


Abstract
It is well known that the analgesic potency of morphine is reduced in neuropathic pain. In this study, we demonstrate that the decreased effectiveness of systemic morphine in neuropathic pain might be caused by the loss of morphine analgesia at the periphery. When given s.c. or i.t., the dose-response curves for morphine analgesia in Hargreaves thermal test were shifted rightward in partial sciatic nerve-injured mice compared with control sham-operated mice. The dose-response curves for i.c.v. morphine analgesia, however, were unchanged in nerve-injured mice, indicating no decrease in morphine potency at the supraspinal level. On the other hand, the dose-dependent analgesia produced by intraplantar (i.pl.) morphine in sham-operated mice almost completely disappeared in nerve-injured mice. With the more sensitive algogenic-induced nociceptive flexion test, significant reduction in the analgesic potency of systemic morphine was observed for bradykinin (BK) nociception in nerve-injured mice, and the analgesic effect of i.pl. morphine against BK nociception in sham-operated mice disappeared in nerve-injured mice. In immunohistochemical experiments, we found that, under normal state, μ-opioid receptors (MOPs) were mainly expressed in small-diameter unmyelinated dorsal root ganglion (DRG) neurons and colocalized with bradykinin B2 receptors. When we examined MOP expression in the DRG of nerve-injured mice, we observed a drastic decrease in MOP expression. Altogether, these data suggest that the lower potency of systemic morphine in neuropathic pain could be at least partly caused by the decreased MOP expression in DRG and subsequent loss of peripheral morphine analgesia in such a condition.
In addition to the supraspinal and spinal localization, opioid receptors have been found in peripheral nerves, and peripherally injected opiates produce potent analgesia in acute pain models (Coggeshall et al., 1997; Dionne et al., 2001; Stein et al., 2001). Peripheral morphine is reported to have potent antinociceptive effect under inflammatory conditions (Stein et al., 1988). Moreover, the peripheral opioid receptors contribute to the analgesic effects of systemically administered opiates (Kayser et al., 1991). We have also recently demonstrated the efficacy of peripherally injected morphine to block different peripheral chemical stimuli-mediated nociceptive pain in naive mice by using the novel technique of the peripheral algogenic-induced nociceptive flexion (ANF) test (Ueda et al., 2000).
The opioid compounds, including morphine, have been used for centuries to combat many extremely painful conditions. However, they are reported to have suboptimal therapeutic efficacy against neuropathic pain (Arnér and Meyerson 1988; Kupers et al., 1991; Bleeker et al., 2001). Analgesic potency of both systemic and intrathecal morphine is greatly reduced in animal models of neuropathic pain (Ossipov et al., 1995; Idanpaan-Heikkila et al., 1997). Until now, most studies examined the reasons behind decreased effectiveness of spinal morphine in neuropathic pain, which include reduction in μ-opioid receptor (MOP) expression in the spinal dorsal horn (Porreca et al., 1998), enhanced spinal release of dynorphin A (Nichols et al., 1997) and cholecystokinin (Nichols et al., 1996; Zhang et al., 2000), increased expression of spinal metabotropic glutamate receptor 1 (Fundytus et al., 2001), and activation of tonic descending facilitation pathways from the brain (Vanderah et al., 2001); however, very few studies examined the peripheral changes that may contribute to the reduction in systemic morphine potency in neuropathic pain. Recently, Pertovaara and Wei (2001) reported that peripheral morphine had more analgesic effect in the neuropathic paw than in the contralateral paw in the rat; however, it is well known that MOP expression is greatly decreased in the dorsal root ganglion neurons after peripheral nerve injury (Li et al., 1996; Zhang et al., 1998). This decrease raises the question of how peripheral morphine affects neuropathy states. In the present study, we applied a systematic approach to examine the effects of morphine in different central and peripheral sites in a model of neuropathic pain in mice. We administered morphine through various routes (s.c., i.t., i.c.v., and i.pl.) in control shamoperated and partial sciatic nerve-injured mice and measured the thermal paw withdrawal responses. We also utilized the ANF test to assess the morphine analgesia after peripheral nerve injury. The ANF test was found to be more sensitive than conventional nociception tests (Inoue et al., 2003). Moreover, the use of a single pain-producing ligand such as bradykinin in the ANF test would be helpful in identifying the immunohistochemical colocalization between that pain-producing ligand receptor and the receptor for the applied analgesics. Finally, we identified the expression of MOP in DRG neurons after peripheral nerve injury by an immunohistochemical technique to confirm our behavioral results.
