


Abstract
In rat dorsal root ganglion neurons, activation of κ- and μ-opioid receptors decreases N-type calcium current, whereas a constitutively active form of protein kinase C (PKC; i.e., PKM, a PKC catalytic subunit fragment) increases N-type calcium current. PKC also attenuates inhibition of calcium current by several G protein-linked neurotransmitter systems. We examined the effects of activation of endogenous PKC by 4β-phorbol 12-myristate 13-acetate (PMA) and dialysis of cells with PKM and a pseudosubstrate inhibitor PKC(19–31) (PKC-I) on κ- and μ-opioid-mediated inhibition of calcium current, calcium current amplitude, and rundown. PMA modestly increased peak calcium current and substantially reduced calcium current “rundown,” effects blocked by PKC-I. In contrast, PKC-I decreased calcium current and increased current rundown. PMA attenuated morphine-, dynorphin A-, and U50,488- but not pentobarbitol-related inhibition of calcium current. Similar effects were seen with intracellular dialysis of PKM. Intracellular PKC-I did not block opioid inhibition of calcium current but did reverse PMA and PKM effects on opioid receptor coupling to calcium channels. Because neither PMA nor PKM changed the proportion of ω-CgTX-inhibited current, their effects were not due to a decrease in the proportion of N-type current. After ω-CgTX treatment, there were no differences in the dynorphin A effects on control and PMA- or PKM-treated neurons, suggesting that PKC primarily affected coupling to N-type calcium channels. These data suggest that in acutely dissociated rat dorsal root ganglion neurons, endogenous PKC is required for maintenance of calcium current, may play a role in regulation of neuronal calcium channels, and could be involved in tolerance and/or cross-talk inhibition of opioid responsiveness.
κ- and μ-opioid agonist inhibition of calcium currents (Macdonald and Werz, 1986; Moises et al., 1994a; Tsunoo et al., 1986; Werz and Macdonald, 1984) is thought to mediate opioid reduction of calcium-dependent neurotransmitter release from presynaptic terminals (Werz and Macdonald, 1984). κ- and μ-agonists selectively inhibited the same high threshold calcium currents in rat dorsal root ganglion (DRG) neurons (Moises et al., 1994b; Wiley et al., 1997). Although N-type calcium current accounted for ∼75% of the total opioid-sensitive calcium current, both κ- and μ-agonists also inhibited P- and Q-type calcium currents (Moises et al., 1994a; Wiley et al., 1997). Opioid inhibition of calcium current was mediated specifically by coupling to the G protein α-subunit Gαo (Gross et al., 1990a; Moises et al., 1994b;Wiley et al., 1997). Activation of a number of other inhibitory G protein-linked neurotransmitter receptors, including muscarinic, adrenergic, somatostatin, and γ-aminobutyric acidB receptors, also inhibited calcium channels in a pertussis toxin-sensitive manner (Swartz, 1993). Although receptors demonstrate specificity for G protein α-subunits, the actual inhibition of calcium current may be mediated via the G protein βγ-subunits (Herlitze et al., 1996; Ikeda, 1996), possibly by inhibiting calcium current through a direct interaction with the α1 subunit of the calcium channel (De Waard et al., 1997; Zamponi et al., 1997).
Protein kinase C (PKC) has been implicated in several in vivo studies of opioid action. Intrathecal administration of the phorbol ester 4β-phorbol 12-myristate 13-acetate (PMA) blocked opioid-induced antinociception in rats (Zhang et al., 1990; Narita et al., 1997). Intrathecal administration of PKC inhibitors did not block opioid antinociception but did block acute opioid tolerance (Narita et al., 1995). Furthermore, chronic i.p. morphine increased PKC activity in the pons/medulla in rats (Narita et al., 1994).
Although in vivo tolerance is often receptor specific (i.e., homologous), heterologous (i.e., cross-receptor) tolerance has also been observed in opioid coupling to calcium channels (Kaneko et al., 1997). δ-Opioid receptors recently also have been shown to be heterologously desensitized byN-methyl-d-asparate in a PKC-dependent manner (Fan et al., 1998). Although PKC does not appear to be involved in agonist-induced receptor phosphorylation (Pei et al., 1995) and thus may not be directly involved in homologous desensitization, it may be important in mediating cross-tolerance between different opioid receptors and/or other neurotransmitters such asN-methyl-d-aspartate.
Activation of PKC with phorbol esters reduced calcium current inhibition by inhibitory G protein-linked neurotransmitter receptor agonists in several rat neuronal types (Swartz, 1993; Wilding et al., 1995; Zhu and Ikeda, 1994), but PKC did not appear to be involved in the actual signaling pathway leading to inhibition of calcium current induced by these agonists (Swartz, 1993). Because multiple neurotransmitter systems can activate PKC, PKC attenuation of κ- and/or μ-opioid receptor coupling to calcium channels could be a potential mechanism for the observed PKC effects on systemic opioid action. However, although PKC has been shown to attenuate inhibition of calcium currents by other Gi-linked neurotransmitter, it is not known whether activation of PKC can block opioid-induced inhibition of calcium currents. To investigate this possibility, we examined the effects of PKC on μ-opioid (morphine) and κ-opioid (dynorphin A and U50,488) inhibition of calcium channel currents in rat DRG neurons.
Materials and Methods
Preparation of Acutely Dissociated Neurons.
DRG neurons were prepared from Sprague-Dawley rats, 14 to 50 days of age, of both sexes using a technique similar to that described previously (Gross et al., 1990b). Briefly, after removal from the spinal column, thoracic DRG neurons were minced, incubated with collagenase and trypsin, and then triturated to separate the cells. Cells were then plated onto uncoated 35-mm culture plates or plates coated with rat-tail collagen in minimal essential media supplemented with 16.5 mM NaHCO3, 28.2 mM glucose, and 10% fetal calf serum. Cultures were incubated at 37°C in 95% air, 5% CO2. Recordings were made at room temperature, typically within 10 h of plating.
