


Abstract
We tested the assumption that nifedipine blocks L-type calcium current [ICa(L)] at +10 mV and unmasks Na+/Ca2+ exchange-triggered contractions in guinea pig isolated ventricular myocytes. Voltage-clamp pulses elicited ICa(L) at +10 mV and evoked contractions in myocytes superfused with Tyrode's solution (35°C). Nifedipine blocked ICa(L) with an IC50 of 0.3 μM; this decreased to 50 nM at a holding potential of −40 mV, indicating preferential block of inactivated L-type Ca2+ channels. Use-independent block ofICa(L) increased with concentration (10–100 μM) and application time when nifedipine was rapidly applied (t1/2 = ∼0.2 s) during rest intervals (5–30 s). The fraction of use-dependent block ofICa(L) diminished with increasing drug concentration. Nifedipine also acceleratedICa(L) inactivation on the first test pulse. The combination of 30 μM nifedipine/30 μM Cd2+ (Nif 30/Cd 30) was as effective as 100 μM nifedipine to suppressICa(L) on the first test pulse at +10 mV. The incidence of complete block of contractions, as for complete block of ICa(L), increased as a function of nifedipine concentration and application time. Neither nifedipine nor Nif 30/Cd 30 affected Na+/Ca2+ exchange current at +10 to +100 mV. Contractions at +100 mV, although as large as those at +10 mV, were delayed in onset and resistant to nifedipine or Nif 30/Cd 30. We conclude that nifedipine-sensitiveICa(L) triggers contractions at +10 mV, whereas nifedipine-resistant Na+/Ca2+ exchange current initiates those at +100 mV.
Contraction depends on calcium-induced calcium release (CICR) from the sarcoplasmic reticulum (SR) in mammalian heart cells (Fabiato, 1985). The usual trigger for CICR is Ca2+ influx through the L-type Ca2+ channel [ICa(L)]. Calcium entry through reverse mode operation of the Na+/Ca2+ exchanger (Nuss and Houser, 1992; Sham et al., 1992; Levi et al., 1994) and through T-type Ca2+ channels [ICa(T)] also can trigger CICR (Sipido et al., 1998). Calcium entry via T-type channels was a much less efficient trigger than entry via L-type channels, inasmuch as at comparable Ca2+ influx, there was less Ca2+ release from the SR withICa(T) than withICa(L).
The trigger function of reverse mode Na+/Ca2+ exchange (INa/Ca) relative toICa(L) is debated. For example, reverse mode Na+/Ca2+exchange is thought to account for phasic intracellular Ca2+ transients and contractions observed at test potentials when ICa(L) is reduced by Ca2+ channel antagonists. In ventricular myocytes from rat (Wasserstrom and Vites, 1996), rabbit (Levi and Issberner, 1996), and guinea pig (Levi et al., 1996), nifedipine (10–32 μM) suppressed 90 to 99% of ICa(L) but did not eliminate either the intracellular Ca2+transient or contractions. Other studies detected a trigger function of reverse mode INa/Ca, particularly at very positive potentials, have questioned the physiological role ofINa/Ca-triggered release of SR Ca2+. In cat ventricular myocytes, reverse mode INa/Catriggered phasic contractions in the presence of either 1 μM verapamil or nifedipine or 0.2 mM Cd2+, yet a more important role of INa/Ca was to load the SR with Ca2+ (Nuss and Houser, 1992). Other studies interpreted similar results at +80 mV in rat ventricular myocytes to indicate that reverse modeINa/Ca had slower kinetics to induce CICR (Sham et al., 1992). Verapamil (20 μM) or Cd2+ (0.3 mM) only partially reduced contractions when ICa(L) was suppressed at a test potential of +2 mV (Vornanen et al., 1994). They considered that reverse mode INa/Ca contributed to triggering contraction at 35°C but not at 23°C because of the marked temperature dependence ofINa/Ca. Although reverse mode Na+/Ca2+ exchange triggered intracellular Ca2+ transients during steady-state block of ICa(L) by either 10 μM nifedipine (Grantham and Cannell, 1996) or 20 μM nisoldipine (Sipido et al., 1997), its contribution during an action potential was estimated as small and inefficient. Model calculations indicate that Ca2+ influx through L-type channels would reduce Ca2+ entry through reverse mode Na+/Ca2+ exchange. This accords with Na+/Ca2+exchange having a variable efficiency such that it can provide a larger Ca2+ influx whenICa(L) is diminished by Ca2+ channel antagonists.
In general, those who report an important trigger function of reverse mode Na+/Ca2+ exchange have used rapid solution switching to deliver l-type Ca2+ channel antagonists; those who report a low triggering function have used steady-state conditions forICa(L) block. Dihydropyridine (DHP) Ca2+ channel antagonists have been favored in these experiments because they are lipid soluble, very potent, and block the channel preferentially in the inactivated state. Channel block by DHPs is a function of drug concentration and assumed to be largely use-independent. However, L-type Ca2+channel block by rapidly applied nifedipine in frog ventricular myocytes includes a small component of use-dependent block (Méry et al., 1996). That rapidly applied DHP antagonists may not be efficient blockers of L-type Ca2+ channels was raised in experiments with 20 μM nisoldipine and 10 to 20 μM nifedipine (Sipido et al., 1995). In rat ventricular myocytes, nifedipine (10 μM) blocked ICa(L) by ∼70% in 2 min and completely at 4 min (Wasserstrom and Vites, 1996).
Consequently, we reexamined the use-independent and use-dependent components of DHP action on mammalianICa(L). We tested nifedipine action onICa(L) as a function of concentration, exposure time, and stimulus number to ascertain the extent and completeness of blockade. The inorganic ligand Cd2+ was used to standardizeICa(L) block. In some experiments, we also tested the effects of these compounds on contractions to evaluate the trigger functions of ICa(L) andINa/Ca. A preliminary account of some of these findings has been presented (Shen et al., 1999).
Materials and Methods
Isolation of Ventricular Myocytes.