Materials and Methods
Experimental Animals. Male ddY mice weighing 20-30 g were used throughout the experiments. The mice were housed in a room maintained at 21 ± 2°C, 55 ± 5% relative humidity, and an automatic 12-h light/dark cycle with free access to standard laboratory diet and tap water. The animals were adapted to the testing environment (maintained at 21 ± 2°C, 55 ± 5% relative humidity, and 12-h light/dark cycle) by keeping them in the testing room 24 h before the experiments. Experiments were performed during the light phase of the cycle (10:00 AM to 5:00 PM). All procedures used in the present study were approved by the Nagasaki University Animal Care Committee and complied with the ethical guidelines of the International Association for the Study of Pain (Zimmermann, 1983).
Drug Administration. Morphine hydrochloride (Takeda Chemical Industries, Osaka, Japan) and bradykinin (Sigma-Aldrich, St. Louis, MO) were dissolved in physiological saline, which was used for control injections. Intraplantar injections were given using a Hamilton microsyringe connected to polyethylene tubing with a 30-gauge hypodermic needle at the tip. The volume of injection was 20 μl in thermal paw withdrawal tests and 2 μl in algogenic-induced nociceptive flexion tests. The i.t. injections were performed freehand between spinal L5 and L6 segments according to the method of Hylden and Wilcox (1980). The exact placement of the drug substances was checked by a quick flicking motion of the mouse's tail upon entry of needle. The i.c.v. injections were carried out into the left lateral ventricle of mice. Injections were performed using a Hamilton microsyringe fitted with a 26-gauge i.c.v. needle according to the method of Haley and McCormick (1957). The site of injection was 2-mm caudal and 2-mm lateral to the bregma and 3-mm in depth from the skull surface. Both i.t. and i.c.v. injections were given in a volume of 5 μl in unanesthetized conscious animals. The mice received the s.c. injections in a volume of 0.1 ml/10 g of body weight.
Partial Ligation of Sciatic Nerve. Partial ligation of the sciatic nerve of mice was performed under pentobarbital anesthesia (50 mg/kg i.p.) following the methods of Malmberg and Basbaum (1998). Briefly, the common sciatic nerve of the right hind limb of mice was exposed at high thigh level through a small incision and dorsal 1/3 to 1/2 of the nerve thickness was tightly ligated with a silk suture. The wound was closed with a single muscle suture, and antibiotic powder was dusted over the wound area following surgery. Sham operation was performed similarly, except that the sciatic nerve was not touched. Immediately following surgery, the animals were kept in a soft bed cage with some food inside so that they could feed themselves without having difficulty standing. The wound healed within 1 to 2 days, and the mice behaved normally. Most of the experiments were conducted in mice at 7 days postligation. In some experiments, mice at 14 and 21 days after nerve ligation were used.
Hargreaves Thermal Paw Withdrawal Test. Analgesia was measured from the latency to withdrawal evoked by exposing the right hind paw to a thermal stimulus (Hargreaves et al., 1988). Mice were placed in Plexiglas cages on top of a glass sheet. The thermal stimulus (IITC Life Science Inc., Woodland Hills, CA) was positioned under the glass sheet to focus the projection bulb exactly on the middle of the plantar surface of mice. A mirror attached to the stimulus permitted visualization of the undersurface of the paw. After 1 h of adaptation, morphine was injected s.c., i.t., i.c.v., or i.pl., and the paw withdrawal latencies were measured at every 10-min interval until 60 min. A cutoff thermal latency of 20 s was set to prevent tissue damage. AUC analgesia was measured by deducting the area under the time-response curve of saline (AUC-saline) from the area under the time-response curve of morphine (AUC-morphine). In some experiments, the paw withdrawal latencies at 10 or 30 min after drug administration were measured.