Drugs and Enzymes.
Constitutively active PKC catalytic subunit (PKM) purified from bovine brain (Woodgett and Hunter, 1987) was a kind gift of Dr. Michael Browning (University of Colorado, Denver, CO). Protein kinase C inhibitory peptide (PKC-I), PKC19–31, and dynorphin A were obtained from RBI (Natick, MA). Fetal calf serum was from Life Technologies, Inc. (Grand Island, NY). Morphine, U50,488, PMA, nifedipine, ω-conotoxin fromConus geographus (GVIA), collagenase, trypsin, and all other chemicals were from Sigma Chemical Co. (St. Louis, MO).
Solutions and Drug Application.
Stock solutions of PKM (1 μM) and PKC-I (4 mM) were stored at −80°C. Just before use, solutions of PKM, PKC-I, and/or vehicle were prepared. PKM and PKC-I were diluted into internal pipette solution (see below) at 40 nM or 4 μM, respectively, and kept on ice until use. To prepare a solution of both PKM and PKC-I, the stock solution of PKC-I was diluted to 4 μM into the working solution of PKM, and the mixture was incubated at 37°C for 15 min to inactivate PKM. When using solutions containing PKM or PKC-I, the recording pipette tip was filled with internal recording solution, and then back-filled with the peptide-containing solution. Stock solutions of all other drugs were diluted into external solution on the day of use. Nifedipine was dissolved in dimethyl sulfoxide (DMSO) (10 mM) on the day used (final DMSO concentration was less than the 1:1000). Stock solutions of dynorphin A and ω-conotoxin GVIA (1 mM) were dissolved in filtered distilled water, lyophilized in aliquots, and stored at −20°C until the day used. Stock solutions of morphine were prepared in distilled water (10 mM) and of PMA in DMSO (100 μM) and were stored at −20°C until used. Opioids were applied to cells using a modified U-tube application system (Greenfield and Macdonald, 1996) using a solenoid-controlled 10-s application of drug, followed by vacuum reuptake. PMA and ω-conotoxin GVIA were applied by pressure ejection from a blunt-tipped (20–40-μm opening) pipette positioned ∼25–50 μm from the neuron.
Whole-Cell Patch Clamp Recording Techniques.
Voltage-clamp recordings were made using the whole-cell variant of the patch-clamp recording technique (Hamill et al., 1981) using an Axopatch 1-B amplifier (Axon Instruments, Foster City, CA), and glass recording pipettes, micropipette tip resistances of 1–2 megaohms, and seal resistances of greater than 1 gigaohm. Patch clamp micropipettes were pulled from Labcraft micro-hematocrit capillary tubes (Curtin Matheson Scientific, Inc., Houston, TX) using a P-87 Flaming-Brown micropipette puller (Sutter Instrument Co., San Rafael, CA). Signals were low pass filtered at 2 kHz using an 8-pole Bessel filter then digitized, recorded, and analyzed using pCLAMP6 software (Axon Instruments).
Cells were removed from the 5% CO2 incubator, and the medium was replaced with external recording medium (67 mM choline chloride, 100 mM tetraetylammonium chloride, 0.8 mM MgCl2, 5.3 mM KCl, 5.6 mM glucose, 10 mM HEPES, and either 5 mM BaCl2 or CaCl2, 325–330 mOsM) with a pH of 7.35. Patch clamp micropipettes of 3–10 megaohms were filled with internal solution (140 mM CsCl, 5.3 mM KCl, 1 mM MgCl2, 10 mM HEPES, 10 mM EGTA, 5 mM MgATP, and 0.1 mM LiGTP). The pH was adjusted to 7.3 with CsOH after addition of ATP and GTP, and the final osmolality was 300–310 mOsM, 10–15% lower than the external solution.
Data Analysis.
Leak current was estimated as the inverse of the current generated by hyperpolarizing commands of equal value to those used to depolarize the neurons. These were digitally subtracted from total currents to give leak-subtracted barium or calcium currents. Statistical comparisons of the effects of drugs, peptides, and PKM on peak current and on current rundown were performed using a two-tailed Student’s t test. Comparisons between dynorphin A inhibition of calcium currents before and after treatment with drugs in the same cell were analyzed using a paired-sample t test.
Results
PMA Increased Calcium Channel Current.
Effects of phorbol esters and PKM on calcium channels and κ-opioid signaling were studied using a tight-seal, whole-cell voltage clamp protocol on the somata of acutely dissociated rat DRG neurons. Barium was used as a charge carrier through calcium channels, and internal calcium was buffered with EGTA. Currents were elicited by voltage steps to 0 mV from a holding potential of −80 mV. Under these conditions, primarily transient high-threshold, voltage-dependent calcium channel currents were activated (Moises et al., 1994a). External application of 250 nM PMA to the neurons increased total barium current, with maximal increase within 3 min (Fig. 1, A and B). In contrast, after treatment of neurons with vehicle (1:1000 DMSO) or 250 nM 4α-phorbol 12,13-didecanoate (4α-PdBu), which does not activate PKC, barium current decreased or “ran down” slightly (Fig.1, A and B). The average maximal increase in current was to 116 ± 4% of current before application of PMA (Fig. 1C). The actual increase in barium current induced by PMA appeared to be somewhat larger than this, because currents in both vehicle- and 4α-PdBu-treated neurons ran down about 10% within 3 min (Fig. 1C). The largest PMA-induced increase recorded was to 136% of control current, and increases in current were seen in 8 of 11 neurons tested. Thus, the PMA-induced increase in calcium current reported here was consistent with effects reported previously of PMA in DRG neurons (Swartz, 1993), although the magnitude of the increase was smaller than that reported in other rat neuronal types such as cerebral cortical and superior cervical ganglion neurons (Swartz, 1993). The observed effect of PMA was also smaller than that produced by internal application of PKM, which we have previously shown maximally increased peak calcium current to >200% of control in DRG neurons (Hall et al., 1995).

