 year old woman with a history of psychosis from the age of 52, probably related to benzedrene addiction and alcoholism.
Patient details and mean spine densities (spines/mm dendrite) on layer III pyramidal neurons in the cerebral cortex of schizophrenic patients and non-schizophrenic controls
Multiple cortical blocks (table 1), each about 1 cm3, were removed and fixed in 4% buffered paraformaldehyde, except for the archive group, which had been fixed in 10% formalin. The fixation time varied from a few hours (CX group), mostly from 1–2 months, to a few years (archive group). Twenty five blocks of neocortical tissue were removed from the temporal and frontal association cortex of the schizophrenic brains and 21 from the controls and prepared for morphological examination (table 1). Details of the areas studied are given in the results section and in table 1. The brains were coded so that the nature of each specimen was unknown to the investigator; the examination was therefore conducted blind to eliminate assessment bias.
DIAGNOSIS
The diagnosis of the CX patients was based on clinical assessment and a judgement made from the case notes using DSM III-R criteria; the diagnosis of the archive cases was as reported from the donating hospital.
PATHOLOGY
The CX schizophrenic brains were examined at necropsy for abnormal gyral patterns, ventricular enlargement, and vascular and degenerative lesions and tumours, and none were found, except that in brain 14 there was evidence of fresh subdural haemorrhage over the left vertex, so frontal lobe blocks were not taken.
The CX patients were under the clinical care of two of us (TB, AM). None of these patients had clinical dementia of the Alzheimer type, although one (No 19) had a history of dementia. Gross examination of the brains postmortem did not show evidence of Alzheimer’s disease. In six of the CX schizophrenic brains sufficient tissue was available for a modified CERAD procedure to be used to control for histological evidence of Alzheimer’s disease (table 2). This involved taking blocks from cingulate, hippocampal, frontal, occipital, and parietal cortex and staining them immunocytochemically for plaques with an antibody to β-amyloid (1E8) and for tangles with an antibody to Tau protein (AT8).
Results of modified CERAD control for Alzheimer’s disease (AD)
Eleven control brains were obtained from routine necropsies or archival sources, fixed respectively in 4% paraformaldehyde and 10% formalin (table 1). None were known to have neurological or psychiatric disease, and all died from acute causes (mostly myocardial infarction, pulmonary embolism, or head injury), although one (No 11) had a chronic history of cardiomyopathy.
CRITERIA FOR ALZHEIMER TYPE PATHOLOGY
We followed CERAD criteria for Alzheimer’s disease but used immunostaining, which is more sensitive than silver. Evidence of plaques and tangles was rated (table 2), but the final diagnosis took into account age, and when available, clinical information.
GOLGI TECHNIQUE
The blocks of tissue were prepared for microscopical study using the rapid Golgi technique. Each block was fixed in osmium tetroxide/potassium dichromate mixture for 5 days in the dark. They were then rinsed with distilled water and placed in 0.75% silver nitrate solution for a further 48 hours in the dark, after which they were shelled in paraffin wax and sectioned at 100 μm using a base sledge microtome. The sections were cleared in xylene and mounted on microscope slides. The relative thickness of the sections helped to maintain the integrity of the impregnated neurons, and allowed more complete analysis of the dendritic arbors of the impregnated cells by focusing to follow the dendritic arborisation to different depths.
