Article Text

Abstract
OBJECTIVE To characterise the phenotype of a family with paroxysmal exercise induced dystonia (PED) and migraine and establish whether it is linked to the paroxysmal non-kinesigenic dyskinesia (PNKD) locus on chromosome 2q33–35, the familial hemiplegic migraine (FHM) locus on chromosome 19p, or the familial infantile convulsions and paroxysmal choreoathetosis (ICCA syndrome) locus on chromosome 16.
METHODS A family, comprising 30 members, was investigated. Fourteen family members in two generations including three spouses were examined. Haplotypes were reconstructed for all the available family members by typing several microsatellite markers spanning the PNKD, FHM, and ICCA loci. Additionally, the four exons containing the known FHM mutations were sequenced.
RESULTS Of 14 members examined four were definitely affected and one member was affected by history. The transmission pattern in this family was autosomal dominant with reduced penetrance. Mean age of onset in affected members was 12 (range 9–15 years). Male to female ratio was 3:1. Attacks of PED in affected members were predominantly dystonic and lasted between 15 and 30 minutes. They were consistently precipitated by walking but could also occur after other exercise. Generalisation did not occur. Three of the affected members in the family also had migraine without aura. Linkage of the disease to the PNKD, FHM, or ICCA loci was excluded as no common haplotype was shared by all the affected members for each locus. In addition, direct DNA sequential analysis of the FHM gene (CACNL1A4) ruled out all known FHM point mutations.
CONCLUSIONS This family presented with the classic phenotype of PED and is not linked to the PNKD, FHM, or ICCA loci. A new gene, possibly coding for an ion channel, is likely to be the underlying cause of the disease.
- paroxysmal exercise induced dystonia
- migraine
- genetics
Statistics from Altmetric.com
Paroxysmal dyskinesias are a rare group of movement disorders that are currently classified into three different groups: paroxysmal non-kinesigenic dyskinesia (PNKD), paroxysmal kinesigenic dyskinesia (PKD), and paroxysmal exercise induced dyskinesia (PED).1
Paroxysmal non-kinesigenic dyskinesia, formerly termed paroxysmal dystonic choreoathetosis (PDC) was first described by Mount and Reback.2 Attacks are mainly choreoathetotic, can last from 5 minutes to 4 hours, and are usually precipitated by alcohol, fatigue, coffee, tea, or excitement, but not by sudden movement.3Between attacks neurological examination is usually normal. The disease is usually inherited in an autosomal dominant fashion,2 4and sporadic cases are rare.5 It has been linked to a 4 cM area on chromosome 2q33–35,6-9 but the causative gene has not yet been identified.
Paroxysmal kinesigenic choreoathetosis (PKC), now termed PKD1 was first described by Kertesz.10Typically the paroxysms consist of dystonic posturing and choreoathetotic or ballistic movements. All attacks are brief (usually less than 2 minutes) and are precipitated by sudden movements.3 10 11 Frequency may be as high as 100 attacks each day. Patients with PKC usually respond to anticonvulsant drugs, particularly carbamazepine.11 In familial cases, the transmission is usually autosomal dominant, but so far no genes have been linked to the disease. However, an autosomal dominant syndrome characterised by familial infantile convulsions and paroxysmal choreoathetosis (ICCA syndrome) has been linked in four French families and one Chinese family to the pericentromeric region of chromosome 16.12 13 Closer inspection of the limited clinical details suggests that the paroxysmal dyskinesias in these families resemble PKC.
Lance was the first author to draw attention to PED.3 He described a family with three affected members. Subsequently, Plantet al reported another family with this disorder14 and recently a third family was described by Kluge et al. 15 In all only 22 patients, seven from the three families mentioned3 14 15and 15 sporadic cases1 5 16 17 have been described. Attacks are mainly dystonic and in most cases predominantly involve the legs.16 They are precipitated only by exercise and are not brought on by sudden movements. Response to medication is generally poor. No gene has been identified for this condition but it has suggested that PED may represent a type of PNKD which, as mentioned earlier, is known to be linked to chromosome 2q33–35. Recently, a family with an autosomal recessive syndrome characterised by rolandic epilepsy, PED, and writer's cramp has been linked to the same region on chromosome 16 as the families with the ICCA syndrome.18
In this report we describe the clinical features of a new family with PED associated with migraine. To test whether this family is linked to either the PNKD locus on chromosome 2 or to the ICCA locus on chromosome 16 we performed haplotype reconstruction in all the available family members, typing several microsatellite markers spanning these loci. As there was associated migraine we similarly studied the familial hemiplegic migraine (FHM) /episodic ataxia 2 (EA2) locus. Additionally, we sequenced the four exons of the FHM / EA2 gene (exons 4, 16, 17, and 36) that were previously found to contain mutations.19
Material and methods
SUBJECTS
A family comprising 30 members was investigated. Fourteen family members in two generations including three spouses were examined. After giving informed consent, all subjects underwent complete on site examination; a detailed family and personal history including previous illnesses and drug history was obtained. Videotape recordings were made by AM and reviewed by KPB. The diagnosis of dystonia and migraine was made according to published standard criteria.20 21Venous blood samples were taken for DNA analysis from all examined members except III:7.