PMA increased current through calcium channels in rat DRG neurons. Calcium channel currents were evoked with a 200-ms pulse to 0 mV from a holding potential of −80 mV with barium as the charge carrier. Currents from three separate neurons treated with either PMA (▪), 4α-PdBu (▴), or vehicle (1:1000 DMSO, ■) are shown in A as peak currents plotted as a function of time after patch rupture and in B as superimposed traces at 0–4 min after treatment. Peak barium current amplitude was measured at 15 ms after pulse. In C, relative change in peak amplitude was plotted as a function of time after treatment with PMA (▪), 4α-PdBu (▴), or vehicle (1:1000 DMSO, ■). Points represent means ± S.E.M. Change in peak amplitude was calculated by dividing the (-Ibarium) at each point by the current before treatment (-Ibariumt = 0).
PMA Decreased and PKC-I Increased Rate of Calcium Channel Current Rundown.
We reported previously that calcium currents elicited from DRG neurons routinely ran down over time; currents reached a peak at 2–5 min and then progressively decreased for the remainder of the recording (Hall et al., 1995). This phenomenon appeared to be due to dialysis out of the neuron of cellular components required for maintaining functional calcium channels. As rundown was slowed by inclusion of ATP in the recording pipette (Hall and Macdonald, unpublished data), it appeared likely that a loss of protein kinase activity was involved. All data reported here were obtained from recordings with 5 mM ATP in the pipette. In control neurons, current declined to 50% of peak value within ∼20 min of patch rupture and was usually <10% of peak within 40 min of patch rupture (Fig.2, ■). We have shown previously that intracellular PKM increased peak current without affecting rundown. In contrast, while external PMA only modestly increased “peak” calcium current, it substantially decreased the rate of current rundown. Current from neurons treated with external PMA ran down at a slower rate than from control neurons (Fig. 2, ▪), typically with currents not reduced to 50% of peak until 30–40 min after patch rupture. In some neurons treated with PMA, currents as large as 50% of peak were recorded as long as 70 min after patch rupture. This suggested potential differences in effects of activation of endogenous PKC by PMA and introduction of a constitutively activated kinase into the neuron.

PMA decreased and PKC-I increased the rate of calcium channel current rundown. Barium currents were evoked by a 200-ms pulse to 0 mV from a Vh of −80 mV with barium as a charge carrier in the presence of 3 μM nifedipine in control (○,n = 6), PMA-treated (●, n = 5), PKC-I-treated (■, n = 5), and PKC-I and PMA-treated (▪, n = 5) DRG neurons. Rates of current rundown were compared by dividing inward currents at each time point (-Ibarium) by the maximum inward current (-Ibariummax) attained during the experiment. The rundown period was defined as the period following maximum current. PMA treatment caused a significant decrease in the rate of rundown in control and PKC-I treated neurons, whereas PKC-I increased the rate of rundown in control neurons. (Data were analyzed using a two-tailed Student’s t test. Values were shown as mean ± S.E.M.
The effect of PMA to protect the currents from rundown was abolished by intracellular application of PKC-I (Fig. 2, ●). As we have reported previously, intracellular application of PKC-I alone increased the rate of current rundown (Fig. 2, ○). Currents from neurons treated with PKC-I ran down faster than those from control cells in the absence of PMA (50% reduction in 9–10 min). These data suggested a role for endogenous PKC in maintaining functional calcium channels in acutely dissociated DRG neurons under whole-cell patch clamp conditions. Rundown of calcium channel current was also observed when 5 mM calcium was used as the charge carrier, and application of PMA slowed rundown to a similar extent (data not shown). PMA also had a similar effect on rundown of peak calcium currents recorded from neurons in the presence of external solution containing 150 mM sodium chloride (data not shown).
PMA Attenuated Dynorphin A Inhibition of Calcium Channel Currents.
Morphine and Dynorphin A, through interaction with μ- and κ-opioid receptors, respectively, inhibit calcium channel currents through a rapid, reversible, voltage-dependent, pertussis toxin-sensitive mechanism (Taussig et al., 1992; Moises et al., 1994b). Acutely dissociated DRG neurons are a heterogeneous preparation, with populations of dynorphin- and/or morphine-sensitive neurons, and furthermore, dynorphin A can have effects through μ- as well as κ-opioid receptors. However, populations of dynorphin-sensitive and morphine-insensitive DRG neurons have been observed (Moises et al., 1994a); and studies using μ-specific H-d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2(CTAP), β-funaltrexamine) and κ-specific (norbinaltorphimine) antagonists also indicated that the effects of dynorphin A and morphine on calcium currents at the concentrations used in this study were due primarily to interactions with κ-opioid and μ-opioid receptors, respectively (Moises et al., 1994a; Wiley et al., 1997; King and Macdonald, unpublished observation). κ-Opioid receptor inhibition of calcium channel currents was probably mediated through pathways similar or identical with those of μ-opioid receptors, which have been shown to be rapid and membrane delimited (Wilding et al., 1995).