LIGHT MICROSCOPY AND SPINE DENSITY MEASUREMENTS
Many densely stained neurons and neuroglia were present in the impregnated sections. From at least one section of each block, 10 adjacent pyramidal cells that complied with the following sampling criteria were chosen for analysis. All had their cell body in layer III; they had complete or near complete dendritic arbors, and they were unobscured by surrounding cells. Any cell devoid of spines, or nearly so, was excluded in case this was due to poor impregnation. Each cell was drawn at 400× magnification using a microscope fitted with a drawing tube, and the total length of its dendrites measured. The magnification was then increased to 1000×, and the number of spines on all the dendrites counted under oil immersion. As some of the spines were obscured by their own dendrite, and to keep the counting procedure consistent, only those spines that protruded laterally from the shafts of the dendrites into the surrounding area of clear neuropil were counted. The spine density of a pyramidal neuron was calculated by dividing the total number of spines on a neuron by the total length of its dendrites, and was expressed as the number of spines/mm dendrite. The mean spine density for each block was then calculated. As the measurements were carried out by several investigators, we corrected for individual counting differences by using a Z score analysis. This was done for each block by calculating a Z score for that block equal to the mean raw spine count for that block, minus the observer’s mean over all the blocks he or she measured, all divided by the SD for these values. This gave a range of spine densities and a range of Z scores for each observer, from which could be determined the Z score value for a single spine for that observer, which was then used to correct raw data from each observer for individual variability.
Results
Details of the number of blocks examined are given in table 1—namely, the area examined and the average number of spines counted/mm dendrite in the area listed.
SPINE DENSITY MEASUREMENTS (TABLE 1 AND FIG1)
For the presentation of results, the schizophrenic brains are described in two groups: eight from the Prospective Project (the CX group) for which precise demographic data are available, and five archive brains (table 1) for which some details were missing and which had been fixed for longer. In the five archive brains impregnation was as good as in the CX group, but only one block in each temporal polar cortex (area 38) was examined.

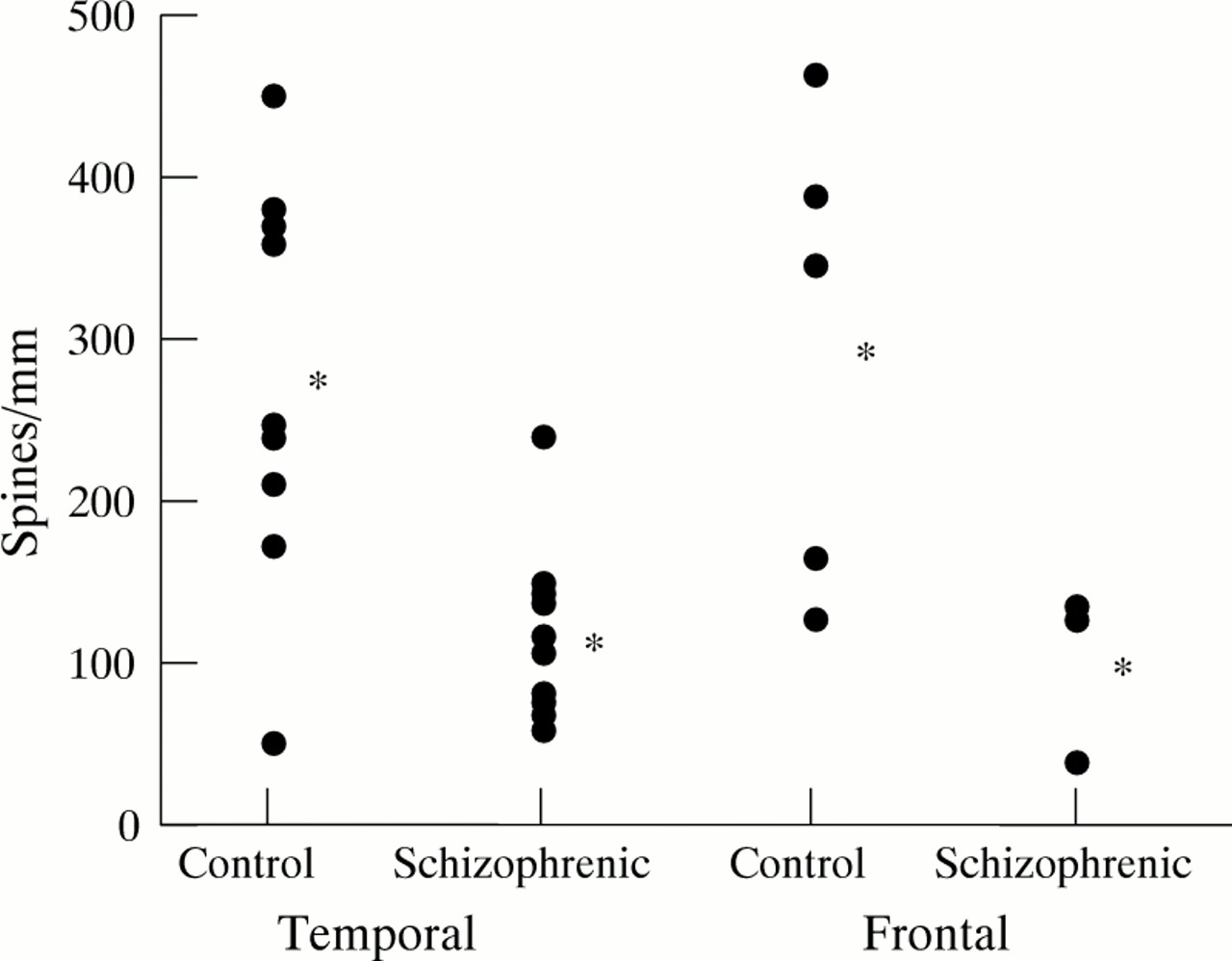
Scatter plots to show spine densities on layer III pyramidal neurons in brains from schizophrenic patients and non-schizophrenic controls. Each point represents the mean number of spines/mm dendrite in temporal and frontal areas of a single brain. The asterisks indicate the means for each of these groups of areas. The difference is highly significant in the temporal cortex, but in the frontal cortex the few cases do not reach significance, although the trend is clearly similar to that in temporal cortex. Details of cortical areas examined are given in the Results section and in table1.