ANF Test. The ANF test or peripheral nociception test was performed as described previously (Inoue et al., 1998, 2003; Ueda 1999). Briefly, mice were lightly anesthetized with ether and held in a cloth sling with their four limbs hanging free through holes. The sling was suspended on a metal bar, and all limbs were tied with soft thread strings. Then three limbs were fixed to the floor while the other one (the right hind limb) was connected to an isotonic transducer and recorder. All experiments began after the mice had completely recovered from the light ether anesthesia. A polyethylene cannula (0.61 mm, outer diameter) filled with the algogenic substance bradykinin (BK) was connected to a 50-μl Hamilton microsyringe and then carefully inserted into the undersurface of the right hind paw via a 30-gauge hypodermic needle. The nociceptive flexion responses induced by BK infused in a volume of 2 μl were then measured. Morphine was injected i.pl. (2 μl) through another cannula or s.c. 10 min before the BK injection. The results were represented as the percentage of control BK responses. At the start of each experiment, control BK responses were measured as the percentage of maximal flexion reflex in each mouse. The maximal flexion response was the biggest of the initial flexion reflexes induced immediately after cannulation.
Immunohistochemistry. Immunohistochemistry for MOP in DRG sections was performed according to the following protocol. Mice were deeply anesthetized with sodium pentobarbital (50 mg/kg i.v.) and perfused transcardially with 50 ml of 0.1 M potassium-free phosphate-buffered saline (K+-free PBS, pH 7.4) followed by 50 ml of 4% paraformaldehyde in K+-free PBS. The L4-L5 DRGs were removed, postfixed, and cryoprotected overnight in 25% sucrose in K+-free PBS. The DRGs were fast-frozen in cryo-embedding compound on a mixture of ethanol and dry ice and stored at -80°C until use. With a cryostat, the DRGs were cut at 10 μm, thaw-mounted on a silane-coated glass slide, and air-dried overnight at room temperature (RT). For immunolabeling of MOP, DRG sections were first washed with K+-free PBS three times for 10 min each; then they were incubated with 50 and 100% methanol for 10 min, respectively, washed with K+-free PBS, and incubated with excess blocking buffer A containing 5% normal goat serum in PBST (2% NaCl, 0.1% Triton X-100 in K+-free PBS) for 2 h at RT. The sections were then washed and reacted overnight (24 h) at 4°C with a rabbit polyclonal antibody against the MOP-C12 (a kind gift from Dr. Chris Evans, UCLA, Los Angeles, CA) at a ratio of 1:300 (Monteillet-Agius et al., 1998) in blocking buffer A. After