κ-Opioid agonists inhibited voltage-activated calcium currents by 20–40% in DRG neurons and produced delayed activation kinetics (Moises et al., 1994a). In the experiments reported here, dynorphin A reduced calcium current an average of 26 ± 4% in the presence of the L-type calcium channel blocker nifedipine. Because nifedipine has been demonstrated not to decrease opioid-sensitive currents (Moises et al., 1994a), 3 μM nifedipine was routinely added to increase the relative proportion of opioid-sensitive current in the neuron tested. Dynorphin A inhibition of total barium current in rat DRG neurons was attenuated by extracellular application of 250 nM PMA (Fig.3). In the neuron shown, an initial application of 3 μM dynorphin A for 10 s rapidly and reversibly reduced total peak barium current from 1.9 to 1.4 nA (26% inhibition) (Fig. 3A). This inhibition of the barium current was associated with two distinct kinetic components, slowing of the activation rate and inhibition of steady-state current. After the current completely recovered from the effect of dynorphin A, 250 nM PMA was applied to this neuron, and after 3 min of treatment with PMA, 3 μM dynorphin A was applied again for 10 s. This application of dynorphin A decreased the current from 1.9 to 1.8 nA, i.e., a 5% inhibition, and the attenuated dynorphin A inhibition in the presence of PMA did not result in a slowing of the activation rate (Fig. 3A). PMA reduced the inhibition of subsequent applications of dynorphin A in the same neuron an average of 80% (p < .005, Fig. 3C). This effect of PMA was not due to a tachyphalaxis resulting from multiple applications of dynorphin A, because multiple applications of dynorphin A after 1 min or more recovery in the absence of PMA showed <10% reduction in efficacy (data not shown). This effect was also not due to effects of time or rundown, because the percentage of inhibition of current by dynorphin A in control neurons (not treated with PMA) was not significantly different at 2–4 min and 10–15 min after seal rupture (data not shown).

PMA attenuated μ- and κ-opioid inhibition of calcium channel current. In A, barium currents evoked by a 200-ms pulse to 0 mV from a Vh of −80 mV were shown as peak current plotted as a function of time after patch rupture and as superimposed traces before and immediately after 10-s applications of 3 μM dynorphin A (arrows marked by Dyn) and 250 nM PMA (bar). B, inhibition (shown as percentage of change in amplitude from currents immediately before application of agonists) of barium current by 3 μM dynorphin A (Dyn, n = 11), 3 μM U50,488 (U50,n = 4), 3 μM morphine (Morph,n = 8), and 100 μM pentobarbital (PentoB,n = 4) before (light column) and 3 min after (dark column) treatment of neurons with 250 nM PMA.
As has been shown previously (Moises et al., 1994a), 1 μM morphine and U50,488 also inhibited barium current (Fig. 3B) and exhibited apparent slowing of activation (data not shown). Similar to its effects on dynorphin, PMA attenuated the inhibitory effects of morphine and U50,488 by 80% and 78%, respectively (Fig. 3B). In contrast, 100 μM pentobarbitol, which appears to act as an open-channel blocker of calcium channels (Gross and Macdonald, 1988), inhibited barium current by 35% and did not slow apparent activation rate (data not shown). PMA had no effect of pentobarbital inhibition of barium currents (Fig. 3B).
To verify that the PMA-induced attenuation of dynorphin A action was due to activation of endogenous PKC, PKC-I peptide (4 μM) was included in the recording pipette to inhibit cellular PKC. This technique has been shown to block various cellular effects of PMA, including attenuation of norepinephrine and baclofen inhibition of calcium channel currents in rat neurons (Swartz, 1993). Neurons were treated with intracellular PKC-I and then tested for dynorphin A inhibition of barium currents before and after external application of 250 nM PMA (Fig. 4A). The initial application of dynorphin A reduced barium current by 21% (Fig. 4A), which indicated that activation of PKC was not required for coupling of κ-opioid receptors to calcium channels. After a 1-min recovery, the neuron was treated with 250 nM PMA, which did not appear to slow current rundown. After 2 min of PMA treatment, 3 μM dynorphin A was reapplied to the neuron, which led to a reversible 18% inhibition of barium current (Fig. 4A). Four neurons treated with intracellular PKC-I and tested in this manner showed no significant difference in dynorphin A inhibition before or after PMA treatment (Fig. 4B).

PKC-I blocks PMA attenuation of dynorphin A inhibition of calcium channel current. In A, barium currents evoked by a 200-ms pulse to 0 mV from a Vh of −80 mV from neurons dialyzed with 4 μM PKC-I in the recording pipette were shown as peak current plotted as a function of time after patch rupture and as superimposed traces before and immediately after 10-s applications of 3 μM dynorphin A (arrows marked by Dyn) and 250 nM PMA (bar). B, inhibition of barium current (shown as percentage of change in amplitude from currents immediately before application of dynorphin) in neurons dialyzed with 4 μM PKC-I by 3 μM dynorphin A before (light column) and 3 min after (dark column) treatment of neurons with 250 nM PMA. (n = 4, NS.)
Effect of Blockade of N-Type Current on Reduction by PMA of Calcium Channel Current Inhibition by Dynorphin A.