Single ventricular myocytes were enzymatically isolated from the hearts of male and female guinea pigs (250–450 g) anesthetized with sodium pentobarbital (30 mg/kg i.p.) and anticoagulated with heparin (1000 I.U. i.p.). The heart was retrogradely perfused with Tyrode's solution for 5 min at a rate of 8 to 10 ml/min through an aortic cannula in a Langendorff apparatus. The composition of Tyrode's solution was 135 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 0.33 mM NaH2PO4, 10 mM HEPES, and 20 mM glucose; pH was adjusted to 7.4 with NaOH. After disruption of the extracellular matrix with collagenase and protease, the enzymes were washed out by perfusion with 50 ml of Recovery solution. Recovery solution contained 130 mM potassium aspartate, 5 mM K2ATP, 5 mM HEPES, and 20 mM glucose; pH was adjusted to 7.4 with KOH. The ventricles were removed and the cells were dispersed in Recovery solution and kept at 4°C for at least an hour. An aliquot of cell suspension was placed in a recording chamber (500-μl volume) mounted on the stage of an inverted microscope. After 10 min, it was superfused with Tyrode's solution (2 ml/min); the glucose concentration was 10 mM for experiments. Temperature was 35°C.
Electrophysiology.
An EPC 7 patch-clamp amplifier (List Electronics, Darmstadt, Germany) was used to deliver voltage pulses in whole-cell mode. Voltage commands and current data acquisition were controlled by an IBM-compatible computer equipped with pClamp software (version 5.5; Axon Instruments, Burlingame, CA) and a Labmaster TL-1 interface (Axon Instruments). Glass capillary electrodes (1.1 mm i.d.; 1.3 mm o.d.) were filled with a pipette solution whose composition was 120 mM potassium aspartate, 30 mM KCl, 5 mM Na2ATP, 10 mM MgCl2, and 5 mM HEPES; pH was adjusted to 7.3 with KOH. The resistance was 2 to 4 MΩ. In initial experiments, the pipette was filled with a Cs+-rich solution with EGTA containing 135 mM cesium aspartate, 10 mM NaCl, 5 mM MgATP, 5 mM EGTA, and 10 mM HEPES 10; pH was adjusted to 7.3 with CsOH. Accordingly, 10 mM CsCl was added to the bath solution. The pipette was connected to the amplifier by a Ag-AgCl wire, and the tip was gently pushed against the cell surface. Negative pressure was applied to the pipette interior until a gigaohm seal was formed. After the electrode capacitance was compensated electronically in the cell-attached mode, the cell membrane was ruptured by additional negative pressure.
Drugs and Application.
Calcium channel-blocking drugs were applied to myocytes by rapid superfusion from a reservoir via solenoid-controlled delivery. The time for complete solution change, estimated from the membrane current response to doubling the extracellular K+ concentration, was <1 s with at1/2 of ∼200 ms. The applied solutions were warmed and the outlet of the rapid solution device brought within 50 μm of the cell. After recording conditions for the cell had stabilized, the rapid solution device was turned on and Tyrode's solution, identical with the bath solution, applied to the cell. The temperature at the cell's position transiently changed by 0.2–0.3°C when first switching on the Tyrode's solution and by ≤0.2°C when switching from Tyrode's solution to a test solution. Nifedipine (Sigma, St. Louis, MO) was dissolved in dimethyl sulfoxide and prepared fresh daily from the stock solution. All nifedipine solutions were protected from light during preparation, storage, and use.
Cell Contraction.
A video-edge detector system (Crescent Electronics, Sandy, UT) tracked cell edge motion. A microscope-magnified (400×) cell image was continuously observed on a high-resolution TV monitor via a sequential scanning video camera attached to a side port of the microscope. The camera position was rotated so that the video monitor raster lines were parallel with the long axis of the cell. The video dimension analyzer monitored a selected raster line for light intensity differences between the end of the myocyte and the surrounding field. The signal from the detector was sent to a strip chart recorder and to a videocassette recorder for storage and off-line analysis.
Data Analysis.
Steady-stateICa(L) block by nifedipine could be readily measured in the presence of Cs+-containing solutions. In this case, the extent of block was taken as absolute peak inward current. When K+-containing solutions were used,ICa(L) block by nifedipine was standardized against that caused by 0.1 to 0.3 mM Cd2+. The latter has been shown to blockICa(L) completely (Hobai et al., 1997). Measurements are reported as mean ± S.E.
Results
Concentration-Dependent Block of ICa(L)by Nifedipine in Steady State
Initially, we determined the concentration-dependent block of ICa(L) by nifedipine in the absence of K+ currents (see Materials and Methods). Membrane voltage was stepped from −80 to −40 mV for 350 ms to inactivate the fast Na+ and theICa(T) currents. A second voltage jump to +10 mV for 300 ms elicited ICa(L); the clamp protocol was repeated at 0.1 Hz. Block ofICa(L) by Cd2+(0.1, 0.3, 1.0 mM) appeared complete because the peak of early inward current was positive after Cd2+ application for at least 2 min. The average Cd2+-sensitive currents of eight cells are essentially equal and amounted to 854 ± 126 (0.1 mM), 857 ± 131 (0.3 mM), and 856 ± 133 pA (1 mM). The effect of 0.1 mM Cd2+ was taken as the standard for 100% block of ICa(L).
Nifedipine (0.1–100 μM) was cumulatively applied to the same myocyte; each concentration was present for at least 3 min. Half-maximal inhibition (IC50) ofICa(L) occurred at 0.3 μM nifedipine when the holding potential was −80 mV between test pulses (Fig.1, filled squares). In steady state, nifedipine blocked ICa(L) by 94 ± 2.3 (30 μM) and 99 ± 0.8% (100 μM), respectively. When nifedipine was applied at single rather than cumulative concentrations,ICa(L) block averaged 91 ± 3.5% for 30 μM (n = 5 cells) and 99 ± 4.0% with 100 μM nifedipine (n = 4 cells). A mixture of 30 μM nifedipine plus 30 μM Cd2+ (Nif 30/Cd 30) blocked ICa(L) by 97 ± 1.6% (n = 4 cells). The IC50for nifedipine block of ICa(L)decreased to 50 nM when the holding potential was maintained at −40 mV between test pulses (Fig. 1, filled circles).

