


Abstract
Anthracyclines are commonly used chemotherapeutics, and in some models enhance p44/42-mitogen-activated protein kinase (MAPK) pathway signaling by effects on upstream kinases. To evaluate the impact of anthracyclines on p44/42-MAPK in breast cancer, A1N4-myc human mammary and BT-474 and MDA-MB-231 breast carcinoma cells were studied. Treatment with doxorubicin or epirubicin resulted in increased phospho-p44/42-MAPK levels in a time- and concentration-dependent manner. This was associated with p44/42 activation, as reflected by increased p90 ribosomal protein S6 kinase and Bad phosphorylation. Activation of p44/42 appeared to be antiapoptotic, since MAPK stimulation with epidermal growth factor or a dominant-positive p42 construct inhibited apoptosis. Modest activation of the upstream MAPK kinase MEK was noted under some conditions, but inhibition of MEK did not abolish p44/42 activation, suggesting a contribution from another mechanism. Anthracyclines were found to decrease expression of MAPK phosphatase-1 (MKP-1) both in vitro and in vivo. MKP-1 mRNA levels were decreased in anthracycline-treated cells, and transcription from the MKP-1 promoter was repressed. Inhibition of MKP-1 expression through the use of small interfering RNAs decreased the ability of anthracyclines to induce phospho-p44/42. Wild-type mouse embryo fibroblasts (MEFs) treated with doxorubicin showed increased phospho-p44/42-MAPK levels, but MEFs from MKP-1 heterozygous and homozygous knockout mice had blunted p44/42 activation. These studies support the ability of anthracyclines to activate antiapoptotic p44/42-MAPK phosphorylation in breast cancer, and indicate that this occurs in part through the novel mechanism of repression of MKP-1 transcription.
Many drugs used in cancer therapy have pleiotropic effects, and although they activate apoptosis they can also activate antiapoptotic signal transduction pathways that promote survival, possibly limiting their own antitumor efficacy. One notable example of this is the nuclear factor κB pathway, whose activation by many DNA damaging agents results in enhanced transcription of Bcl-2 homologs such as Bcl-xL (recently reviewed by Orlowski and Baldwin, 2002). Another is the p44/42-mitogen-activated protein kinase (MAPK) pathway whose activation, with some exceptions, generally results in an increase in the threshold for cell death (reviewed by Dent and Grant, 2001). The survival signals induced by p44/42 activation may in part be communicated through phosphorylation of p90 ribosomal protein S6 kinase (RSK) and then Bad, which in its phosphorylated form cannot translocate to mitochondria and form proapoptotic heterodimers with Bcl-xL (Bonni et al., 1999; Scheid et al., 1999). Anthracycline-based antitumor antibiotics have been described to activate p44/42-MAPK in some cell systems. These include primary rat ventricular myocytes (Adderley and Fitzgerald, 1999; Zhu et al., 1999), neuroblastoma cells (Guise et al., 2001), rat hepatoma cells (Kim et al., 2001), human cervical carcinoma cells (Yeh et al., 2002), and monoblasts (Mas et al., 2003). As a result of the cardiotoxic effects of anthracyclines the consequence of this activation on apoptosis has best been studied in cardiac myocytes, where it seems to be protective since MEK blockade increased cell death (Zhu et al., 1999). In other models, however, p44/42 activation may be proapoptotic, in that MEK blockade reduced apoptosis of SK-N-SH cells (Guise et al., 2001), and in still others such blockade had no impact on programmed cell death (Kim et al., 2001; Yeh et al., 2002). Although anthracyclines are used in a variety of human cancers, one of their more common applications is in the therapy of patients with breast cancer. Signaling by the erbB receptor tyrosine kinase family, which occurs in part through p44/42, has been implicated in the development and progression of breast cancer, and elevated expression of c-erbB-2 (HER-2/neu) and the epidermal growth factor (EGF) receptor (c-erbB-1) is a poor prognostic sign (reviewed by Dickson and Lippman, 1995). Elevated activity of p44/42-MAPK alone, also referred to as the extracellular signal-regulated kinases (ERK-1/2), has been suggested to have prognostic significance for survival of breast cancer patients (Mueller et al., 2000). The impact of anthracyclines on p44/42-MAPK in breast cancer, or the influence of any activity changes on apoptosis, however, has not been well studied.
The mechanism by which anthracyclines activate p44/42 may involve generation of oxygen free radicals, in that scavengers such as dimethyl sulfoxide, catalase, and N-(2-mercaptopropionyl)-glycine suppressed phospho-ERK levels (Zhu et al., 1999). Once generated, these radicals seem then to activate the Ras-Raf-MEK-MAPK pathway, since dominant negative Ras (Zhu et al., 1999) and Raf-1 constructs (Zhu et al., 1999; Mas et al., 2003), as well as pharmacologic inhibitors of Raf (Mas et al., 2003) and MEK (Zhu et al., 1999; Yeh et al., 2001; Mas et al., 2003) suppressed ERK activation. Raf-1 stimulation may also be mediated through phosphatidylcholine-derived diacylglycerol production and phosphoinositide-3 kinase stimulation products that converge toward protein kinase C zeta (Mas et al., 2003). Since free-radical scavengers do not completely block ERK activation, however (Mas et al., 2003), the possibility that other mechanisms contribute needs to be considered.
In the current report we present data demonstrating that anthracycline treatment of breast epithelial and breast carcinoma cells resulted in a time- and concentration-dependent activation of signaling through ERK-1/2. This increase appeared to be antiapoptotic, in that stimulation of p44/42 signaling in combination with anthracyclines protected cells from apoptosis. Although modest activation of MEK by anthracyclines was observed in some experiments, others did not demonstrate this finding, and MEK inhibition did not abolish p44/42 activation. Anthracyclines were found to decrease expression of the MAPK phosphatase MKP-1 at both the protein and mRNA levels, and to specifically repress transcription from the MKP-1 promoter. Cells in which MKP-1 expression was decreased through the use of small interfering (si) RNAs, and cells derived from MKP-1 knockout mice, had blunted activation of p44/42 in response to doxorubicin. These studies support the hypothesis that anthracyclines activate the p44/42-MAPK pathway in breast epithelial and carcinoma cells, that this activation may inhibit their own antitumor activity, and that this occurs in part through repression of transcription of MKP-1.