DRG neurons contain multiple high-threshold calcium current subtypes, including N-, L-, Q-, P- and R-types (Moises et al., 1994a; Wiley et al., 1997). N-type current accounted for about 75% of the total dynorphin A-sensitive current in rat DRG neurons, although dynorphin A also inhibited a portion of the Q-, P-, and R-current (Wiley et al., 1997). To gain information as to the specific calcium channels being affected by PMA, we examined the effect of the N-channel blocker ω-conotoxin GVIA on the ability of PMA to attenuate dynorphin A inhibition of calcium currents (Fig. 5). Dynorphin A (3 μM) was applied to DRG neurons to determine the total dynorphin A-sensitive current (Fig. 4A, traces 1 and 2). Dynorphin A reduced the current from 2.7 to 2.2 nA (21%). After recovery, the neuron was treated with 250 nM PMA for 2 min, and dynorphin A was reapplied, which reversibly inhibited current from 2.5 to 2.4 nA (a 7% decrease) (Fig.5A, traces 3 and 4). After recovery for 1 min, N-channels were blocked with ω-conotoxin GVIA, which reduced the basal current by 52% from 2.5 to 1.2 nA, and then dynorphin A was reapplied. Dynorphin A only reduced the current from 1.2 to 1.1 nA (Fig. 5A, traces 5 and 6), a decrease which represented 5% of the original current and a 9% inhibition of the residual current. A similar protocol was followed to determine the levels of dynorphin A- and ω-conotoxin GVIA-sensitive currents in neurons not treated with PMA. In the absence of conotoxin, dynorphin A inhibited an average of 22 ± 2% of the current before and 5 ± 2% of the current after PMA treatment (n = 11); after ω-conotoxin GVIA treatment, dynorphin A inhibited an average of 6 ± 1% of the current in control (n = 4) and 6 ± 2% of the current in PMA treated (n = 4) neurons (Fig. 5B). Although the effects of dynorphin A on the residual calcium currents after PMA were small, there appeared to be no difference in dynorphin sensitivity in control and PMA-treated neurons after ω-conotoxin GVIA treatment (Fig. 5B). ω-Conotoxin GVIA also did not appear to further decrease the dynorphin A-sensitive current when applied after PMA (Fig. 5B, compare striped columns). Furthermore, when PMA was applied after ω-conotoxin GVIA, the small residual dynorphin-sensitive current was not further reduced (data not shown).

Effect of PMA and ω-conotoxin GVIA on dynorphin A inhibition of calcium channel currents. In A, barium currents evoked by a 200-ms pulse to 0 mV from a Vh of −80 mV in the presence of 3 μM nifedipine were shown as peak current plotted as a function of time after patch rupture and as superimposed traces at times indicated by arrows. Application of dynorphin A (3 μM) for 10 s reversibly reduced total high-threshold barium current. Application of PMA (250 nM) for 5 min had little effect on total barium current but substantially reduced the fraction of current inhibited by dynorphin A. Application of ω-conotoxin GVIA (3 μM) for 30 s irreversibly reduced total whole-cell barium current but had little effect on the fraction of current inhibited by dynorphin A. B, dynorphin A inhibition (shown as percentage of change) of barium current in control (n = 4) and PMA-treated (n = 4) neurons before and after treatment with ω-conotoxin GVIA (3 μM). PMA treatment significantly (p < .01) decreased dynorphin A inhibition of barium current. There was no difference in dynorphin A inhibition in control and PMA neurons after treatment with ω-GVIA, and ω-GVIA did not further reduce the fraction of current inhibited by dynorphin A in PMA-treated neurons.
These data suggested that PMA did not decrease the efficacy of dynorphin A to inhibit the residual conotoxin-insensitive components (e.g., P-, Q-, and R-types) of barium current. These data suggested that PMA primarily affected κ-opioid receptor coupling to N-type calcium channels. The observed PMA-induced decrease in dynorphin A inhibition of barium current was not due to a decrease in the proportion of N-type current present. There was no difference in ω-conotoxin GVIA inhibition of barium current in control neurons and neurons treated with 250 nM PMA (data not shown).
PKM Attenuated Dynorphin A Inhibition of Calcium Channel Current.
In addition to activating endogenous PKC, phorbol esters could be accompanied by other “nonspecific” effects on calcium channels, receptors, or other cellular proteins. For example, phorbol esters are known to alter the cycling and down-regulation rate of multiple receptors and ion channels and have been shown to recruit covert or previously inactive calcium channels in Aplysia neurons (Strong et al., 1987). Phorbol esters activate multiple isoforms of PKC, which may be differentially regulated in normal cells, and there is evidence that a phorbol ester-insensitive PKC isoform was involved in norepinephrine regulation of calcium channels in chick DRG neurons (Boehm et al., 1996). Furthermore, there have been conflicting reports of the effects of phorbol esters and other activators of PKC to either increase (Swartz, 1993; Zhu and Ikeda, 1994), decrease (Diverse-Pierluissi and Dunlap, 1993; Werz and Macdonald, 1987), or have no effect (Boehm et al., 1996) on calcium channel currents. Because of these concerns, we examined the effect of intracellular application of purified, constitutively active PKC, PKM, on dynorphin A inhibition of calcium channels. As demonstrated previously with DRG neurons (Hall et al., 1995), intracellular application of 20 nM PKM increased peak calcium currents compared with those from control neurons (3.4 ± 0.4 nA PKM treated, n = 6versus 2.5 ± 0.4 nA control, n = 7,p < .05); whereas intracellular application of excess PKC-I (4 μM) with 20 nM PKM, decreased peak currents (1.5 ± 0.3 nA, n = 6, p < .05), likely as a result of inhibition of endogenous PKC.