Concentration-dependent block ofICa(L) by nifedipine in steady state. Pipette and bath solutions contain Cs+. Two-step voltage-clamp pulses (see text for details) were applied at 0.1 Hz. The curve conforms to a 1:1 binding relation between nifedipine and receptor; the IC50 for nifedipine was 0.3 μM when voltage was held at −80 mV between test pulses (▪). The IC50 for nifedipine was reduced to 0.05 μM when the membrane was held at −40 mV (●).
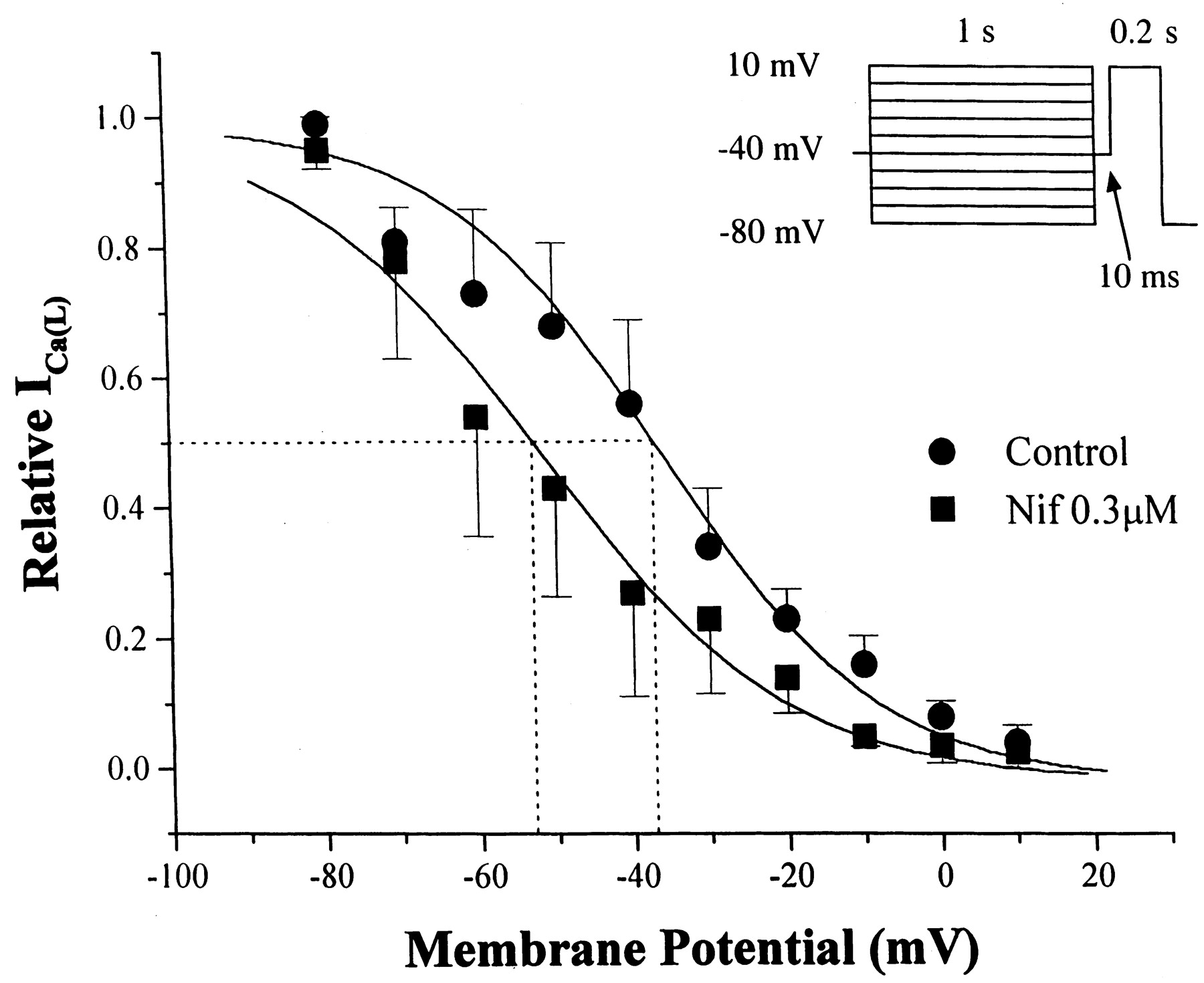
Steady-state inactivation of ICa(L)was determined in the absence and presence of 0.3 μM nifedipine (n = 4 cells). A 1-s command changed the conditioning potential from −80 to +10 mV in 10-mV steps. Afterward, a 10-ms return to −40 mV was inserted before applying a 200-ms jump to the test potential of +10 mV. Inactivation ofICa(L) was described by a Boltzmann relation (Fig. 2). Voltage-dependent inactivation of ICa(L) at 50% was shifted from −36 ± 3.3 (control) to −53 ± 1.6 mV (nifedipine). The slope factor was 14 ± 1.5 and 15 ± 1.5 in control and nifedipine, respectively. Steady-state block by nifedipine of ICa(L) in the presence of K+-rich pipette solution was essentially the same as in the presence of Cs+-rich pipette solution (vide infra).

Steady-state inactivation ofICa(L) in the absence (●) and presence of 0.3 μM nifedipine (▪). The experimental protocol is shown as an inset. Ordinate, ICa(L) as fraction of maximum; abscissa, conditioning membrane potential. The curves are drawn according to Boltzmann relations; see text for details.
Concentration- and Time-Dependent Block ofICa(L) by Rapidly Applied Nifedipine and/or Cadmium
Nifedipine Alone.
In some studies of excitation-contraction (E-C) coupling (see the Introduction), the ability to suppressICa(L) completely on the first test pulse after rapid application of a blocking agent has been emphasized. Thus, ICa(L) block should be maximal and complete on the first test pulse and be invariant during a train of test pulses. The protocol to test this hypothesis included two trains of 200-ms voltage-clamp pulses (−40 to +10 mV at 0.5Hz) separated by a 10-s rest interval at −40 mV. The test-blocking agent was applied by rapid superfusion and present throughout the 10-s interval and the second train of test pulses. Membrane voltage was held at −40 mV to promote the blocking effect of nifedipine; the pipette solution was K+-rich (see Materials and Methods).