QUALITATIVE FINDINGS
Typical pyramidal neurons were found with somata in all layers except layer I, but most commonly in layers II, III, and V. Those of which the soma was in layer III (fig 2 A) tended to form a more homogeneous population than those in other layers, and there was less fragmentation of their dendrites, particularly the apical dendrite that was, of necessity, shorter than that of layer V pyramidal cells. The soma was not always triangular, being often more rounded, but a single apical dendrite was always identified extending from the superior pole, usually as far as layer I where it ramified. Sometimes it branched earlier, with branches ascending almost parallel to the main apical trunk. Several basal dendrites typically formed a skirt around the inferior pole of the soma. All selected dendrites bore clearly impregnated spines (fig 2 B), but various degrees of loss of spines were detectable qualitatively on the pyramidal neurons in most specimens of schizophrenic cortex (fig 2C).

Micrographs of pyramidal cells in the cerebral cortex stained with the Golgi method. (A) A layer III pyramidal cell from a control brain, showing its morphology and spiny dendrites. (B) A higher power view of spines on an apical dendrite of a control pyramidal neuron. (C) Smooth, spineless segment on an apical dendrite in schizophrenic cortex. Scale: (A) 30 μm; (B) 20 μm; (C) 15 μm.
SPINE COUNTS
Table 1 and fig 1 show the mean spine count/mm dendrite in pyramidal cells in layer III for blocks taken from the temporal and frontal lobes. The mean spine count for the temporal lobe of CX patients (areas 20, 21, 22, and 38 of Brodmann22) in 15 blocks was 125 (SD 58), compared with 276 (SD 126) spines/mm from 13 blocks from non-schizophrenic controls. In the five archive brains the count was 90 (SD 33) spines/mm dendrite. On this measure CX schizophrenic patients differed significantly from controls (p<0.001) and the CX plus the archive groups combined were also significantly different from controls.
If only the spine counts for area 38 were compared with area 38 controls, the mean count was 98 spines for 13 blocks from eight schizophrenic patients and 284 for 10 blocks from eight controls. There was no significant difference in spine counts from area 38 between the CX group and the archive group, the mean of the second being 90. The mean count from five blocks from the frontal lobe of three patients of the CX schizophrenia group (areas 10, 11, and 45) was 101. If patients 13 and 19, who had high plaque and tangle counts in the hippocampus, were excluded the difference between schizophrenic patients and controls was not significantly affected. There was considerable consistency in the frontal and temporal cortices.