DNA ANALYSIS
DNA was extracted from peripheral blood leucocytes using standard techniques. To perform haplotype analysis in the family, the following microsatellite markers spanning the critical regions on chromosome 2 (PNKD), 19 (FHM/EA2) and 16 (ICCA) were analysed:
PNKD:cen-D2S164-1.1cM-D2S295- 0.2cM-D2S173-0.5cM-D2S2250-2.1cM-D2S2359-2.1cM-D2S377-tel
FHM/EA2:cen-D19S432-3.9cM-D19S899-0.6cM-D19S1150-0.4cM-D19S199-tel
ICCA:cen-D16S3046-5.6cM-D16S3133-0.9cM-D16S3068-1.9cM-D16S3131-0.9cM-D16S3093-0.5cM-D16S685-3.9cM-D16S298-tel
Map distances are according to the Genetic Location Database at:http://cedar.genetics. soton.ac.uk/public_html/.
For the PNKD locus, the markers D2S164, D2S295, and D2S377 are located outside the linked area, whereas the other four markers span the 4 cM linked region. The gene responsible for FHM/EA2 has been identified and sequenced (calcium channel CACNL1A4). The microsatellite marker D19S1150 is located within the gene, whereas D19S899 and D19S199 are placed at very short distance on either side of the gene. For the ICCA locus, the flanking markers D16S3046 and D16S298 are just outside the 10 cM linked region.
Microsatellite markers were amplified from genomic DNA using the polymerase chain reaction and analysed as previously described.22 Haplotypes were reconstructed in all members using all the typed markers spanning the three regions.
For automated DNA sequencing, three sets of oligonucleotide primers were used to amplify the four exons containing the known FHM mutations (exons 4, 16, 17, and 36). The primers employed were tagged with M13 tails to facilitate subsequent DNA sequencing. The sequences of the primers are as published elsewhere.19
Abnormal clinical features in family members
{kind=link}
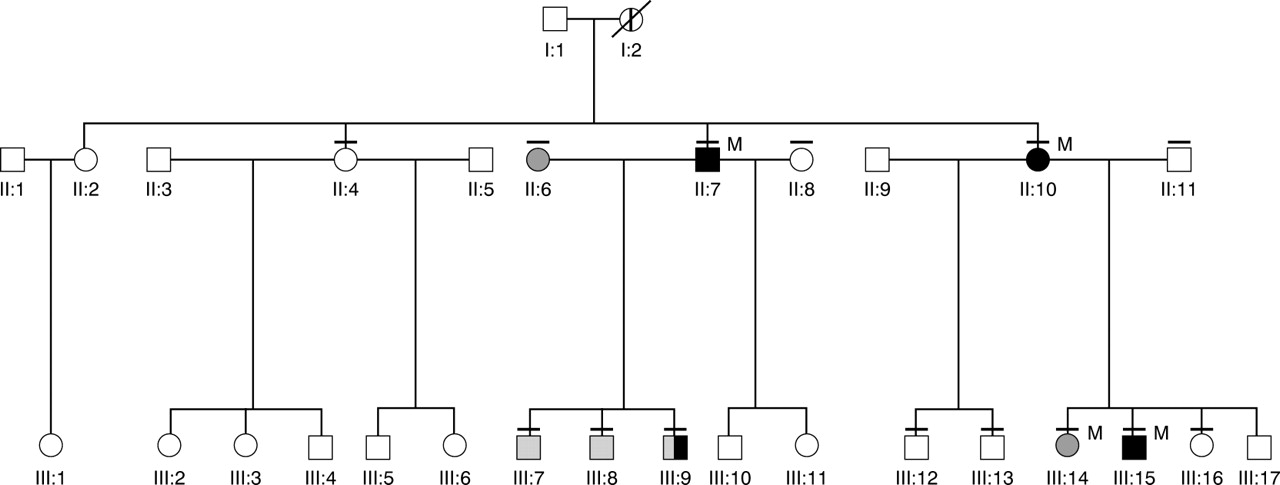
Extended family tree. Black symbols denote those affected by PED; hatched symbols signify those with dystonia or chorea on clinical examination; the member affected by history is shown by a thick vertical bar. She is deceased (indicated by a diagonal bar). A thin horizontal bar above symbols indicates members of the family who were examined clinically. M=Migraine.