The results in Fig. 3A illustrate the effects of nifedipine (30 and 100 μM) and Cd2+(300 μM) in one ventricular myocyte. The nifedipine-sensitiveICa(L) was standardized against the complete block of ICa(L) that is caused by Cd2+. The control currents on the 1st and 10th test pulses are labeled “0”. The Cd2+-sensitiveICa(L) was practically the same on the 1st (−1375 pA) and 10th (−1430 pA) test pulses (traces labeled “3”). Suppression of ICa(L) by 30 μM nifedipine (traces labeled “1”) was incomplete on the 1st test pulse, increased with successive test pulses, yet was still less than maximum on the 10th test pulse (Fig. 3A). With 100 μM nifedipine (traces labeled “2”), suppression ofICa(L) was greater than that by 30 μM on the 1st test pulse and almost equaled that by 300 μM Cd2+ on the 10th test pulse.

Development of ICa(L)block by nifedipine (30 and 100 μM) or 300 μM Cd2+; K+-rich pipette solution. After 10 conditioning pulses (see inset at top of A for protocol), nifedipine or Cd2+ was applied during a 10-s rest interval and 10 subsequent test pulses. A, membrane currents in one cell in control (0), 30 (1), or 100 μM (2) nifedipine or 300 μM Cd2+ (3) on the 1st (top) and 10th test pulse (bottom) after the 10-s interval. B, summary of results with unblocked ICa(L) (a), use-dependent (b), and use-independent (c) block of ICa(L).
The results of eight experiments of this type are shown in Fig. 3B.ICa(L) is divided into three components: nonblocked, use-dependent, and use-independent block. The percentage block of ICa(L) on the first test pulse is attributed to use-independent inhibition (Fig. 3B, c). The difference of percentage block between 1st to 10th pulses gives the use-dependent inhibition (Fig. 3B, b), whereas the difference between 10th nifedipine-sensitiveICa(L) and 10th Cd2+-sensitiveICa(L) is the nonblockedICa(L) by nifedipine (Fig. 3B, a). For 30 μM nifedipine, use-independent inhibition ofICa(L) was 85 ± 4.4%, use-dependent inhibition of ICa(L) was 8%, and the nonblocked ICa(L) was 7%. Raising nifedipine to 100 μM yielded use-independent block of 95 ± 2.3%. For Cd2+, there is a greater use-independent inhibition of ICa(L)amounting to 99 ± 1.0% and a small use-dependent inhibition of 0.6%. The probability of completely blockingICa(L) on the first test pulse after 10-s application is greatest with Cd2+, intermediate with 100 μM, and least with 30 μM nifedipine.
Our aim was to ascertain the optimal conditions to achieve completeICa(L) block by nifedipine on the first test pulse because this is desirable for evaluating E-C coupling triggers. With the same protocol and standardization as previously used (Fig. 3), different concentrations of nifedipine (10, 30, 100 μM) were applied during selected rest intervals (5, 10, 15, 20, 30 s) plus test periods. These results are summarized in Fig.4. Use-independent inhibition ofICa(L) progressively increased with nifedipine concentration. After 5-s application, use-independent block of ICa(L) averaged 70 ± 9.7, 80 ± 4.4, and 93 ± 3.6% at 10, 30, and 100 μM nifedipine, respectively. After 30-s application, 10, 30, and 100 μM nifedipine produced 93 ± 4.3, 93 ± 2.8, and 95 ± 2.5% use-independent inhibition ofICa(L), respectively. Thus, the likelihood of blocking ICa(L)completely on the first test pulse increased as a function of time as well as concentration of nifedipine. The decline of use-dependent fraction of block was monoexponential in the first 20 s with rate constants of −0.13 s−1 and −0.09 s−1 at 10 and 30 μM nifedipine, respectively. At 100 μM nifedipine, the use-dependent inhibition ofICa(L) declined at a rate of −0.04 s−1; this estimate is less reliable because use-dependent block varied from 6 to 4% at 5- to 20-s application times. However, at 100 μM nifedipine, the fraction of nonblockedICa(L) was practically abolished an indication that under steady-state conditions (10th test pulse), 100 μM would completely suppress ICa(L).

Concentration and time dependence forICa(L) block by nifedipine. Experiments done as in Fig. 3 with K+-rich pipette solution;ICa(L) block standardized with 0.3 mM Cd2+. Each column represents data from increasing application intervals of 5, 10, 15, 20, and 30 s for a nifedipine concentration shown at top. In each column, fractional block by use-independent (unfilled) and use-dependent (filled) actions of nifedipine is shown; N, number of cells at each interval. Unblocked fraction of ICa(L) indicated by cross-hatching.
Nifedipine and Inactivation of ICa(L)
Nifedipine increased the rate ofICa(L) inactivation (Lee and Tsien, 1983). We evaluated the hypothesis that some block ofICa(L) occurs during the first test pulse in experiments with 3 μM nifedipine. The voltage-clamp protocol was the same as described in Fig. 3. In control,ICa(L) inactivated in a biexponential manner with time constants (in milliseconds) τ1and τ2 of 5.5 ± 0.4 and 51.3 ± 6.0 on the 1st test pulse and 5.2 ± 0.4 and 59.0 ± 3.3 on the 10th test pulse, respectively (n = 11 cells). In nifedipine, τ1 decreased by 20% to 4.4 ± 0.5 ms (1st test pulse; P = .006) and by 33% to 3.5 ± 0.4 ms (10th test pulse; P = .002). There was a tendency of τ2 to decrease in nifedipine on the 1st (46.8 ± 3.8 ms) and 10th (53.3 ± 8.2 ms) test pulses, respectively. However, these reductions were not statistically significant (P = .3 and .36, respectively). These averages are from all 11 cells in which τ1 and τ2 were reduced in seven cells. It was difficult to discern two phases of inactivation accurately at higher nifedipine concentrations. We measured the half-time for inactivation (t1/2) in experiments with 30 μM nifedipine (n = 8 cells) and found that the controlt1/2 was reduced from 11.5 ± 1.8 to 8.5 ± 1.2 ms on the 1st test pulse (P = .01) and from 12.0 ± 2.2 to 8.5 ± 1.2 on the 10th test pulse (P = .01).