Measurements were made in the frontal cortex of the patient with benzedrene addiction and alcoholism (No 25), and the spine count was close to the mean found in the other controls (238/mm dendrite).
EFFECT OF AGE AND POSTMORTEM INTERVAL
An analysis of variance (ANOVA) was carried out on the CX, archive, and control groups for those patients with a complete data set. The analysis included nine controls, the eight schizophrenic patients from the CX cohort, and four from the archive group.
There was a significant inverse correlation (p<0.01) between age and spine density in the non-schizophrenic controls but the correlation was not significant for postmortem interval and spine density, whether this was calculated by parametric (Pearson) or non-parametric (Spearman) tests (table 3 and fig 3). In the first analysis, schizophrenic patients with coexisting Alzheimer’s disease pathology were not excluded, as their results were in line with the other schizophrenic patients. Analysis of variance (SPSS) taking out the effect of age and postmortem interval on the difference between groups in spine density in area 38 gave an F value of 5.57 (p=0.015). Individual variables were nor significantly skewed (skew=0.9 for spines, 1.55 for PMI, and –0.29 for age), but a Kruskal-Wallis non-parametric test one way ANOVA also showed a significant difference between groups (Kruskal-Wallis test χ2=8.159, p=0.0169). Excluding the archive group and taking out the effect of postmortem interval and age there was a significant difference between non-schizophrenic and CX schizophrenic patients (F=5.581, p=0.015).
Correlation between spine density versus age and postmortem interval (PMI) in subgroups of schizophrenic and control brains

{kind=link}
{kind=link}
{kind=link}
Regression analysis of spine density v age in the temporal cortex. The solid circles represent control subjects and show a correlation of lowering spine density with increasing age (R2=0.64). However, in the schizophrenic group (open circles) there is no correlation of spine loss with age (R2=0.13).
EVIDENCE OF ALZHEIMER’S DISEASE (TABLE 2)
In the CX schizophrenic group, patients 13 (age 56) and 19 (age 83) showed heavy staining for amyloid plaques, and patient 17 had slight to moderate staining for plaques. This suggests coexisting Alzheimer’s disease in 13 and 19, but this is doubtful in patient 17 as the age was 71. Nevertheless, all three were excluded in the next analysis. Patient 25 had a history of alcohol and drug misuse and was rediagnosed after detailed examination of the notes, but before knowledge of the spine count, as having a non-schizophrenic psychosis. The presence of plaques and tangles was not associated with a lower spine count; thus the three schizophrenic patients from the CX group (patients 12, 15, and 18) with little or no evidence of plaques or tangles had a mean spine count in area 38 of 81 whereas patients 13, 17, and 19 with plaques had a mean count of 128. Patients 14 and 16, for whom there was no tissue to examine for Alzheimer’s disease, had a mean count of 188. If Alzheimer’s disease pathology lowers the spine count then patients with plaques and tangles should have a lower count, not higher, than that of patients without plaques or tangles. Even when we include the spine counts of patients 14 and 16, who were not examined for Alzheimer’s disease, with those for whom it was ruled out, the total mean count is 150, significantly less than the control group.
Finally, we repeated the ANOVA for spine counts of CX schizophrenic patients with possible Alzheimer’s disease cases removed (n=5) and controls (n=9), taking out the effect of age and postmortem interval. A one tailed test predicting that spine counts would be significantly different was confirmed.
Discussion
In this pilot study of dendritic spines of cortical pyramidal neurons, we have found a significantly lower spine count in schizophrenic patients than in controls. As a reduction in the number of spines has been reported in the cerebral cortex of elderly subjects,24 we took the effect of age into consideration by comparing the schizophrenic patients as a group with age matched controls, and by comparing matched pairs of schizophrenic patients and controls. Although in the present study the age range of the control subjects was restricted (50 to 78 years), there was significant reduction in spine density with age (fig 3) consistent with previous findings,24 but not in schizophrenic patients. Analysis of variance taking into account age and postmortem interval, separately and together, still found a large difference between schizophrenic patients and controls. We found no evidence that low spine counts in schizophrenia are related to age because spine counts in schizophrenia were low at all ages (fig 1).