Results
CLINICAL FEATURES
Of 14 members examined, four were definitely affected and one member who is deceased was affected by history. Three of the definitely affected members also had migraine. The transmission pattern in this family seemed to be autosomal dominant with reduced penetrance. Of the four definitely affected members, interictal examination was normal in one patient, postural tremor of the arms was evident in two, and dystonic posturing and chorea of the arms in one. The remaining 10 examined members of the family were not affected by PED, but two had dystonic posturing and mild chorea in the arms, one had dystonic posturing and position specific arm tremor, and one had epilepsy. Additionally, one unrelated spouse (II:6) had mild dystonia and tremor in the arms. Table 1 summarises the abnormal clinical findings in the examined family members. Mean age of onset in members affected by PED was 12 (range 9–15 years). Male to female ratio was 3:1. Figure 1 shows the extended pedigree of the family.
CASE REPORTS
Figure 1 shows the family pedigree of members definitely affected with PED.
Patient III:15 (index case)
This 25 year old patient's symptoms started at the age of 12. He develops stiffness and spasms in his leg muscles after walking for 1 hour. His legs tend to “turn in”, his toes “curl in”, and he has to sit down. Spasms in his leg muscles last 10–15 minutes. He has to rest for at least 30 minutes before being able to walk a reasonable distance. If he gets up too soon he is able to walk only for several yards before developing spasms in his legs again.
His hands sometimes “tighten up” when he is using tools for some time—for example, holding a paintbrush. When he is talking for a long time or chewing gum his speech becomes slurred. On one occasion he also developed spasms in his pectoralis muscles after lifting barrels. He has several attacks a week. After an attack he usually feels light headed and exhausted. Other than exertion there are no precipitating events. Before an attack he sometimes gets tingling and mild aching in his thigh muscles.
Sometimes he has cramping in his calf muscles at night. Now and again, usually after some exercise, he has a pounding bifrontal headache, and feels irritable and light headed. This headache lasts only for 15–30 minutes. He tries to go to sleep and usually feels fine after a short nap.
Apart from mild jerky postural tremor in the outstretched arms neurological examination was normal. Nerve conduction studies were normal. Routine blood investigations including CK, lactate, liver enzymes, and renal function were normal. Autoantibodies including ANA, ENA, striated muscle antibodies, and thyroid and antivoltage gated potassium channel antibodies were negative. An ischaemic exercise test showed a normal response. Electromyography and muscle biopsy were normal. Treatment with acetazolamide did not prevent attacks of PED.
Patient II:7
This 41 year old maternal uncle of the index case developed symptoms at the age of 15. Typically after walking for 1 mile (or cycling for several miles) his leg muscles gradually become very stiff and twisted. His feet and legs turn inwards and he becomes unable to walk. He loses control over his legs and has to sit down to prevent falling. He has to rest for at least 10 to 15 minutes before the spasms subside and he is able to walk again but then usually gets another attack after walking for 10–20 yards. To prevent another attack occurring after brief exercise he has to rest for at least 35–40 minutes. Before an attack he has tingling in his thighs. During a typical attack his speech sometimes becomes slurred. He has not noticed any other precipitating events apart from exercise and thinks that neither cold nor heat has an influence on his symptoms.
Frequency of attacks solely depends on physical activity. On average he has three to four attacks a week. On several occasions he developed similar stiffness and cramps in his right hand after writing for a long time. He often has painful muscle spasms between his shoulder blades and in the lower spine without radiation to the legs. These spasms can be brought on by walking, bending, and sometimes by sudden movements.
Intermittently he has double vision with horizontal separation of images. This can happen at any time and is not provoked by exercise or fatigue. Once or twice a week he has a right sided severe pulsating headache that usually lasts for 6 hours and is accompanied by sensitivity to light. He thinks that when having these headaches he is more likely to get attacks of muscle stiffness. As a child he was prone to develop painful muscle spasms in his calf muscles at night. At the age of 7 there was one episode, lasting 35 minutes, when he woke up in the morning and could not get up, as his legs were floppy. He has never tried any medication.