Test of Combination of Nifedipine and Cadmium
Cadmium inhibited ICa(L) with an IC50 of ∼2 μM (Hobai et al., 1997). The kinetics of block by Cd2+ is rapid (Lansman et al., 1986) and there is little use-dependent inhibition ofICa(L) at 10 s (Fig. 3). After 5-s application, Cd2+ inhibited 98 + 2.0% ofICa(L) elicited on the first test pulse (n = 13). The residuum of 2% is less than that seen with nifedipine and was abolished at ≥10-s application intervals (<1%). Thus, 0.3 mM Cd2+ inhibitedICa(L) nearly completely on the first test pulse at ≥10 s. Because 0.3 mM Cd2+inhibits INa/Ca by almost 50%, it cannot be used to distinguish the triggering roles ofICa(L) versusINa/Ca in E-C coupling (Hobai et al., 1997).
Inhibition of INa/Ca at 30 μM Cd2+ is predicted to be ≤3%, whereas suppression of ICa(L) is estimated at 88% by these authors. At 30 μM, Cd2+inhibited ICa(L) by 83 ± 2.2 (n = 5), 90 ± 2.2 (n = 6), and 95 ± 1.3% (n = 4) on the first test pulse after 5-, 10-, and 15-s rest intervals, respectively. Inhibition by 30 μM Cd2+ amounted to 96 ± 1.2% at the 10th test pulse, which was steady state (n = 15). We tested the effects of Nif 30/Cd 30 to increase the use-independent inhibition of ICa(L). On the 1st and 10th test pulses after 10-s application of Nif 30/Cd 30 (Fig.5A, traces labeled “1”), peak inward current was essentially the same as that seen after 0.3 mM Cd2+ alone (Fig. 5A, traces labeled “2”). The inward shift of holding current at −40 mV with 0.3 mM Cd2+ may result fromINa/Ca suppression. The end-of-pulse currents were the same in the presence and absence of the test agents. A summary of experiments after 10-s application is given in Fig. 5B. Nif 30/Cd 30 produced a use-independent inhibition ofICa(L) of 95 ± 4.5% at 5-s, 97 ± 1.8% at 10-s, and 96 ± 4.1% at 15-s application. Thus, combining low concentrations of Cd2+ and nifedipine produced use-independent inhibition ofICa(L) at 10 s that was equivalent to that seen with 100 μM nifedipine.

Concentration and time dependence forICa(L) block by a mixture of 30 μM nifedipine plus 30 μM Cd2+ (Nif 30/Cd 30). Experimental conditions are as in Fig. 3. A, membrane currents in one cell in control (0), Nif 30/Cd 30 (1) or 300 μM Cd2+ (2) on the 1st (top) and 10th test pulse (bottom) after a 10-s application period. B, summary of results (n = 5 cells) for 10-s application with unblocked ICa(L) (a), use-dependent (b), and use-independent (c) block ofICa(L).
Relationship between Block of ICa(L) and Contraction by Test Ligands
Experiments at +10 mV.
We repeated the experiments with ligands used to block ICa(L) to ascertain the coupling between the L-type Ca2+current and contraction at a test potential of +10 mV. The protocol is shown in the upper portion of Fig. 6B. Six 200-ms conditioning pulses from −80 to 30 mV were applied at 1 Hz to maintain a relatively constant SR Ca2+content. During the 10-s pause, membrane voltage was held at −40 mV; a single 200-ms pulse to +10 mV elicitedICa(L) and a contraction. Results from an experiment on a single ventricular myocyte are shown in Fig. 6. The control ICa(L) and its corresponding contraction are shown in Fig. 6, A and B, respectively. The current and contraction obtained on the first test pulse after 10-s application of 0.3 mM Cd2+ are indicated by the filled squares; both variables are completely suppressed. The records just after washout of Cd2+ are not shown. Subsequently, 30 μM nifedipine was tested; ICa(L) and its accompanying contraction was greatly, but not completely blocked (filled circles). Like 0.3 mM Cd2+, Nif 30/Cd 30 completely blocked the test ICa(L) and its contraction on the first test pulse after 10 s. Test contraction recovered essentially completely after washout of Nif 30/Cd 30 (Fig. 6B, bottom). Figure 6A (bottom) shows amplified, superimposed current traces taken from the cell. A transient inwardly directed current is evident in the test of 30 μM nifedipine. In contrast, the current traces at +10 mV in either 0.3 mM Cd2+ or Nif 30/Cd 30 do not show this inwardly directed transient. The inward shift of current in 0.3 mM Cd2+ is consistent with suppression of INa/Ca, which is outward at +10 mV. The end-of-pulse currents at +10 mV are the same in 30 μM nifedipine and Nif 30/Cd 30.

Coupling between ICa(L)and contraction in one ventricular myocyte in the absence and presence of L-type Ca2+ channel antagonists. After six conditioning pulses (−80 to +30 mV at 1 Hz), membrane was held at −40 mV for 10 s during which time antagonist was rapidly superfused. Test pulse to +10 mV followed. A, ICa(L) in control (○) and in 300 μM Cd2+ (▪), or 30 μM nifedipine (●) or Nif 30/Cd 30 (▾). Lowest trace in A shows superimposed currents symbolized as above with control as ○. B, experimental protocol shown above. Cell contraction records taken from control (○), 300 μM Cd2+ (▪), 30 μM nifedipine (●), and Nif 30/Cd 30 (▾). The lowest trace is a record after washout of Nif 30/Cd 30 solution. Washout records between (▪) and (●) and between (●) and (▾) not shown.