Materials and Methods
Materials. Doxorubicin was from Sigma-Aldrich (St. Louis, MO); epirubicin (Ellence; Pharmacia and Upjohn, Peapack, NJ) was from the University of North Carolina at Chapel Hill Memorial Hospital Pharmacy; and pegylated liposomal doxorubicin (Doxil) was from Ortho Biotech Products, L.P. (Bridgewater, NJ). Phosphatase inhibitors deltamethrin, nodularin, and okadaic acid were from Calbiochem (San Diego, CA), as was the MEK inhibitor 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD 98059), while sodium orthovanadate was from Sigma-Aldrich. The protease cocktail Complete was from Roche Applied Science (Indianapolis, IN), and phenylmethylsulfonyl fluoride was from Fisher Scientific Co. (Fair Lawn, NJ). Stock solutions were prepared in isopropanol for phenylmethylsulfonyl fluoride, Dulbecco's PBS (from the Lineberger Comprehensive Cancer Center Tissue Culture Facility (LCCC TCF, Chapel Hill, NC)) for sodium orthovanadate, or dimethyl sulfoxide for all others, and stored at –20°C. These reagents were used at concentrations indicated in the text, with a final vehicle concentration that did not exceed 0.5% (v/v). All other chemicals, unless otherwise indicated, were obtained from Fisher Scientific Co.
Cell Lines and Cell Culture. A1N4-myc cells were grown in Richter's modified Eagle's medium (MEM) with insulin, gentamicin, and 20 mM Hepes (LCCC TCF), and further supplemented with 10 ng/ml human recombinant EGF, 9% fetal bovine serum, 100 units/ml penicillin G sodium, 100 μg/ml streptomycin sulfate (these reagents from Invitrogen, Carlsbad, CA), and hydrocortisone (Sigma-Aldrich). MDA-MB-231 and BT-474 human breast carcinoma cells (LCCC TCF) were grown in Richter's MEM as above except without EGF and hydrocortisone. Mouse embryo fibroblasts from heterozygous and homozygous MKP-1 knockout mice (Dorfman et al., 1996), as well as wild-type controls, generously provided by Dr. Naomi Laing and the Bristol Myers Squibb Research Institute (Princeton, NJ), were cultured in Dulbecco's MEM. All cells were propagated in incubators providing a humidified atmosphere with 5% CO2 in air.
Western Blotting. Adherent cells were detached by exposure to trypsin (Invitrogen), counted using a hemacytometer (Hausser Scientific; Horsham, PA), and 2 × 106 cells were seeded onto 60 mm Falcon 3002 tissue culture plates (BD Labware; Lincoln Park, NJ) in complete medium. These cells were allowed to reattach overnight and subjected to conditions described in the text, along with the addition of fresh medium. At the completion of an experiment all plates were placed on ice, washed with 5 ml of ice-cold PBS, and collected by scraping into eukaryotic lysis buffer as described previously (Orlowski et al., 2002). Relative protein concentrations of each sample were determined using the BCA protein assay kit (Pierce Chemical, Rockford, IL). Equivalent protein amounts were subjected to Western blotting and immunoreactive bands were detected as described (Orlowski et al., 2002). For repeated analyses of the same filter, blots were stripped for 45 min using Western Re-Probe (Geno Technology, Inc., St. Louis, MO) following the manufacturer's specifications.
Activation status of the p44/42-MAPKs was determined using murine monoclonal antibodies recognizing active, dually phosphorylated p44/42 (Thr202/Tyr204), with rabbit p44/42 antibodies used as loading controls. The activation status of the mitogen-activated protein kinase kinase (MEK) was determined using rabbit polyclonal antibodies recognizing active, dually phosphorylated MEK-1/2 (Ser217/Ser221), with rabbit MEK-1/2 antibodies used as loading controls. p90 RSK phosphorylation status was evaluated using rabbit polyclonal antibodies recognizing active, dually phosphorylated (Thr359/Ser363) RSK, and Bad phosphorylation status was evaluated using a murine monoclonal antibody recognizing phospho-Ser112-Bad (all from Cell Signaling Technology, Inc., Beverly, MA). Total RSK levels were determined by Western blotting with a murine monoclonal anti-RSK antibody (BD Biosciences, San Jose, CA), and Bad levels were evaluated with a rabbit polyclonal anti-Bad antibody (Cell Signaling Technology, Inc.). Expression levels of the MKP-1 phosphatase were determined with the rabbit polyclonal C-19 antibody to MKP-1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Additional loading controls were provided by a rat monoclonal anti-HSC-70 antibody (StressGen Biotechnologies, San Diego, CA).
To quantify protein bands autoradiographs were scanned with an Agfa Duoscan T2500 scanner (Agfa Corp., Ridgefield Park, NJ) into Adobe Photoshop 5.0 (Adobe Systems, Inc., San Jose, CA), and densitometry was performed using NIH Image version 1.61.
Apoptosis Assays. For apoptosis experiments cells were seeded onto Costar 3595 96-well plates (Corning Glassworks, Corning, NY) at a density of 1 × 104 cells/well. Fresh medium was added containing the agents whose impact was being tested, and cells were analyzed for apoptosis using the Cell Death Detection ELISAPLUS kit (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's specifications. The enhancement of apoptosis was calculated in relation to parallel control cells that received vehicle alone, and tabulated in KaleidaGraph version 3.0.1 (Synergy Software, Reading, PA). Mean percentages and standard errors of the mean were then calculated. For experiments with EGF, cells were treated for 18 h with 0.75 μM anthracyclines in the absence or presence of EGF at 15 ng/ml. Dominant positive constructs and controls were treated with 1 μM doxorubicin for 18 h.
Cloning and Cell Line Preparation. The hyperactive allele of ERK-2 (D319N) tagged with hemagglutinin was cloned into the vector pLPCX (BD Biosciences Clontech, Palo Alto, CA) as described (Orlowski et al., 2002). Briefly, to prepare A1N4-myc- and MDA-MB-231-based cell lines, which must be transduced with retroviral-based vectors, the indicated ERK clones were transfected into AmphoPack-293 cells using the CalPhos Mammalian Transfection Kit (both from BD Biosciences Clontech) according to the manufacturer's specifications. Supernatants with retroviral particles were collected and used to infect the target cells in medium containing 8 μM Polybrene. Selection was performed in media containing puromycin (Calbiochem-Novabiochem, San Diego, CA) and screening of clones was performed using a murine anti-HA monoclonal antibody (F7; Santa Cruz Biotechnology, Inc.). Stable cell lines in BT-474 cells containing this construct were prepared by direct transfection using the Gene-PORTER reagent according to the manufacturer's specifications (Gene Therapy Systems, San Diego, CA), followed by selection and screening as described above.