On neurological examination there was a mild, jerky postural tremor in the arms. When asked to imitate a typical attack, the posture of his legs and arms seemed dystonic.
Patient II:10
This 44 year old woman is the mother of the index case. Her symptoms started at the age of 11. After walking for more than half a mile her legs start feeling like jelly before beginning to twist involuntarily. She then becomes unable to walk and has to sit down or else she would fall. Cramps last for up to 30 minutes. She has to rest for at least 30 minutes before she is able to walk again without immediately getting another attack. Usually she feels very exhausted after an attack. She also occasionally gets muscle spasms in her hands after carrying a heavy grocery bag or in her right hand after writing for a while. She also has cramps in her abdominal muscles after exercise. When chewing gum for some time her tongue starts to feel heavy and her speech becomes slurred. During pregnancy or during her periods she did not notice any change in the attacks. She tried phenobarbital without success.
She now avoids any excessive physical exertion and rarely has an attack but used to have attacks two to three times a week. Once a week she has left sided pounding headaches that usually start in the morning and last for a few hours, during which she is sensitive to light and noise. Neurological examination was normal.
Patient III:9
This 12 year old boy is a maternal cousin of the index case. At the age of 9 he had his first episode of involuntary movements in his legs after exercise. After walking for several hours he suddenly lost control over his legs, which started to “jerk”. His whole body was then swaying, he lost his balance and was forced to sit down. He had to rest for 30 minutes before he was able to walk normally. So far he has had three attacks. The last attack was brought on after playing for about 15 minutes. For several days before this event he had been very inactive. He also gets painful spasms in his right hand after writing for an hour. Neither he nor his mother had noticed precipitating events other than exercise. On neurological examination he had mild dystonic posturing and chorea in his arms.
Family member affected by history: patient I:2
This is the maternal grandmother of the index case, who died at the age of 71. She had developed spasms in her legs after walking for a mile at the age of 12. She mentioned to her daughter (mother of the index case) that the severity of her attacks improved as she got older. No other details are known.
Family members affected with dystonia, chorea or tremor: patient II:6
This is the 33 year old ex-wife of II:7. She mentioned having tremor in both arms since the age of 17. On examination there was bilateral dystonic posturing of her arms and hands and mild to moderate irregular postural and kinetic tremor in her hands that was more marked on the left.
Patient III:7
This 7 year old maternal cousin of the proband did not report any symptoms. However, on examination he had dystonic posturing and chorea in his arms.
Patient III:8
This 14 year old maternal cousin of the index case did not report any symptoms. However, on examination he had definite dystonic posturing of both arms. There was also mild chorea of the outstretched arms.
Patient III: 14
This is the 19 year old sister of the index patient. She did not mention involuntary movements. She complained of occasional left sided headache of a rather dull character. This headache usually lasts for several hours and responds well to paracetamol. On examination she had a dystonic posture of both arms when holding out the arms and position specific tremor of the left arm on supination.
Family member affected with epilepsy: patient II: 4
This maternal aunt of the index case started having generalised tonic-clonic seizures at the age of 18. These were successfully treated with phenytoin. Several years later she was also diagnosed as having frontal lobe seizures with attacks of bizarre behaviour, confusion, and hallucinations. Treatment with sodium valproate resulted in some improvement. She also mentioned intermittent episodes of muscle spasms starting in one arm or one leg with spread to other body regions. These attacks could happen at any time at rest or after brief exercise. She also complained of painful cramps in her hands after writing for a long time. There was no clear description of dystonia during these attacks. Neurological examination was normal. She was not considered to be affected with PED.
DNA ANALYSIS
The haplotype reconstruction for PNKD, FHM/EA2, and ICCA areas failed to show a common haplotype shared by all four definitely affected members, hence allowing us to exclude linkage between the disease and all the tested loci.
Direct DNA sequencing of the exons 4, 16, 17, and 36 of the FHM/EA2 gene did not disclose the previously described mutations R192Q, T666M, V714A, and I1811L, nor any other mutation.