A summary of all such experiments is shown in Fig.7. The controlICa(L) (999 ± 150 pA) diminished by 690 ± 100 pA on the first test pulse in 10 μM nifedipine (72 ± 5.5%); contractions decreased by 61 ± 6.8% from an initial value of 3.7 ± 0.50 to 1.6 ± 0.52 μm. At 30 μM, nifedipine reduced ICa(L) from 1352 ± 249 pA at control to 265 ± 93 pA (83 ± 3.6% block) and contraction from 4.0 ± 0.53 to 1.0 ± 0.47 μm (79 ± 7.8% decrease) in nine cells. In three of these nine cells (33%), the contraction was abolished on the first test pulse. In the remaining six cells, the latency between the onset ofICa(L) and contraction averaged 30 ± 6.5 ms in control and 30 ± 4.9 ms in 30 μM nifedipine, respectively. After 10 s in 100 μM nidedipine, theICa(L) decreased from 1052 ± 352 pA at control to 89 ± 28.6 pA (92 ± 2.1% block) and contraction diminished from 2.9 ± 0.20 to 0.14 ± 0.10 μm (96 ± 2.8% decrease) in 14 cells. Contractions on the first test pulse were eliminated completely in 12 of these 14 cells (86%). Adding Nif 30/Cd 30 significantly decreasedICa(L) at control from 1395 ± 212 to 65 ± 21 pA (94 ± 2.2% block) and abolished contraction in six of seven cells (86%). The average contraction of all the cells was reduced from 4.4 ± 0.43 to 0.1 ± 0.09 μm (98 ± 2% decrease). The results indicate that the greater the inhibition of ICa(L), the more likely contraction will be abolished at +10-mV test potential.

Summary of blocking effects of test ligands onICa(L) and contraction. The ordinate shows results as percentage of decrease (mean ± S.E.) ofICa(L) (■), percentage of decrease of cell shortening (CS, ▩), and complete block of CS (▪) grouped according to ligand concentration on the abscissa. Number of cells shown in parentheses. See text for details.
Membrane Current and Cell Contraction at ≥+50 mV
Reverse mode INa/Ca increases andICa(L) decreases as membrane voltage becomes more positive. We tested the hypothesis thatICa(L) may be present at +50 mV. From a holding potential of −40 mV, membrane voltage was jumped in 10-mV steps to +100 mV at 0.33 Hz before and after addition of 100 μM nifedipine (n = 6 cells). The reversal potential of nifedipine-sensitive ICa(L) was 68 ± 2.7 mV, which is comparable to that seen with nisoldipine (Sipido et al., 1997).
We next evaluated the effects of 100 μM nifedipine (Fig.8A) or Nif 30/Cd 30 (Fig. 8B) on membrane current and contraction at +50 and +100 mV with the same conditioning protocol as in Fig. 6. At +50 mV, the average contraction amplitude was 4.0 ± 0.59 μm (n = 13 cells), essentially the same as at +10 mV (3.6 ± 0.21 μm; n = 38;P = .42). Initial membrane current at +50 mV shifted outward by 120 pA in 100 μM nifedipine (Fig. 8A, left, inset). In seven such experiments, nifedipine shifted peak membrane current at +50 mV outward by 212 ± 30.0 pA (P < .01) yet the end-of-pulse current differed by only 14 ± 15.1 pA (P = .38). There was no significant change in membrane currents at either −40 (14 ± 18.1 pA; P = .48) or −80 mV (9 ± 14.3 pA; P = .81). These results can be explained by block of inwardly directedICa(L) at +50 mV with no effect onINa/Ca. In the example shown in Fig.8A, cell shortening decreased from 3.2 (control) to 1.9 μm in nifedipine; contraction latency increased from 45 to 75 ms. Nifedipine completely blocked contraction at +50 mV in only one cell. In the remaining six cells, cell shortening was reduced from 4.4 ± 0.67 to 2.5 ± 0.67 μm (43% decrease; P = .04) and the latency to contraction onset was 67 ± 4.9 ms in 100 μM nifedipine compared with 48 ± 3.0 ms in control (P = .01).

Effect of nifedipine (100 μM) or Nif 30/Cd 30 on membrane current and contraction at positive potentials. Voltage-clamp conditioning protocol same as in Fig. 5 with 10-s rest interval during which time test drugs were applied by rapid superfusion. In all traces, membrane current is uppermost and cell shortening is lowermost. A, nifedipine action (trace 1) at +50 mV (left) and +100 mV (right) test potentials. B, Nif 30/Cd 30 effects (trace 2) at +50 and +100 mV, respectively. See text for details.
The combination of Nif 30/Cd 30 was tested in the remaining six cells. In the example shown (Fig. 8B, left), peak current shifted outward by 225 pA. Control contraction amplitude and latency were 7.0 μm and 60 ms, respectively; these values changed to 2.2 μm and 100 ms in Nif 30/Cd 30. Nif 30/Cd 30 had the same actions on membrane current as 100 μM nifedipine. Thus, Nif 30/Cd 30 (n = 6) did not change current at −40 mV (−12 ± 6.8 pA; P = .72) or at −80 mV (33 ± 18.1 pA; P = .30). However, peak current at +50 mV shifted outward by 218 ± 30.4 pA (P < .01) and not at end of pulse (9 ± 26.7 pA;P = .95). Contractions ceased in three of six cells with Nif 30/Cd 30 and decreased from 3.5 ± 1.0 to 1.2 ± 0.70 μm (66% decrease; P = .01) in the remainder. In these three cells, contraction onset was delayed from 53 ± 11.5 to 107 ± 11.5 ms by Nif 30/Cd 30 (P < .01). Thus, at +50 mV, there is an inwardly directedICa(L) whose block by nifedipine or Nif 30/Cd 30 not only decreased the extent of cell shortening but also delayed the onset of contraction.
Addition of 5 mM Ni2+ shifted membrane current inward at +50 and −40 mV and outward at −80 mV. In six experiments with Ni2+, membrane current shifted inward by −238 ± 42.2 pA at −40 mV (P < .01) and outward by 158 ± 46.5 pA at −80 mV (P < .01). Thus, Ni2+suppressed a current whose reversal potential is between −40 and −80 mV, consistent with its beingINa/Ca. Nickel had no effect on peak current at +50 mV (−33 ± 86.1 pA; P = .19) presumably because the outward shift fromICa(L) block was offset by an inward shift due to INa/Ca suppression. Evidence for the latter is the inward shift in end-of-pulse current by −273 ± 53.5 pA (P < .01). Contractions were completely eliminated by 5 mM Ni2+(n = 6 cells).