Immunofluorescence. The in vivo impact of anthracycline therapy was evaluated using a BT-474-based xenograft model system developed as described previously (Somasundaram et al., 2002). All experiments were performed under a protocol approved by the Institutional Animal Care and Use Committee. Three mice each were assigned to the two treatment groups and received either an injection of vehicle or a single injection of pegylated, liposomal doxorubicin at 2 mg/kg. Twenty-four hours later the groups were euthanized using guidelines established by the American Veterinary Medical Association's Panel on Euthanasia. Tumors were then excised, and fixed in Tissue-Tek O.C.T. (Sakura Finetek U.S.A., Inc., Torrance, CA) while freezing in liquid nitrogen. For detection of phospho-ERK-1/2 and MKP-1 sections were washed in 6:1 methanol/water, air-dried, and after blocking they were stained using the antibodies identified above. Secondary antibodies consisted of either cy3-fluorescent-conjugated goat anti-mouse antibody or fluorescein-conjugated goat anti-rabbit antibody (Chemicon International, Temecula, CA). Sections were incubated for 15 min, washed in PBS, and mounted with a 4,6-diamidino-2-phenylindole nuclear stain (Vector Laboratories, Burlingame, CA). Slides were visualized using an ultraviolet Zeiss Axioplan fluorescence microscope (Carl Zeiss Inc., Thornwood, NY). Separate photographs were taken with appropriate filters for blue nuclear staining, red phospho-ERK-½ staining, and green MKP-1 staining, overlaid using Adobe Photoshop software, and displayed as a fusion image at 10× magnification.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR). Cell cultures were treated as above for Western blotting, after which total RNA was isolated using RNeasy (QIAGEN, Valencia, CA) according to the manufacturer's specifications. RNA yields were assessed by spectrophotometric analysis, and specific gene expression was examined using the Access RT-PCR System (Promega, Madison, WI). Briefly, first-strand cDNAs were synthesized starting with 10 ng of total RNA from each sample using avian myeloblastosis virus reverse transcriptase at 48°C for 45 min with the appropriate primers. Second-strand cDNA synthesis and amplifications were then performed with Tfl DNA polymerase using an automated thermal cycler (PerkinElmer 480; PerkinElmer Instruments, Norwalk, CT) set to the following parameters: 94°C for 2 min (1 cycle); 94°C for 30 s, 57°C for 2 min, 68°C for 2 min (40 cycles); and 68°C for 7 min (1 cycle). The primers used for MKP-1 were 5′-GCTGTGCAGCAAACAGTCGA-3′ (upstream), 5′-CGATTAGTCCTCATAAGGTA-3′ (downstream) (Kojima et al., 2000); for c-myc 5′-CTGGTGCTCCATGAGGAG-3′ (upstream), 5′-AGGTGATCCAGACTCTGAC-3′ (downstream); and for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 5′-CCATGGAGAAGGCTGGGG-3′ (upstream), 5′-CAAATGTGTCATGGATGACC-3′ (downstream). Samples of the reaction products were then subjected to electrophoresis on a 5% polyacrylamide gel (19:1 acrylamide: bisacrylamide ratio) in 1% Tris-borate-EDTA running buffer at 50 V, visualized by ethidium bromide staining, and images were acquired using the FluorChem 8000 imaging system with Alpha-EaseFC software (Alpha Innotech Corp., San Leandro, CA). Densitometry was performed using NIH Image version 1.61. Statistical analyses of the results were performed using the Wilcoxon signed-rank test. A p value of <0.05 was considered statistically significant.
Luciferase Assays. The pGL3-MKP-1-luc reporter plasmid containing the MKP-1 promoter in a genomic fragment spanning positions –2975 to +247 cloned in front of the firefly luciferase (luc) gene was kindly provided by Dr. Yusen Liu (National Institutes of Health; Baltimore, MD) (Li et al., 2001). pHRL-CMV containing the cytomegalovirus (CMV) immediate early enhancer/promoter cloned in front of the Renilla luciferase gene was from Promega. Stable cell lines containing these reporter vectors were constructed by cotransfection of BT-474 cells with either pGL3-MKP-1-luc and pLPCX (BD Biosciences Clontech), or pHRL-CMV and pLPCX as described above.
Luciferase reporter assays were conducted by seeding these cells at 2 × 104/well into Costar 3595 96-well tissue culture plates, which were then allowed to recover overnight. Following the indicated treatments, cells were harvested using the Promega dual-luciferase reporter assay system according to the manufacturer's specifications. Cell lysates were transferred to an opaque 96-well plate and luciferase activity was measured using an Lmax microplate luminometer with SOFTmax PRO 1.1L software (Molecular Devices Corp., Sunnyvale, CA) set for an injection of 75 μl of detection reagent and a 2-s preread delay, followed by a 20-s signal integration time. MKP-luciferase activity was then normalized against pHRL (CMV-luciferase) activity and expressed relative to an untreated control. Statistical analysis of the results was performed as described above.
Silencing of MKP-1. The human MKP-1 gene was analyzed to identify possible sequences for small interfering (si) RNAs using the siRNA Target Finder and Design Tool (Ambion, Austin, TX). Potential targets were evaluated by BLAST sequence analysis, and for those specific for MKP-1 complementary oligonucleotides with overhanging ApaI and EcoRI restriction sites were synthesized. These were cloned into pSilencer 1.0-U6 (Ambion) that had been linearized with ApaI and EcoRI, and the resulting vectors were cotransfected with pLPCX into BT-474 cells and selected as described above. Clones were screened by Western blotting to identify those that had lowered basal levels of MKP-1 expression, and also those induced to synthesize lower amounts of MKP-1 upon addition of a proteasome inhibitor (Orlowski et al., 2002). Of the analyzed sequences, siRNA constructs targeting the region from nucleotides 106–123 were most active. The corresponding oligonucleotides were 5′-CGCCGGCCACATCGCCGGCG-AGTACTGGCC-GGCGATGTGG-CCGGCGTTTTTT-3′ (sense) and 5′-AATTAAAAAA-CGCCGGCCAC-ATCGCCGGCCAGTACTCGCC-GGCGATGTGG-CCGGCGGGCC-3′ (antisense). As controls, cell lines were prepared that contained pLPCX and pSilencer 1.0-U6 with cloned oligonucleotides that had an identical nucleotide composition to the siRNA construct, but a scrambled sequence (ss) that was not complementary to any known genes.
Results
Anthracyclines Increase Phosphorylation of p44/42-MAPK. To evaluate the impact of anthracyclines on the p44/42-MAPK pathway in breast epithelium-derived cell lines, A1N4-myc human mammary epithelial cells and BT-474 and MDA-MB-231 breast carcinoma cells were exposed to doxorubicin at 5 μM for 4 h. This anthracycline induced an increase in dually phosphorylated (Thr202/Tyr204), activated p44 and p42 in all three cell lines (Fig. 1A). Total levels of p44 and p42 were unaffected under these conditions, as were the levels of another control protein, HSC-70, indicating that doxorubicin indeed changed the phosphorylation status of ERK-1/2. Under similar conditions, the related anthracycline epirubicin also increased phospho-ERK-1/2 levels in these cells (Fig. 1B). The anthracycline-enhanced signaling was seen in the presence of medium containing serum-derived growth factors that stimulate p44/42 (BT-474 and MDA-MB-231 cells), and even in the presence of medium supplemented with both serum and EGF (A1N4-myc cells). Both anthracyclines induced an increase in phospho-p44/42-MAPK levels in a time-dependent fashion (Fig. 1C for MDA-MB-231 cells and doxorubicin; others not shown), with the first evidence of an increase seen at 2 h. At 4 h phospho-ERK-1/2 levels reached their maximum, when they were increased by up to 5.9-fold compared with vehicle controls. Subsequently, the magnitude of the increase declined somewhat, but phospho-p44/42 levels remained elevated throughout the time course examined. Doxorubicin and epirubicin also increased phospho-ERK-1/2 levels in a concentration-dependent fashion (Fig. 1D for MDA-MB-231 cells and epirubicin; doxorubicin not shown), beginning at 0.5 μM. Further titration of the anthracycline concentration generally led to a plateau, with the maximal effect being noted in the 2- to 5-μM range.