Discussion
Attacks in all affected family members fulfil the criteria of PED as outlined in earlier reports.1 3 16
The clinical presentation of the affected members was remarkably stereotyped. The age of onset varied little (9–15 years), the duration of an attack was always between 15 and 30 minutes and attacks were consistently precipitated by walking. All members had attacks involving the legs, which were mainly dystonic. However, depending on the exercised part attacks could also occur in other body regions—for example, in the hand after writing for a long time in all affected members, in the jaw after chewing (patients II:10 and III:15), or in the pectoralis muscles after heavy lifting (patient III:15). Generalisation did not occur. Speech was slurred during an attack in three patients and these patients also reported prodromi, mainly sensory symptoms in their legs. Between attacks two members had a postural tremor in the arms and one had dystonic posturing and chorea in the arms, a feature that has also been mentioned by Nardocciet al.5 Interestingly, three other family members unaffected by PED also had mild dystonia in their arms (two also had mild chorea and one had tremor in the arms). It is possible that the two maternal cousins of the index patient (patients III:7 and III:8) might have inherited their symptoms from their mother (patient II:6 and spouse of II:7) who also had arm dystonia and it is therefore unclear whether an association with PED can be made.
Each of the families with PED reported by Lance,3 Plantet al, 14 and Klugeet al 15 also presented a characteristic phenotype. In the three family members reported by Lance,3 spasms and cramps were almost exclusively confined to the legs, lasted for 5–30 minutes, and could be precipitated by walking or other strenuous exercise. Attacks in the family described by Plant et al 14 involved legs, arm, and trunk and, apart from exercise, could also be induced by passive movements and vibration. In the family reported by Klugeet al 15 the attacks, which usually involved the legs and sometimes the arms after prolonged manual work, sometimes became generalised.
In a review of PED, Bhatia et al 16 pointed out that in sporadic cases attacks were also mostly dystonic and most commonly involved the legs.
In addition to paroxysmal dyskinesias, three of the affected members in our family also had migraine without aura. The association of PED and migraine has only been reported once before, in a girl also affected with learning impairment, ataxia, and epilepsy.16 23 The occurrence of different paroxysmal disorders in the same family is intriguing, and raises the question of a similar underlying cause. As other paroxysmal neurological disorders including FHM and episodic ataxia with interictal myokymia (EA type 1) are known to be caused by mutations in ion channels, it seems likely that the defect underlying PED is also a channelopathy. However, our family was not linked to the FHM locus.
Recently, two novel syndromes characterised by PED plus other paroxysmal disorders have been linked to a 10-cM region on chromosome 16.12 13 18 One of them, named the ICCA syndrome, is inherited as an autosomal dominant condition and is associated with benign infantile convulsions.12 13 Closer inspection of the clinical features mentioned by Lee et al 13 suggests that patients with the ICCA syndrome had brief frequent attacks which were either induced by sudden movement or exertion resembling PKD or PED but not PNKD (PDC).
The other syndrome is characterised by the association of PED, rolandic epilepsy, and writer's cramp.18 It is an autosomal recessive condition and interestingly is also linked to the same region on chromosome 16 as in the family with the ICCA syndrome. However, the haplotype reconstruction with markers spanning this region showed that the affected members in our family did not share a common haplotype, thus excluding linkage of the disease with this novel locus.
Although clinically the family under discussion differed from PNKD, historically it has been suggested that PED may be a type of PNKD.3 Indeed, there have been reports of families with features of both PNKD and PED. In the family with PNKD reported by Schloesser et al 24 two affected female members described exercise as a precipitant of attacks that were longer than usual attacks of PED and more typical of PNKD. Prolonged exertion induced PNKD attacks in three of five family members with PNKD reported by Kurlan et al.25 We therefore looked for linkage to the PNKD locus on chromosome 2. Haplotype reconstruction allowed us to exclude linkage of the disease with this locus, thereby suggesting that typical PED is not a type of PNKD linked to chromosome 2.
In conclusion, we have reported a new family with PED in which three of four affected members also had migraine. Linkage to the PNKD locus on chromosome 2, to the FHM/EA2 locus on chromosome 19p and the ICCA locus on chromosome 16 was excluded. The number of affected members in the family was too small for linkage analysis to identify the gene locus. A mutation in an as yet unidentified ion channel gene could be the underlying cause of the paroxysmal attacks found in this family and further work is needed to characterise this disorder genetically.
Acknowledgments
AM was supported by the Ernst Jung-Stiftung für Wissenschaft und Forschung in Hamburg, Germany and the Eugene Brehm Bequest, United Kingdom. EMV was supported by Telethon, grant No E499, and LHE by the Brain Research Trust and the Epilepsy Research Foundation.