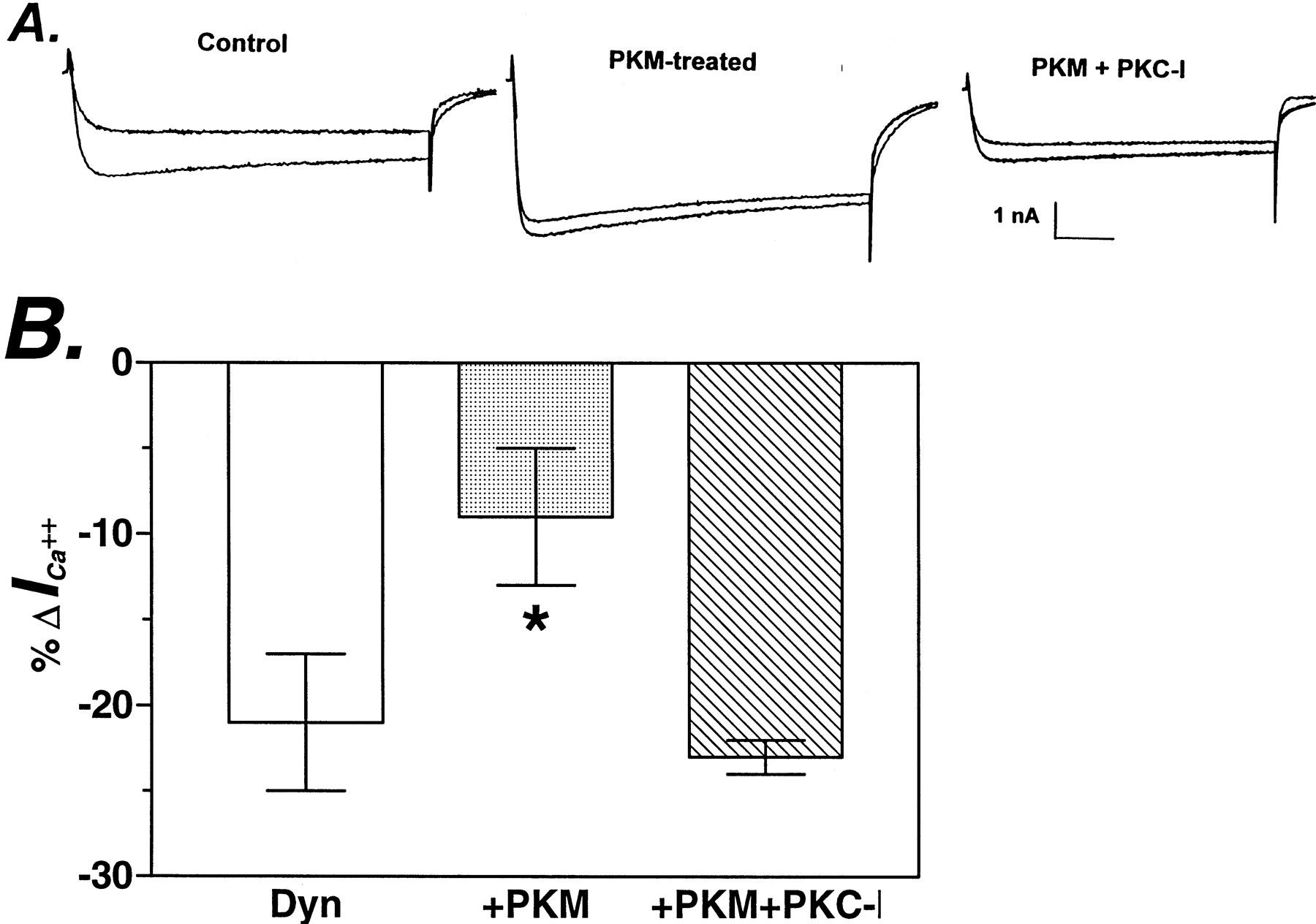
Dialysis of neurons with 20 nM PKM decreased dynorphin A-mediated inhibition of calcium currents from 21 ± 4% to 9 ± 4% (Fig. 6, control (n = 7) and PKM (n = 6), p < .005). Thus, intracellular treatment of neurons with purified PKC, like activation of endogenous PKC with PMA, attenuated dynorphin A inhibition of calcium current. When 4 μM PKC-I was included in the recording pipette with 40 nM PKM, the basal calcium current amplitude was reduced, but dynorphin A inhibited calcium current by 23 ± 1% (Fig. 6, PKM + PKC-I, n = 14), not significantly different from Control and p < .005 from PKM-treated neurons. Thus, the effect of PKM on dynorphin A inhibition of calcium current was reversed by PKC-I.

Dynorphin A inhibition of calcium current was decreased in PKM-treated neurons. A, superimposed current traces before and immediately after 10-s treatment with dynorphin A (3 μM) in control neurons (Control), neurons dialyzed with PKM (40 nM) (PKM), and neurons dialyzed with PKM (40 nM) and PKC-I (4 μM) (PKM+PKC-I). Currents were evoked by a 200-ms pulse to 0 mV from aVh of −80 mV, with calcium as the charge carrier, in the presence of 3 μM nifedipine. B, inhibition (shown as percentage of change) of barium current by 3 μM dynorphin A in control (n = 7, light column), PKM-treated (n = 7, dark column), and PKM + PKC-I-treated (n = 6, striped column) neurons. Data were normalized to the amplitudes of currents immediately before application of dynorphin A.
Effect of Blockade of N-Type Current on PKM Attenuation of Dynorphin A Inhibition of Calcium Channel Current Inhibition.