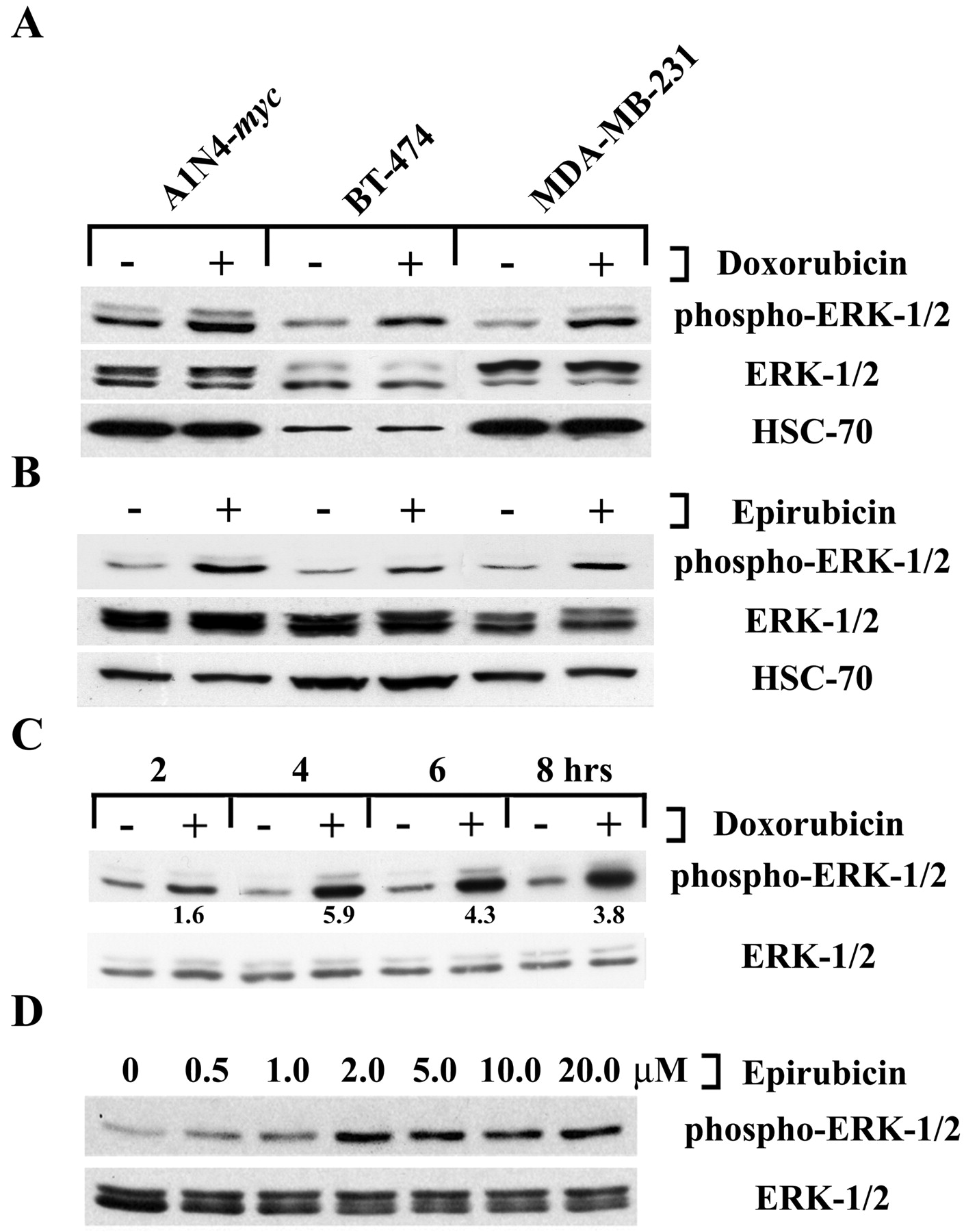
Phospho-p44/42-MAPK in cells exposed to anthracyclines. A, A1N4-myc, BT-474, or MDA-MB-231 cells were exposed to vehicle (–) or to 5 μM doxorubicin (+), in complete medium containing fetal calf serum (all cell lines) or serum supplemented with EGF (A1N4-myc cells) for 4 h. The impact on p44/42-MAPK signaling was evaluated by Western blotting using antibodies directed against the dually phosphorylated (Thr202/Tyr204) activated forms of ERK-1 and -2. Equal loading was confirmed by reprobing with antibodies that recognized ERK in a phosphorylation status-independent manner, and then with antibodies directed against HSC-70. All of the panels in this figure depict one representative result of two independent experiments. B, A1N4-myc, BT-474, or MDA-MB-231 cells were exposed to vehicle or epirubicin at 5 μM for 4 h and analyzed as in panel A above. C, MDA-MB-231 cells were exposed either to vehicle or to doxorubicin at 5 μM for the indicated time periods. The -fold increase in phospho-MAPK abundance is shown in relation to vehicle-treated cells, and is adjusted for loading of ERK. D, MDA-MB-231 cells were exposed to the indicated concentrations of epirubicin for 4 h, and phospho-ERK-1/2 levels were analyzed.