A proportion of elderly schizophrenic patients develop a form of dementia25 but only some of these have Alzheimer’s disease pathology and a decreased spine density has also been reported in Alzheimer’s disease.26-28 Although patients had a form of intellectual and cognitive deterioration, none of the CX group were thought clinically to have Alzheimer’s disease. No suggestion of Alzheimer’s disease pathology, such as cortical atrophy or ventricular enlargement, was seen on gross examination of the brains. One brain (No 18) was reported as being heavily gliosed and initially queried for Alzheimer’s disease, but plaques and tangles were not detected using 1E8 and AT8 immunostaining. This brain had temporal cortical spine counts close to the mean for schizophrenic patients and above the mean for the frontal areas.
Overall there was no obvious correlation of Alzheimer’s disease-type histology and spine density. When patients 13, 17, and 19 (with possible Alzheimer’s disease-type pathology) were excluded from the CX schizophrenic group the mean spine count was 106, lower than when they were included. There was insufficient material from the control brains to immunostain for Alzheimer’s disease pathology, but none was reported either clinically or pathologically. Moreover, Alzheimer’s disease pathology in controls would be expected to be associated with decreased spine density, and decrease the difference between schizophrenic patients and controls, thus acting against the hypothesis that spine loss in schizophrenia might be induced by Alzheimer’s disease (or other) pathology. That the difference in spine counts between schizophrenic patients and controls is not an artefact of age, Alzheimer’s disease, or postmortem interval is supported by the ANOVA. This question is being further considered in our current follow up studies of patients assessed in life.
Neuroleptic drugs may have toxic effects in animals,29 and this might lead to loss of dendritic spines30 in some areas but not in others.31 Two of the patients described here (14 and 19) were not on neuroleptic treatment at the time of death but still had low spine counts compared with controls. The possibility that spine loss occurs as a result of neuroleptic treatment will be studied further in our current prospective sample. The effect of an altered social environment must also be considered. In the CX group, the schizophrenic patients had spent long periods in institutions, which was not the case with controls, except for one schizophrenic patient (14) who had relatively late onset schizophrenia and who had been out of hospital for many years. His spine count was close to the mean of the group. The effects of social deprivation in humans are unknown, although rats housed in isolation showed no difference in cortical spine densities compared with rats kept in a social environment.32 Our next cohort includes non-schizophrenic patients from an institutionalised environment to further test this possible confounding factor, and also younger patients without prolonged stays in hospital.
Other factors which could affect spine density in schizophrenic patients include the effects of alcohol.33 There was one alcoholic patient in the CX group (No 14), whose condition was sufficiently severe to have produced clinical encephalopathy and liver cirrhosis, and who died from ruptured oesophageal varices. His spine count (138) was not low in comparison with patients in the schizophrenic group, which were as low as 58. Another alcoholic patient was No 25, who was diagnosed as having a non-schizophrenic psychosis related to alcohol and benzedrene misuse. Her spine count was 238, well into the normal range. This last patient also illustrates that in non-schizophrenic psychosis, spine density was normal, paralleling a similar normal result in bipolar depression.15
Long postmortem interval and long fixation could result in tissue damage and loss of dendritic spines, but we found no relation between postmortem interval (death to fixation) or fixation time and the number of spines, and this variable had no effect on the significance of the difference between schizophrenic patients and controls. Analysis of variance in a subsample of patients confirmed a large and significant difference in spine density between archive schizophrenic patients and controls after taking out the effect of age and postmortem interval. The longest times to fixation were from the archive patients, who had three of the lowest spine counts, but patients 5,7, 8, and 9 were the longest fixed of the control group and all had relatively high counts. Braak and Braak34 have drawn attention to various problems in using the Golgi technique in neuropathology. However, our quantification shows that, although there is considerable variation in spine count between individual brains, when considering a large enough cohort of cases there is no significant correlation with postmortem interval and fixation time, and that neither of these factors could explain the diminished spine count in schizophrenia.