At +100 mV, current through L-type Ca2+ channels should be outward and carried by K+ (Lee and Tsien, 1983). Nifedipine (100 μM) shifted peak membrane current slightly inward by −35 pA at +100 mV (Fig. 8A, right). Neither contraction amplitude (3.2 μm) nor latency (70 ms) changed in nifedipine. On average, nifedipine shifted peak current inward by −180 ± 44 pA (n = 6 cells). With Nif 30/Cd 30 (Fig. 8B, right), current shifted inward by −100 pA. Cell shortening increased slightly from 5.8 to 6.0 μm, whereas latency remained constant at 70 ms. The Nif 30/Cd 30-sensitive current of −152 ± 38 pA (n = 6) was indistinguishable from that of nifedipine. Before drug addition, the average amplitude of contraction at +100 mV (4.9 ± 0.64 μm; n = 12) is larger than at +50 mV (4.0 ± 0.59 μm), and the latency to onset of the contraction at +100 mV (81 ± 4.5 ms) is greater than at 50 mV (50 ± 2.9 ms). Neither 100 μM nifedipine nor Nif 30/Cd 30 significantly changed contraction amplitude at +100 mV. In six cells, contraction amplitude averaged 4.8 ± 0.80 μm in control versus 4.3 ± 0.80 μm with 100 μM nifedipine (P = .12). In another six cells, cell shortening averaged 5.0 ± 1.0 and 5.3 ± 1.0 μm in control and Nif 30/Cd 30, respectively (P = .14). Contraction onset at +100 mV is slightly delayed by 100 μM nifedipine from 75 ± 6.2 to 88 ± 8.7 ms (P = .08) and by Nif 30/Cd 30 from 87 ± 6.7 to 95 ± 8.8 ms (P = .09), respectively.
Discussion
Rapidly applied nifedipine displayed use-dependent and use-independent components of ICa(L)block. The steady-state IC50 was 0.3 μM at a holding potential of −80 mV and 50 nM when the membrane was held at −40 mV (Lee and Tsien, 1983; Yamamoto et al., 1990). Depolarization promotes ICa(L) block because DHP affinity for receptor is greatest as L-type channels shift toward the inactivated state (Carmeliet and Mubagwa, 1998). TheV0.5 for steady-state inactivation ofICa(L) shifted by 17 mV to more negative potentials in 0.3 μM nifedipine as predicted (Sunami et al., 1995; for review, see Carmeliet and Mubagwa, 1998).
Use-Independent Block of ICa(L).
Fractional block of ICa(L) on the first test pulse increased with nifedipine concentration and application time, a finding favorable for studying contraction triggering mechanisms. Neutral DHPs rapidly partition into the plasma membrane lipid bilayer (Herbette et al., 1989), binding at hydrophobic amino acids ∼11 to 14 Å from the external plasma membrane surface (Bangalore et al., 1994) of the sixth transmembrane segments of domains III and IV in the L-type Ca2+ channel α1-subunit (Hockerman et al., 1997). Membrane partitioning did not limit kinetics of nifedipine action in frog ventricular myocytes (Méry et al., 1996).
Use-Dependent Block of ICa(L) by Nifedipine.
Use-dependent block diminished as nifedipine concentration and time increased, and was least with Nif 30/Cd 30. Use-dependent block by nifedipine after 10 test pulses underestimates the magnitude of this component at steady state. DHPs exert use-dependent block of ICa(L) in mammalian and amphibian ventricular myocytes even when present during 3- to 15-min rest intervals (Lee and Tsien, 1983; Uehara and Hume, 1985; Sunami et al., 1995) and when rapidly applied at saturating concentrations (Levi and Issberner, 1996; Méry et al., 1996).
Some block of ICa(L) by nifedipine (≤30 μM) appears during the first test pulse because the early inactivation phase, but not the second, decreased significantly. Our experimental conditions cannot distinguish it from inactivated-state block (for review, see Carmeliet and Mubagwa, 1998). DHP agonist and antagonists accelerate inactivation ofICa(L) orIBa(L (Lee and Tsien, 1983; Hess et al., 1985); this is attributed to a transition from mode 1 gating to mode 0 gating where the Ca2+ channel is stabilized and unavailable for opening (Hess et al., 1985). Nitrendipine (Lee and Tsien, 1983), nisoldipine (Sanguinetti and Kass, 1984), and nifedipine (Alvarez and Vassort, 1992) acceleratedICa(L) inactivation during the first test pulse.
Nifedipine Effects on ICa(L) and Contraction at +10 mV.
In steady state, nifedipine suppressed the slow inward current and contraction force by 50% at 0.3 and 0.5 μM, respectively (Bayer et al., 1977). We find contraction eliminated when either nifedipine or Nif 30/Cd 30 suppressedICa(L) by ≥95% on the first test pulse after a 10-s rest exposure. The advantage of Nif 30/Cd 30 is the presence of ligands that use hydrophobic and hydrophilic pathways to their receptor sites. Both ligands are largely use-independent, unlike nifedipine combined with the verapamil analog D600 (Howarth and Levi, 1998); D600 is very use-dependent (for review, see Carmeliet and Mubagwa, 1998).