Induction of Phospho-p44/42 Is Associated with MAPK Activation. To determine whether the increased levels of phospho-ERK-1/2 were an indication of enhanced activity of the p44/42-MAPK pathway, the phosphorylation status of p90 RSK was evaluated. Activation of the downstream target p90 RSK occurs by ERK-mediated phosphorylation at residues Thr359 and Ser363 (Dalby et al., 1998). Treatment of A1N4-myc, BT-474, and MDA-MB-231 cells with doxorubicin (Fig. 2A) or epirubicin (Fig. 2B) resulted in increased levels of dually phosphorylated (Thr359/Ser363) RSK-1 in all three cell lines compared with controls that received fresh medium containing serum and vehicle only. Since activated RSK phosphorylates the downstream target Bad at Ser112 (Bonni et al., 1999), this protein's phosphorylation status was then studied. As for p44/42 and RSK-1, phospho-Ser112-Bad levels increased in all three cell lines after treatment with doxorubicin (Fig. 2A) or epirubicin (Fig. 2B). RSK and Bad phosphorylation was enhanced without any changes in either total ERK-1/2 (Fig. 1, A and B) or in total RSK, Bad, or HSC-70 levels (Fig. 2, A and B). These effects of doxorubicin occurred in a time-dependent manner, but were somewhat delayed compared with that for phospho-p44/42-MAPK. Although ERK phosphorylation peaked at 4 h (Fig. 1C), the increase in phospho-RSK, which peaked at an up to 4.9-fold increase, occurred at 6 h (Fig. 2C), and for phospho-Bad this occurred at 8 h, when Bad phosphorylation was increased by up to 2.7-fold (Fig. 2C). Phospho-RSK and phospho-Bad also increased in a manner that was dependent on the concentration of doxorubicin (not shown). These results support the conclusion that anthracyclines enhanced not only the phosphorylation of ERK-1/2, but also the activity of this pathway, as evidenced by increased levels of the phosphorylated forms of downstream target proteins.

Phospho-RSK and -Bad in cells exposed to anthracyclines. A, A1N4-myc, BT-474, or MDA-MB-231 cells were exposed to vehicle (–) or to doxorubicin (+) at 5 μM for 4 h. The impact on phospho-RSK (Thr359/Ser363) and phospho-Bad (Ser112) levels was evaluated by Western blotting using antibodies directed against the indicated phosphoproteins. Equal loading was confirmed by reprobing with RSK and Bad antibodies that recognize these proteins in a phosphorylation state-independent manner, and also with HSC-70 antibodies. All of the panels in this figure depict one representative result of two independent experiments. B, A1N4-myc, BT-474, or MDA-MB-231 cells were exposed to vehicle (–) or to epirubicin (+) at 5 μM for 4 h, and analyzed as in panel A above. C, MDA-MB-231 cells were exposed either to vehicle or to doxorubicin at 5 μM for the indicated time periods. The impact on phospho-RSK (Thr359/Ser363) and phospho-Bad (Ser112) was evaluated as above. For phospho-RSK and phospho-Bad, the fold increase in phosphorylated proteins is shown in relation to vehicle-treated cells, and is adjusted for loading of HSC-70.
MAPK Activation Is Antiapoptotic. The p44/42-MAPK pathway plays a role in apoptosis, and its activation generally results in an increase in the threshold for cell death (Dent and Grant, 2001). It was therefore of interest to examine the possibility that the anthracycline-induced increase in phospho-ERK-1/2 was antiapoptotic in breast-derived epithelial cells. To evaluate the impact of stimulation of p44/42-MAPK, cells were treated with anthracycline either in the presence of EGF, which activated ERK-1/2 phosphorylation (not shown), or in its absence. For all three cell lines both anthracyclines induced less apoptosis in the presence of EGF. In the case of epirubicin, for example, EGF reduced the induction of apoptosis by 40% in A1N4-myc and BT-474 cells, and by 54% in MDA-MB-231 cells (Table 1). EGF can also activate other antiapoptotic pathways in some breast cancer cells, however, such as the protein kinase B/Akt serine/threonine kinase (Mao et al., 2000). Cell lines expressing the dominant positive D319N ERK-2 sevenmaker mutant were therefore prepared and treated with vehicle or anthracycline. Expression of this dominant-positive allele in A1N4-myc cells inhibited apoptosis by 62% when these were treated with epirubicin compared with vector-containing controls, and by 39% and 45% in BT-474 and MDA-MB-231 cells, respectively (Table 1). This finding supports the hypothesis that activation of signaling through p44/42-MAPK by anthracyclines is antiapoptotic.
Stimulation of p44/42-MAPK and apoptosis*
Anthracyclines Decrease Expression of MKP-1. Activation of the p44/42-MAPK pathway by anthracyclines has previously been described to occur as a result of effects on the upstream kinases, which can be blocked by inhibitors of the MAPK kinase MEK. Evaluation of A1N4-myc, BT-474, and MDA-MB-231 cells did occasionally reveal a modest increase in dually phosphorylated (Ser217/Ser221) MEK, but this was not consistently seen either at 4 h (Fig. 3, A and B) or in time course experiments (not shown), suggesting a contribution from another mechanism. To further investigate a possible role for upstream events, cells were preincubated with the MEK inhibitor PD 98059 at 25 and 100 μM for 1 h (Fig. 3C). Although this agent decreased overall p44/42-MAPK phosphorylation, subsequent treatment with doxorubicin still resulted in higher levels of phospho-ERK-1/2 compared with controls. Experiments with the structurally unrelated MEK inhibitor 1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenyl mercapto)butadiene (U0126) yielded comparable results (not shown). Cellular MAPK activity is also modulated through the action of protein phosphatases, including both phosphoprotein phosphatase-2A (PP2A) and -1 (PP1) (English et al., 1999; reviewed by Camps et al., 2000). To determine whether these might be involved, cells were preincubated either with okadaic acid alone at concentrations that would inactivate both PP2A and PP1, or okadaic acid in combination with PD 98059 (Fig. 3D). Doxorubicin was able to enhance phospho-ERK-1/2 levels under all of these conditions, suggesting that inhibition of either PP1, PP2A, or both played no role in this process.