The Golgi technique may only partially impregnate some neurons. This was overcome as far as possible by adhering to strict criteria for selecting and analysing cells and using similar technical procedures in all cases. To avoid the difficulties of counting spines in front of and behind the dendritic shafts, we only counted those in the clear zones of neuropil flanking the shafts. It follows that such counts are underestimates of the true population. However, as a common procedure was used in all brains, including the fact that the observers were blind to the clinical diagnosis until all measurements were completed, the results should be consistent in relation to each other and truly reflect differences between schizophrenic and non-schizophrenic brains. Other difficulties experienced in the counting of spines included detection of two separate but closely spaced spines, spines lying close to the parent dendrite, and the detection of weakly stained spines. These difficulties were minimised by counting at high magnification and following strict criteria across all samples.
The nature of the changes that could lead to a schizophrenia specific spine loss is unknown. The reduced spine density in schizophrenia could be the result of some event at a critical point during development. Cortical spine density increases after birth and then decreases to normal adult levels in the first years of life.35 This pruning of spines in normal development has important implications for pathological loss of spines from pyramidal neurons and could account for a loss of NMDA receptors, which have been reported to be present on spines.17 As pyramidal cells are intercortical association neurons,21 it is also possible that communication between neocortical areas may be disrupted as a result of loss of spines. Spines may be lost as a result of reduced synaptic transmission, suggested by a decrease in the synaptic proteins synapsin and synaptophysin in schizophrenic cortex.36 37 Afferent axon terminals may be lost, and we have seen with electron microscopy “naked” postsynaptic densities on the dendrites in schizophrenic cortex, albeit in a study of a single brain which has, therefore, limited importance.38 We have also found abnormal excitatory synapses with vesicles that clump away from unusually short postsynaptic densities where the glutamate receptor is situated.18 19 38 Our group has reported reduced glutamate receptors in schizophrenic temporal cortex,39 40 and there is reduced expression of glutamate receptor gene in the hippocampus.41 An excessive pruning of prefrontal synapses, perhaps involving excitatory glutamatergic inputs to pyramidal neurons, has been suggested as a possible aetiological factor in schizophrenia.42 Fewer dopaminergic fibres innervate pyramidal cells in the cingulate cortex of schizophrenic patients whereas more innervate GABAergic non-pyramidal cells.43 As spines of pyramidal cells receive dopaminergic synapses as well as glutamatergic ones,44 it is possible that spine loss accounts for at least some reduction of dopaminergic innervation.
Our results are consistent with the concept of a deficit in glutamatergic functions in schizophrenia brought about by a reduction in the number of spines on cortical pyramidal cells. The variability between brains could reflect individual differences in brain structure and the heterogeneity of schizophrenia.23 We are currently studying a new cohort of patients and attempting to remedy some of the deficits of the present pilot study, with more precise matching of cortical areas, more detailed clinical history, and fuller control for Alzheimer’s disease. We shall also relate the spine counts to clinical subtypes, including the prospective neuropsychological data available on these patients. It remains to be determined whether subsyndromes of the disease affect different areas of cortex or are associated with different degrees of change in glutamatergic neurons. We shall also measure and compare other parameters such as dendritic length, branching pattern, and size of neuronal cell bodies. We think that our findings represent the first report of reduced spine density in the neocortex in schizophrenia.
Acknowledgments
This work was supported by grants from the Stanley Foundation and the National University of Singapore. We are indebted to the late Dr CJ Bruton and Drs TJ Crow and MC Royston for providing some early material, and to Dr SM Gentleman for screening for Alzheimer’s disease. One control brain was provided by Drs C Hornstein and M Bauer. We are grateful to Drs A Klugman and W Jamal for help with analysis of clinical notes, Dr K MacRae and Dr B Puri for statistical advice, Drs KA von Bussmann and GZ Mentis for valuable discussion, and SM Ansell, TB Bull, CM Lee, CT Lee, and LB Moran for expert technical assistance.