three 30-min washings in K+-free PBS, the sections were placed in rhodamine-conjugated anti-rabbit IgG secondary antibody (1:200; Chemicon International, Temecula, CA) for 2 h at RT. For double immunolabeling of DRG sections between MOP and N52, sections were then rinsed and first incubated with anti-mouse IgG (1:50; Cappel Laboratories, Durham, NC) for 60 min and then reacted with a mouse monoclonal antibody raised against the N52 clone of the Neurofilament 200, a marker of myelinated fibers (Franke et al., 1991) (anti-N52; 1:30,000; Sigma-Aldrich) overnight at 4°C. The sections were then placed in fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (1:200; Cappel Laboratories) for 60 min at RT. For double labeling between the MOP and bradykinin B2 receptor in DRG, the sections after MOP staining were incubated with goat polyclonal antibody raised against the B2 receptor (1:100; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) for 48 h at 4°C, and the sections were then placed in FITC-conjugated anti-goat IgG secondary antibody (1:200; Rockland, Gilbertsville, PA) for 60 min at RT. Double immunohistochemistry for the B2 receptor and N52 in DRG sections was essentially performed as described previously for vanilloid receptor 1 (Rashid et al., 2003). The DRG sections were washed and blocked with 2% bovine serum albumin in PBST (0.1% Triton X-100 in 0.1 M potassium-free phosphate-buffered saline) and incubated with goat polyclonal antibody for the B2 receptor (1:100; Santa Cruz Biotechnology) for 48 h at 4°C. The sections were then placed in Texas Red-conjugated anti-goat IgG secondary antibody (1:200; Rockland) for 60 min at RT. For double immunolabeling with N52, these sections were rinsed and first incubated with anti-mouse IgG (1:50; Cappel) for 60 min and then reacted overnight with mouse monoclonal anti-N52 (1:30,000; Sigma-Aldrich) at 4°C. The sections were then placed in FITC-conjugated anti-mouse IgG (1:200; Cappel Laboratories) for 60 min at RT. Finally, the sections were rinsed and coverslipped with Perma Fluor (Thermo Shandon, Pittsburgh, PA) and examined under a fluorescence microscope (Olympus, Tokyo, Japan). The specificity of the antibodies for B2 and μ-opioid receptors were confirmed by the absence of staining of the DRG sections in experiments excluding the primary antibodies.
Statistical Analysis. Statistical evaluations of the data were performed using Student's t test following comparison with repeated measures analysis of variance. In the time course experiments, statistical evaluations were also performed using Student's t test with one-way analysis of variance at each time. The criterion of significance was set at p < 0.05. All results are expressed as the mean ± S.E.M.