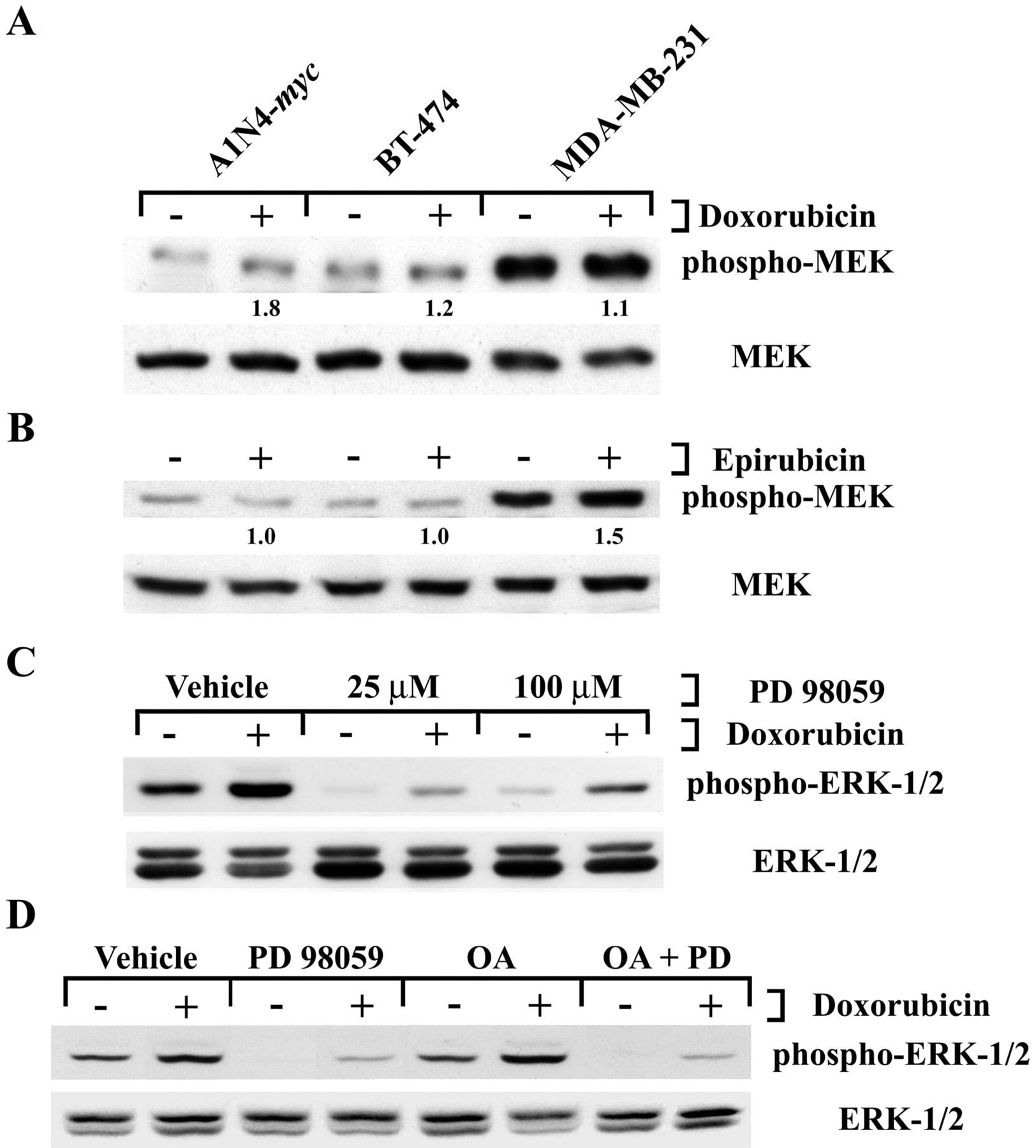
Effects of anthracyclines on factors upstream of ERK. A, A1N4-myc, BT-474, or MDA-MB-231 cells were exposed to vehicle (–) or to doxorubicin (+) at 5 μM for 4 h. The impact on phospho-MEK was evaluated by Western blotting using antibodies directed against the dually phosphorylated (Ser217/Ser221) protein. Equal loading was confirmed by reprobing with antibodies that recognize MEK in a phosphorylation status-independent manner. The -fold increase in phospho-MEK abundance is shown in relation to vehicle-treated cells, and is adjusted for loading of MEK. All of the panels in this figure depict one representative result of two independent experiments. B, A1N4-myc, BT-474, or MDA-MB-231 cells were exposed to vehicle (–) or to epirubicin (+) at 5 μM for 4 h, and the impact on phospho-MEK was evaluated as in panel A above. C, MDA-MB-231 cells were pretreated with vehicle or PD 98059 at either 25 or 100 μM for 1 h. They were then treated either with vehicle or doxorubicin at 5 μM for 4 h. The impact on p44/42-MAPK signaling was evaluated by Western blotting. D, MDA-MB-231 cells were preincubated with vehicle, 25 μM PD 98059, 30 μM okadaic acid (OA), or both (OA + PD) for 1 h. They were then treated either with vehicle or doxorubicin at 5 μM for 4 h, and the phospho-ERK-1/2 content was analyzed by Western blotting.
Another group of phosphatases that have an impact upon p44/42-MAPK activity are the dual specificity phosphatases (English et al., 1999; Camps et al., 2000). Since MKP-1 has been reported to be overexpressed in a high proportion of breast carcinomas (Loda et al., 1996), the impact of anthracyclines on this phosphatase was evaluated. In A1N4-myc, BT-474, and MDA-MB-231 cells treated with doxorubicin, absolute MKP-1 levels declined sharply (Fig. 4A). This occurred in a manner that was dependent on both the anthracycline concentration (not shown) and time (Fig. 4B). In regard to the latter, decreased levels of MKP-1 were seen as early as 2 h, a time at which p44/42 phosphorylation was just beginning to increase (Fig. 1C).

MKP expression in cells treated with anthracyclines. A, A1N4-myc, BT-474, or MDA-MB-231 cells were exposed to vehicle (–)orto5 μM doxorubicin (+) for 4 h. The impact on MKP-1 expression was assayed by Western blotting, and loading was confirmed by stripping and reprobing with HSC-70 antibodies. In addition to MKP-1, which is identified by the arrowhead, the polyclonal antibody used also detects a second, larger-molecular-weight nonspecific protein in MDA-MB-231 cells, which is indicated with an asterisk (★). All panels in this figure are representative results from one of two independent experiments. B, MDA-MB-231 cells were exposed to vehicle or to 5 μM doxorubicin for the indicated time periods, and the impact on MKP-1 expression was analyzed by Western blotting. MKP-1 and the cross-reacting protein detected in MDA-MB-231 cells are labeled as indicated above.
The anthracycline concentrations used in these studies were representative of peak plasma concentrations achieved in patients undergoing chemotherapy (Greene et al., 1983), suggesting that such effects could also occur in vivo. To further evaluate this possibility, a murine xenograft model of human breast carcinoma using BT-474 cells was used, and tumor-bearing mice were treated with a single dose of liposomal doxorubicin. Immunofluorescence studies of tumor tissue harvested 24 h later showed that the abundance of phospho-ERK-1/2 was increased by this anthracycline (Fig. 5B) when compared with vehicle-treated controls (Fig. 5A). In contrast, while MKP-1 protein was detectable in control xenograft tissue (Fig. 5C), after treatment with doxorubicin its expression levels were decreased (Fig. 5D). These results support the hypothesis that the ability of anthracyclines to repress MKP-1 and enhance phospho-ERK-1/2 is seen both in vitro and in vivo.

Effect of anthracyclines on in vivo phospho-ERK and MKP-1 expression. A, immunodeficient nu/nu mice bearing tumor xenografts induced by subcutaneous injection of BT-474 breast carcinoma cells were treated with vehicle. Twenty-four hours later tumor tissue was obtained and evaluated for p44/42-MAPK activation status by immunofluorescence. Phospho-ERK-1/2 appears as red fluorescence, and the background blue is a 4,6-diamidino-2-phenylindole nuclear stain. All panels in this figure are representative results from one of two experiments. B, immunodeficient nu/nu mice bearing the BT-474-based xenograft were treated with liposomal doxorubicin (Doxil), and tumor tissue was analyzed for p44/42-MAPK activation status as indicated above. C, expression of MKP-1 in tumor tissue from vehicle-treated mice was analyzed by immunofluorescence. MKP-1 was detected as green fluorescence. D, MKP-1 expression in tumor tissue from doxorubicin-treated mice was analyzed as indicated above.