Nifedipine blocked ICa(L) less and contractions more on the first test pulse at +10 mV than that reported by other studies. The residual inward current in nifedipine at +10 mV (Fig. 3A) could be remaining ICa(L)(Sipido et al., 1995; Evans and Cannell, 1997). Alternatively, the current could be forward mode INa/Cathat progressively diminishes as SR Ca2+ content decreases during the pulse train. Assuming completeICa(L) block on the first test pulse, some studies have proposed that reverse modeINa/Ca triggered the residual contraction or Ca2+ transient in nifedipine or verapamil (Vornanen et al., 1994; Levi and Issberner, 1996; Levi et al., 1996; Wasserstrom and Vites, 1996). Several lines of evidence implicate unblocked ICa(L) as the residual inward current at +10 mV. First, although 0.3 mM Cd2+ blocksINa/Ca by 50% andICa(L) completely, the early inward current sensitive to 100 μM nifedipine or Nif 30/Cd 30 (Fig. 5) was often the same as the Cd2+-sensitive current on the first test pulse. Neither nifedipine nor Nif 30/Cd 30 inhibitedINa/Ca between +10 and +100 mV. Second, we obtained similar results with 0.1 mM Cd2+, which blocksICa(L) completely yet has a lesser effect on INa/Ca (Hobai et al., 1997). Third, the likelihood of blockingICa(L) completely on the first test pulse increased as nifedipine concentration rose from 30 to 100 μM. Fourth, the residual peak inward current decreased but was not delayed (Fig. 3A) in nifedipine. We ascribe this to a smallerICa(L). InwardINa/Ca is delayed because it follows the intracellular Ca2+ transient (Grantham and Cannell, 1996; Sipido et al., 1997). Nifedipine (20 μM), applied rapidly during a 9-s rest interval, delayed the time to peak of intracellular Ca2+ transients initiated by action potentials in ventricular myocytes from transgenic mice overexpressing the Na+/Ca2+ exchanger (Yao et al., 1998). This delay indicated that Ca2+influx from reverse mode INa/Ca is slower than that by ICa(L). In other experiments with ventricular myocytes from transgenic mice overexpressing the cardiac Na+/Ca2+ exchanger, Ca2+ influx via the exchanger did not trigger CICR at −10 to +20 mV yetICa(L)-triggered CICR caused forward mode INa/Ca (Adachi-Akahane et al., 1997). When nisoldipine completely blockedICa(L), the peak of the intracellular Ca2+ transient was delayed >80 ms (Sipido et al., 1997), an effect attributed to triggering by reversedINa/Ca. In the latter experiments, conditioning pulses to +60 mV maintained SR Ca2+content, defined by caffeine-induced Ca2+transients. In contrast, the residual intracellular Ca2+ transient on the first test pulse at +10 mV in 32 μM nifedipine/10 μM D600 was attributed to reverse modeINa/Ca, yet the time to peak of the Ca2+ transient did not change (Howarth and Levi, 1998). Our results question the assumption that addition of ≤30 μM nifedipine is sufficient to block allICa(L) on the first test pulse at +10 mV so that simultaneous exposure to a ligand such as Ni2+ is unable to block such channels further.
Nifedipine Effects on Membrane Current and Contraction at +50 and +100 mV.
Reverse-mode Na+/Ca2+ exchange can trigger Ca2+ release at voltages ≥50 mV because disabling the SR with caffeine (Sham et al., 1992) or ryanodine (Sipido et al., 1997) blocks Ca2+ release transients at +80 mV (but see Adachi-Akahane et al., 1997; Mattiello et al., 1998).
Nifedipine (100 μM) or Nif 30/Cd 30 partially suppressed contractions evoked at +50 mV. Even at an overshoot potential of +50 mV, the onset and the initial component of contraction appears closely related to the inwardly directed ICa(L). The residual contraction was significantly delayed in onset whenICa(L) was suppressed at +50 mV (Nuss and Houser, 1992; Sham et al., 1992; Vornanen et al., 1994; Sipido et al., 1997). At +70 mV, the intracellular Ca2+transient was essentially unchanged in amplitude and time to peak (Sipido et al., 1997) as expected at the Ca2+reversal potential. At the action potential peak, Ca2+ influx viaINa/Ca is ∼10 to 30% of that byICa(L) (Grantham and Cannell, 1996;Sipido et al., 1997; Litwin and Bridge, 1998). At +100 mV, contraction amplitudes are greater and their latencies longer than at +50 mV. The former indicates that the relation between voltage and contraction is not bell shaped when the pipette solution contains 10 mM Na+ (Vornanen et al., 1994; Wasserstrom and Vites, 1996; Litwin and Bridge, 1998). Blockers ofICa(L) did not affect the amplitude or latency of contractions at +100 mV; this excludesICa(L) and implicates reverse modeINa/Ca.
Limitations.
L-type Ca2+ channel block by nifedipine was indexed with Cd2+. The implications of nonselective block by Cd2+ have been presented (vide supra). A second limitation centers around the relative gain of triggering (Stern et al., 1999) and the synergy of triggers (Litwin et al., 1998). Gain (Ca2+release from SR/L-channel Ca2+ influx) decreases as membrane voltage approaches ECa (Wier et al., 1994; Stern et al., 1999), yet the relative efficacy ofICa(L) increases as Ca2+ influx is blocked by drugs (Cannell et al., 1995). In contrast, Ca2+ entry via reverse modeINa/Ca andICa(L) could interact synergistically with the former amplifying the trigger effect of the latter (Litwin et al., 1998). Our findings at +10 mV do not accord with the synergy hypothesis because ≥95% ICa(L) block prevented contractions and contraction latency did not shift at <95%ICa(L) block. Nifedipine (100 μM) or Nif 30/Cd 30 delayed residual contractions at +50 mV and had no effect on contraction amplitude and latency at +100 mV. We assume reverse modeINa/Ca could trigger Ca2+ transients and contractions at these positive potentials as reported by other studies (Nuss and Houser, 1992; Sham et al., 1992; Sipido et al., 1997; Litwin et al., 1998). However, some studies have not detected this outcome (Adachi-Akahane et al., 1998; Mattiello et al., 1998). Nifedipine-resistant contractions at these positive potentials (Fig. 8) did not relax until repolarization such as can occur with tonic entry of Ca2+ (Nuss and Houser, 1992; Adachi-Akahane et al., 1997). Experiments that disable the SR release mechanism are needed to distinguish between triggered and tonic nifedipine-resistant contractions at positive potentials. Finally, experiments with action potential-initiated contractions should provide evidence about the physiological role of Ca2+ influx via the exchanger on E-C coupling.
Footnotes
-
Send reprint requests to: Achilles J. Pappano, Ph.D., Department of Pharmacology, MC-6125, University of Connecticut Health Center, 263 Farmington Ave., Farmington, CT 06030. E-mail:pappano{at}nso1.uchc.edu
-
↵1 This study was supported by U.S. Public Health Service Grant HL-13339.
- Abbreviations:
- CICR
- calcium-induced calcium release
- SR
- sarcoplasmic reticulum
- ICa(L)
- L-type calcium channel current
- ICa(T)
- T-type Ca2+ channel current
- INa/Ca
- Na+/Ca2+ exchange current
- DHP
- dihydropyridine
- Nif 30/Cd 30
- 30 μM nifedipine plus 30 μM Cd2+
- E-C
- excitation-contraction
- Received November 24, 1999.
- Accepted April 14, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}