We next investigated effects of intracellular PKM on specific calcium current subtypes. Dynorphin A inhibition of calcium currents in control and PKM-treated neurons was compared before and after blocking N-type currents with ω-conotoxin GVIA. Before treatment with ω-conotoxin GVIA, dynorphin inhibition of calcium current was significantly smaller (p < .05) in neurons dialyzed with 20 nM PKM (9 ± 4%, n = 6) than in control (23 ± 3%,n = 7) neurons (Fig. 7A, traces 1 and 2; Fig. 6B). After ω-conotoxin GVIA treatment, however, dynorphin A inhibition of calcium current was not significantly different in control (5 ± 4%) and PKM-dialyzed (6 ± 2%) neurons (Fig. 7A, traces 3 and 4; Fig. 6B). Furthermore, in neurons dialyzed with PKM, treatment with ω-conotoxin GVIA did not appear to further decrease dynorphin A effects (Fig. 7B, compare the striped columns). These findings suggested that, like PMA treatment, PKM appeared to primarily affect κ-opioid receptor coupling with N-type calcium currents. Also similar to PMA, intracellular PKM did not decrease the proportion of N-type current. ω-Conotoxin GVIA inhibited a slightly larger proportion of calcium current in PKM-treated neurons than in control neurons (data not shown).

Effect of PKM and ω-conotoxin GVIA on dynorphin A inhibition of calcium currents. A, superimposed current traces of control and PKM-dialyzed neurons before (trace 1) and immediately after (trace 2) 10-s treatment with 3 μM dynorphin A; after treatment with 3 μM ω-conotoxin GVIA (trace 3); and subsequently with 10-s 3 μM dynorphin A (trace 4). Currents were evoked by a 200-ms pulse to 0 mV from a Vh of −80 mV with calcium as the charge carrier in the presence of 3 μM nifedipine. B, comparison of dynorphin A inhibition (shown as percentage of change) of calcium current (-Icalcium) in control neurons (n = 6) and neurons dialyzed with PKM (n = 7) before and after treatment with ω-conotoxin GVIA (3 μM). Dynorphin A inhibition ofIcalcium was reduced in PKM-treated neurons (p < .05). There was no difference in dynorphin A inhibition in control and PKM-treated neurons after treatment with ω-GVIA, and ω-GVIA did not further reduce the fraction of current inhibited by dynorphin A in PKM-treated neurons.
Discussion
External PMA and Dialysis with PKM Both Attenuated κ- and μ-Opioid Receptor Inhibition of Calcium Channel Currents in DRG Neurons.
We demonstrated that in acutely dissociated rat DRG neurons, both external treatment with the phorbol ester PMA and intracellular dialysis with activated PKC, PKM, attenuated κ- and μ-opioid receptor-mediated inhibition of calcium currents. The PMA attenuation of dynorphin A inhibition of calcium current was similar to effects of PMA on several types of rat neurons on inhibition of calcium currents by agonists of α2-adrenergic, γ-aminobutyric acidB, and muscarinic cholinergic receptors (Swartz, 1993; Zhu and Ikeda, 1994). The effects of PMA on calcium current inhibition appeared to be due to activation of endogenous PKC rather than nonspecific actions on membranes or cellular proteins, because the effects of PMA were blocked by dialysis of the neuron with the PKC-I peptide. Endogenous PKC activity did not appear to be necessary for dynorphin A inhibition of calcium currents because intracellular PKC-I did not block dynorphin A inhibition, consistent with effects of PKC-I on morphine- and other agonist-induced inhibition of calcium currents in rat neurons (Swartz, 1993; Wilding et al., 1995).
To examine directly the effects of activated PKC on calcium currents, we dialyzed neurons with PKM. PKM also attenuated κ-opioid receptor-mediated inhibition of calcium currents in a manner similar to PMA treatment. These data demonstrated that constitutively active PKC was capable of blocking κ-opioid receptor-mediated inhibition of calcium currents, suggesting that endogenous PKC could be involved in the physiological modulation of opioid receptor sensitivity. The finding that both PKM and PMA attenuated κ-opioid receptor inhibition and increased basal calcium current was significant in light of conflicting reports of actions of activators and inhibitors of PKC in various species and neuronal types (Boehm et al., 1996;Diverse-Pierluissi and Dunlap, 1993; Swartz, 1993; Zhu and Ikeda, 1994). The present findings are in contrast to the report ofNomura et al., 1994, which found that 2–3 min pretreatment with 1 μM PMA did not affect inhibition of calcium currents by the μ-agonistd-Ala2,(Me)Phe4,Gly-(ol)5in DRG neurons acutely dissociated from rats, 5–10 days of age. We saw comparable effects of PMA to attenuate dynorphin and morphine action, whether 5 mM barium or calcium was used as the charge carrier (data not shown). The present study used rats 14–50 days of age, which may account for the apparent differences in PMA action.
Differential Effects of External PMA and PKM Dialysis on Peak Calcium Current and Current Rundown.
Although both PKM dialysis and external PMA application increased calcium current, they did so with different efficacy. Treatment of DRG neurons with PMA produced only a modest increase in peak calcium current (15%), which was in contrast to larger effects of PMA (50–100%) in rat cerebral cortical, hippocampal, and superior cervical ganglion neurons (Swartz, 1993; Zhu and Ikeda, 1994). Dialysis of DRG neurons with PKM resulted in a larger increase in basal calcium current (∼100%) than did PMA treatment, i.e., the PKM-induced increase in calcium current in DRG neurons was comparable to that induced by activation of endogenous PKC by PMA in central neuronal types. This could reflect differences in levels of PKC activity or efficacy between DRG and central neurons. However, it is also possible that differences between effects of PMA and PKC may have been due to the relatively high level of kinase activity in neurons dialyzed with PKM, leading to the phosphorylation of nonphysiological substrates or other artifactual causes.
External treatment of neurons with PMA also acted differently than dialysis with PKM in terms of effects on current rundown. As shown previously (Hall et al., 1995), dialysis of DRG neurons with PKM, although increasing basal current, had little or no effect on current rundown. However, PMA treatment substantially reduced the rate of calcium current rundown, whereas dialysis of neurons with PKC-I increased the rate of current rundown. This suggested that at least in whole-cell patch conditions used in these experiments, endogenous PKC activity was necessary for maintenance of functional calcium currents. These findings also strengthened the conclusion that PKC may be involved in regulation of calcium currents in a dynamic fashion.