Anthracyclines Decrease MKP-1 Transcription. The ability of anthracyclines such as doxorubicin to specifically decrease expression of genes, including α-actin, myosin light chain, and muscle creatine kinase in cardiac and skeletal muscles (Ito et al., 1990), suggested the possibility that there might be an impact on MKP-1 expression at a transcriptional level. When RT-PCR was used to evaluate the levels of MKP-1 mRNA after treatment with doxorubicin in a semiquantitative fashion, the abundance of these transcripts was indeed decreased in all three cell lines when compared with GAPDH (Fig. 6A). By densitometry, messenger levels were decreased in A1N4-myc, BT-474, and MDA-MB-231 cells to 80 ± 9, 65 ± 5, and 72 ± 10% of baseline, respectively (p = 0.03) (Fig. 6B). This occurred in parallel with a decreased abundance of c-myc mRNA, which is known to be repressed after doxorubicin treatment. To evaluate the impact on MKP-1 mRNA in a more quantitative fashion, and to determine whether decreased transcription from the MKP-1 promoter contributed to the decline in MKP-1 mRNA, cell lines harboring an MKP-1 promoter-luc reporter construct were treated with doxorubicin or epirubicin. Neither anthracycline induced a decrease in luciferase activity from a control CMV-luc vector, but both repressed absolute (not shown) and relative luciferase activity from the MKP-1-luc construct (Fig. 6C). With respect to the latter, doxorubicin decreased relative activity to 62 ± 16% of baseline (p = 0.02), whereas epirubicin did so to 53 ± 15% of baseline (p = 0.03).

Impact of anthracyclines on MKP-1 mRNA abundance and promoter function. A, A1N4-myc, BT-474, or MDA-MB-231 cells were exposed to vehicle (–), or to 5 μM doxorubicin (+) for 4 h. Total RNA was isolated, and MKP-1, c-myc, and GAPDH messenger RNA expression was evaluated by semiquantitative RT-PCR, with products separated by nondenaturing PAGE. This panel shows a representative result from one of three independent experiments. B, the abundance of MKP-1 and c-myc mRNA detected as described above was determined by densitometry, and is expressed in relation to GAPDH, which was arbitrarily set at 100. The results are depicted as the mean ± standard error of the mean from three independent experiments. C, BT-474 cells stably transfected, either with a plasmid containing an MKP-1 promoter-luciferase reporter construct or a control CMV promoter-luciferase reporter construct, were treated with vehicle, doxorubicin, or epirubicin, with the latter two at 5 μM for 8 h. Luciferase activity was determined for each condition and expressed as a relative reporter activity to vehicle, which was arbitrarily set at 100. MKP-1-luc was then normalized to CMV-luc, and the mean of this ratio is depicted in the graph, along with the standard error of the mean from four independent experiments.
Down-Regulation of MKP-1 Blunts Activation of p44/42-MAPK. To more directly evaluate the role of MKP-1 repression in anthracycline-mediated activation of p44/42 MAPK, cell lines were constructed that stably expressed the Q3 small interfering RNA directed against MKP-1. siRNA-expressing cells had lower basal levels of MKP-1 expression compared with cells expressing a control scrambled sequence (ss) small interfering RNA. In addition, when exposed to proteasome inhibitors that induce MKP-1 (Orlowski et al., 2002), the levels of MKP-1 expressed were lower in the siRNA-expressing cells (not shown). BT-474/Q3 ssRNA control cells treated with doxorubicin had higher levels of phospho-ERK-1/2 (Fig. 7A), as had been the case for parental BT-474 cells (Fig. 1A). However, BT-474/Q3 siRNA cells had lower levels of phospho-p44/42 MAPK after treatment with doxorubicin. As an additional test of this hypothesis, mouse embryo fibroblasts derived from MKP-1 knockout mice (Dorfman et al., 1996) were examined and compared with control MEFs. After treatment with doxorubicin the wild-type MKP-1 +/+ MEFs had increased levels of phospho-p44/42 (Fig. 7B). Activation of p44/42 MAPK in MKP-1 heterozygote (+/–) knockout cells was decreased compared with wild-type, however, whereas activation in homozygous (–/–) MKP-1 knockout cells was almost undetectable (Fig. 7B). Since cells with decreased (Fig. 7A) or absent (Fig. 7B) MKP-1 had blunted levels of phospho-ERK-1/2 after doxorubicin treatment, these findings support the hypothesis that anthracyclines activate p44/42 in part by repressing MKP-1.

Impact of decreased MKP-1 expression on activation of p44/42 by anthracyclines. A, BT-474-based cell lines stably expressing either the Q3 siRNA to MKP-1 or an RNA with the same nucleotide composition but a scrambled sequence were treated with vehicle or 5 μM doxorubicin for 4 h, and phospho-ERK levels were evaluated by Western blotting. Total ERK levels are shown as loading controls. Each panel in this figure shows representative results from one of two independent experiments. B, wild-type (MKP-1 +/+) MEFs, as well as heterozygote knockout (MKP-1 +/–) and homozygote knockout (MKP-1 –/–) MEFs, were treated either with vehicle or 5 μM doxorubicin for 4 h, and phospho-ERK levels were evaluated by Western blotting. The genotype of these cells was confirmed by Southern blotting (data not shown).
Discussion
Some of the more ubiquitous cellular signal transduction cascades are the MAPK pathways, including p44/42-MAPK, which play important roles in cell development, growth, differentiation, and apoptosis (Dent et al., 1998; Schaeffer and Weber, 1999; Cross et al., 2000). With respect to the latter, activation of ERK-1/2 generally results in an increase in the threshold for cell death (Dent and Grant, 2001). The anthracyclines are chemotherapeutics that have been described in some models to activate p44/42-MAPK, including in primary rat ventricular myocytes (Adderley and Fitzgerald, 1999; Zhu et al., 1999), SK-N-SH neuroblastoma cells (Guise et al., 2001), H4IIE rat hepatoma cells (Kim et al., 2001), SiHa human cervical carcinoma cells (Yeh et al., 2002), and U937 monoblasts (Mas et al., 2003). In other cells, however, including the H9 human T-cell leukemia line (Yu et al., 1996) and human KB-3 human epidermal carcinoma cells (Stone and Chambers, 2000), ERK was not activated. Such activation is of interest in that it may inhibit the ability of anthracyclines to induce apoptosis, and we therefore sought to examine the effects of doxorubicin and epirubicin on breast epithelium-derived and carcinoma cell lines, where this has not been well studied. Both anthracyclines were noted to increase the levels of phospho-ERK-1/2 in a time- and concentration-dependent manner (Fig. 1). This was associated with enhanced activity of the p44/42-MAPK pathway in that there was increased phosphorylation of the direct downstream MAPK target p90 RSK-1, and also of Bad (Fig. 2), on whose phosphorylation RSK has an impact. Since Bad phosphorylation at Ser112 is antiapoptotic through its inability to associate with Bcl-xL, the possibility was considered that activation of p44/42 provided the breast-derived cells with survival signals. Consistent with this possibility, stimulation of p44/42 with EGF or a constitutively active ERK-2 construct inhibited apoptosis (Table 1).
Although anthracyclines are active agents in the treatment of breast cancer, many patients do not have complete responses, whereas others suffer relapses after chemotherapy. Novel combination regimens that would enhance the ability of anthracyclines to induce apoptosis of cancer cells could, therefore, improve the prognosis of breast cancer patients. The finding that stimulation of the p44/42 pathway decreases anthracycline-mediated apoptosis suggests that inhibition of p44/42, such as by targeting MEK, may enhance doxorubicin- and epirubicin-mediated programmed cell death. Indeed, preliminary investigations have shown that PD 98059 and U0126 can enhance doxorubicin- and epirubicin-mediated apoptosis in these three cell lines (not shown). One important consideration would be the known potential cardiac toxicity of anthracyclines, which often limits their clinical applications. Since blockade of MEK in primary rat ventricular myocytes enhances apoptosis (Zhu et al., 1999), while an MEK inhibitor/anthracycline combination could have enhanced antitumor activity, it might also increase myofibrillar damage. In this regard it is interesting to note that combination regimens containing anthracyclines and trastuzumab, an anti-HER-2/neu monoclonal antibody that in part decreases activation of p44/42 through upstream effects, have been reported to have enhanced cardiac toxicity (reviewed by Sparano, 2001).
Activation of p44/42-MAPK by anthracyclines has been attributed to the induction of oxygen free radicals, which then stimulate upstream kinases including Ras, Raf, and MEK. Although we could demonstrate modest MEK activation, this was not a consistent finding (Fig. 3), suggesting that another mechanism could be involved. The ability of anthracyclines to decrease MKP-1 protein levels in vitro and in vivo (Figs. 4 and 5), and MKP-1 mRNA and promoter function (Fig. 6), implicated this activity as a possibility. Additional findings that cells with decreased MKP-1 due to expression of siRNAs, and fibroblasts from MKP-1 knockout mice treated with anthracyclines, had lower levels of phospho-ERK (Fig. 7) strongly support the hypothesis that down-regulation of MKP-1 by anthracyclines is a significant contributor to preservation of phospho-p44/42 levels. Our results do not rule out the possibility, however, that other mechanisms may have an impact on this process as well. For example, anthracyclines could stimulate proteasome-mediated degradation of MKP-1, which is known to be subject to proteolysis through this pathway (Brondello et al., 1999; Orlowski et al., 2002). In that anthracyclines such as aclarubicin seem to inhibit the proteasome (Isoe et al., 1992) this may seem less likely. However, in myelomonocytic leukemia cells anthracyclines stimulate proteasome activity (Ciftci et al., 2001), and this possible mechanism may therefore be worthy of further study. Also, since MKP-1 is one of a family of dual specificity phosphatases involved in the control of p44/42 activity, the possibility that anthracyclines may also have an impact on the expression of other family members would merit consideration. In this regard we have found evidence in preliminary studies that MKP-2 expression is decreased by doxorubicin as well (not shown). It is interesting to note that, although the impact of other agents on both MKP-1 and ERK-1/2 has not been widely studied, proteasome inhibitors have the opposite effect of anthracyclines in that they transcriptionally induce MKP-1 and decrease ERK-1/2 activity (Orlowski et al., 2002). Thus, repression of MKP-1 resulting in increased phosphorylation of p44/42-MAPK may be a mechanism unique to anthracyclines.
The ability of anthracyclines to specifically have an impact upon gene expression is a well known property of this class of compounds. Acute doxorubicin toxicity is associated with changes in cardiac-specific gene expression, with suppression of muscle-specific genes including α-actin, myosin light chain, and muscle creatine kinase (Ito et al., 1990; Torti et al., 1998), as well as suppression of the mitochondrial iron sulfur protein, phosphofructokinase, and ATP/ADP translocase (Jeyaseelan et al., 1997b). Several studies have documented that transcription of the gene CARP is repressed by doxorubicin in neonatal rat cardiac myocytes (Jeyaseelan et al., 1997a). Mechanistic investigations revealed that this down-regulation is not due to p44/42-MAPK activation, but rather seems to depend on an H7-sensitive serine/threonine kinase (Aihara et al., 2000). Doxorubicin has also been shown to inhibit ventricular myocyte sarco(endo)plasmic reticulum Ca2+-ATPase 2 by stimulating expression of the transcription factor Egr-1 (Arai et al., 2000). One of these pathways may be involved in doxorubicin's suppression of MKP-1 transcription, although we were unable to document enhanced Egr-1 transcription by RT-PCR (not shown). Additional studies will be needed to further delineate the mechanism by which anthracyclines specifically repress MKP-1.
Acknowledgments
We acknowledge the generosity of Dr. Yusen Liu for the gift of the pGL3-MKP-1-luc reporter plasmid, and Dr. Naomi M. Laing and the Bristol Myers Squibb Research Institute for providing MKP-1 knockout MEFs.
Footnotes
-
This work was supported in part by Department of Defense Breast Cancer Research Program Grant BC991049, the Leukemia and Lymphoma Society Grant R6206-02, and the Ellence Research Fund.
-
DOI: 10.1124/jpet.103.055806.
-
ABBREVIATIONS: MAPK, mitogen-activated protein kinase; RSK, ribosomal protein S6 kinase; MEK, mitogen-activated protein kinase kinase; HSC, heat shock cognate protein; EGF, epidermal growth factor; ERK, extracellular signal-regulated kinase; siRNA, small interfering RNA; MKP, mitogen-activated protein kinase phosphatase; PBS, phosphate-buffered saline; MEM, modified Eagle's medium; LCCC TCF, Lineberger Comprehensive Cancer Center Tissue Culture Facility; RT-PCR, reverse transcription-polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CMV, cytomegalovirus; luc, luciferase; PP1, phosphoprotein phosphatase-1; PP2A, phosphoprotein phosphatase-2A; MEFs, mouse embryo fibroblasts.
-
↵1 Current address: University of Houston-Victoria, School of Arts and Science, 3007 N. Ben Wilson, Victoria, TX 77901-5731.
- Received June 16, 2003.
- Accepted August 28, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}