PKC Primarily Attenuated κ-Opioid Receptor Coupling to N-Type Calcium Currents.
Rat DRG sensory neurons contain low-threshold transient (T-type) calcium current and multiple high-threshold calcium current subtypes. Using toxins that are specific blockers of current subtypes, L-, N-, P-, Q- and “R-” (toxin-resistant) subtypes of high-threshold voltage-dependent calcium currents have been reported in rat DRG neurons (Moises et al., 1994a; Rusin and Moises, 1995; Wiley et al., 1997). Dynorphin A did not inhibit L-type currents but did inhibit N-, P-, and Q-type currents, although N-type current accounted for ∼75% of the total dynorphin A-sensitive current (Wiley et al., 1997). Neither PMA nor PKM decreased the proportion of N-type current, indicating that their effect to decrease dynorphin A inhibition of calcium currents was not simply due to a selective removal of a large part of the dynorphin A-sensitive calcium currents by PKC. Because PMA and PKM did not block dynorphin A inhibition of total calcium current completely, it is possible that PKC selectively had an effect on one or more subtype of calcium channel. Thus, the residual dynorphin A inhibition seen in the presence of activated PKC could have been due to effects of dynorphin A on PKC-insensitive channels. Blocking N-type current with ω-conotoxin did not further decrease dynorphin A inhibition in PMA- and PKM-treated neurons, suggesting that PKC primarily affected κ-opioid receptor coupling to N-type channels.
Potential Mechanisms for PKC Attenuation of κ-Opioid Receptor Coupling to Calcium Channels.
Recently, inhibition of calcium currents by G protein-linked receptors has been concluded to be mediated by G protein βγ-subunits, because coexpression of G protein β- and γ-subunits, but not expression of α-subunits or β- or γ-subunits individually, were capable of inhibiting calcium currents in a manner similar to agonists (Herlitze et al., 1996; Ikeda, 1996). G protein βγ-subunits bind directly to the α1-subunit of the calcium channel at sites on the cytoplasmic “linker” region between transmembrane domains I and II (De Waard et al., 1997; Zamponi et al., 1997), in a region overlapping or adjacent to the putative interaction site for the calcium channel β-subunit (De Waard et al., 1996). Furthermore, there was evidence that PKC directly phosphorylated the calcium channel α1-subunit on serines and threonines within the same region and thus blocked the effect of G protein βγ-subunits binding (De Waard et al., 1997). Thus, these findings point to PKC phosphorylation of the calcium channel as a mechanism for attenuation of G protein inhibition.
However, additional PKC pathways modulating opioid receptor coupling to ion channels must exist, because PKC also attenuated opioid receptor activation of G protein-regulated potassium channels (Chen and Yu, 1994; Ueda et al., 1995), and it is plausible that PKC effects on these other signaling pathways also could affect calcium currents. Ostensibly, PKC phosphorylation of the opioid receptor, G protein subunits, or other associated cellular proteins could affect opioid receptor coupling to calcium channels.
Agonist-dependent phosphorylation of opioid receptors did not appear directly to involve PKC (Pei et al., 1995; Zhang et al., 1996). However, PKC can phosphorylate the opioid receptor, leaving open the possibility that PKC phosphorylation of opioid receptors may be a site of regulation of opioid receptor sensitivity by signals other than opioids. There also has been evidence that PKC phosphorylation of G protein subunits may modulate opioid receptor signal transduction. In acutely isolated dorsal raphe nucleus neurons, inhibition of calcium currents by guanosine-5′-O-(3-thio)triphosphate was not blocked by subsequent treatment with PMA (Chen and Penington, 1997), suggesting that the site of PMA action was not the channel but rather was the G protein or other upstream proteins. Activation of PKC led to phosphorylation of Giαa2 and abolished δ-opioid receptor inhibition of adenylate cyclase in NG-108-15 cells (Strassheim and Malbon, 1994), and PKC was shown to be involved in the functional uncoupling of δ-opioid receptors from G proteins in guinea pig striatum (Fukushima et al., 1994; Ueda et al., 1994).
Calcium channel α1 subunits have putative interaction sites for SNARE-family proteins (Yokoyama et al., 1997). Thus, it is possible that the binding of these or other proteins may be involved in regulation of channel function, and thus phosphorylation of these proteins could also be a site of regulation. Thus, there is evidence that PKC may function at any of these possible sites of ion channel regulation.
Acknowledgments
We thank Dr. Michael Browning and Ellen Dudek for the kind gift of catalytic protean kinase C (PKM). We also thank Nadia Esmaeil for assistance in preparing rat DRG neurons.
Footnotes
-
Send reprint requests to: Robert L. Macdonald, M.D., Ph.D., Neuroscience Laboratory Building, 1103 East Huron Street, Ann Arbor, Michigan 48104-1687. E-mail: rlmacd{at}umich.edu
-
↵1 This study was supported by National Institutes of Health Grant DA 04122 to RLM. APJK is a recipient of a National Institutes on Drug Abuse Postdoctoral Training Grant Fellowship 2T32DA07268.
- Abbreviations:
- PKC
- protein kinase C
- PKC-I
- protein kinase C inhibitory peptide, PKC19–31
- PKM
- protein kinase C catalytic subunit fragment
- PMA
- 4β-phorbol 12-myristate 13-acetate
- 4α-PdBu
- 4α-phorbol 12,13-didecanoate
- DMSO
- dimethyl sulfoxide
- Received August 24, 1998.
- Accepted December 2, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}