Results
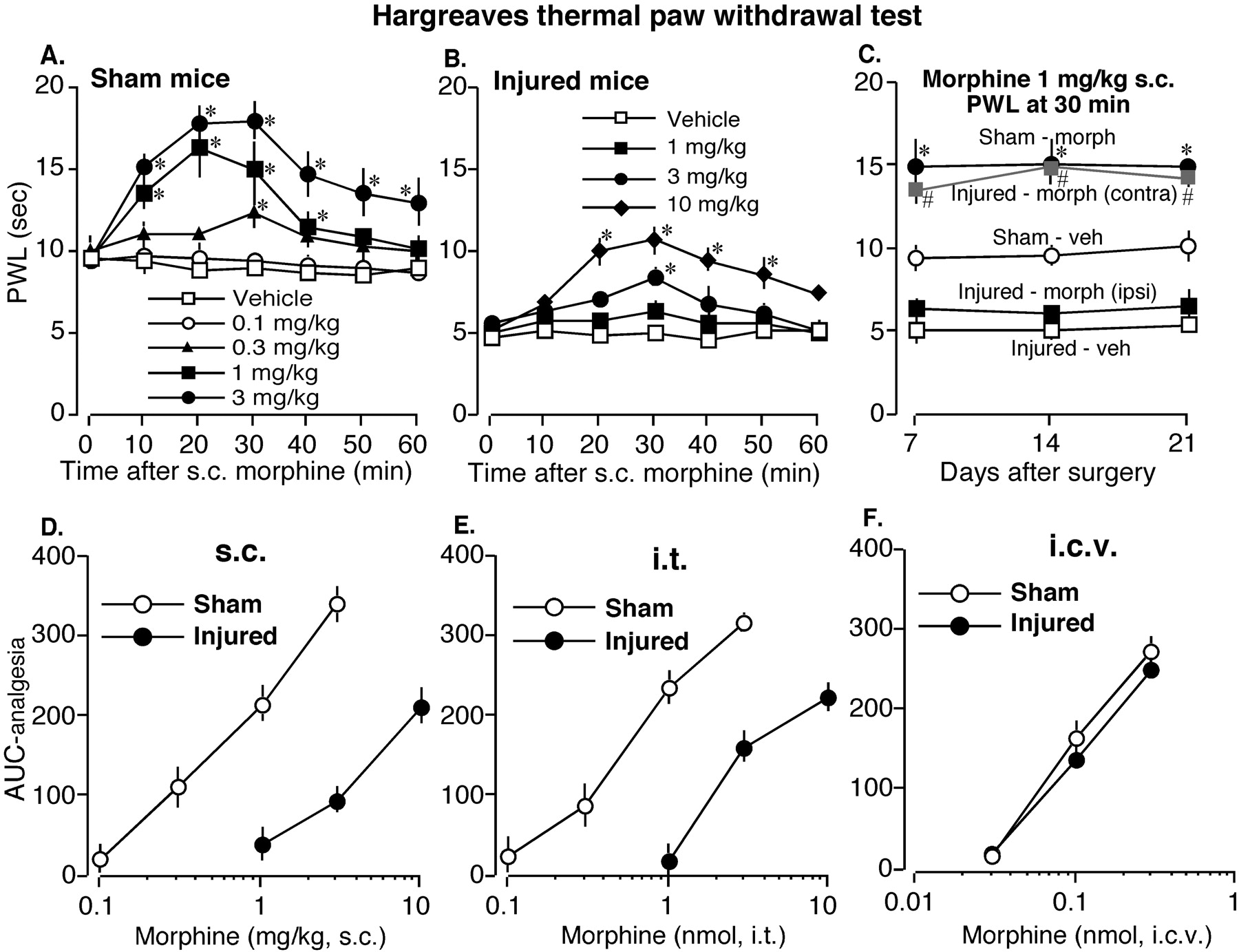
Decreased Analgesic Potency of Subcutaneous and Intrathecal, but Not Intracerebroventricular, Morphine in Partial Sciatic Nerve-Injured Mice. In Hargreaves thermal paw withdrawal test, the analgesic potency of s.c. morphine was considerably reduced in nerve-injured mice compared with sham-operated mice (Fig. 1, A-D). When we measured the paw withdrawal latency with different doses of morphine at 7 days after nerve ligation, we saw that, in control sham-operated mice, s.c. injection of morphine dose-dependently increased the thermal latency producing significant analgesic action at 0.3, 1, and 3 mg/kg (Fig. 1A). In nerve-injured mice, morphine failed to produce significant analgesic effect at 1 mg/kg. At 3 mg/kg, morphine produced significant analgesia only at a single time point (30 min). Morphine produced considerable analgesia in nerve-injured mice only at a higher dose of 10 mg/kg (Fig. 1B). We also examined the effects of s.c. morphine in nerve-injured mice at later time points of 14 and 21 days after nerve ligation. As shown in Fig. 1C, there was a similar level of thermal hyperalgesia in nerve-injured mice at 7, 14, and 21 days after injury, and morphine analgesia was also reduced in nerve-injured mice compared with sham-operated mice. However, morphine analgesia was not affected in the contralateral side of nerve-injured mice. When we plotted the AUC analgesia (AUC-morphine—AUC-saline) versus morphine doses, a clear rightward shifting in the dose-response curve of s.c. morphine was observed in nerve-injured mice (Fig. 1D). Similarly, the dose-response curve for i.t. morphine was also shifted rightward in nerve-injured mice compared with sham-operated mice (Fig. 1E). On the other hand, there was no change in the dose-response curve of i.c.v. morphine between sham-operated and nerve-injured mice (Fig. 1F), suggesting that the analgesic potency of morphine remained unaltered at the supraspinal level after peripheral nerve injury.

Dose-response curves of morphine in sham-operated (sham) and nerve-injured (injured) mice after s.c., i.t., and i.c.v. injection with the Hargreaves thermal paw withdrawal test. A and B, time course of the effects of s.c. morphine in sham and injured mice at 7 days after surgery. Results are presented as paw withdrawal latency (PWL) in seconds. *, p < 0.05 compared with vehicle. C, effects of 1 mg/kg s.c. morphine (morph) on paw withdrawal latency in sham-operated and nerve-injured mice at 7, 14, and 21 days after surgery. In morphine-treated nerve-injured mice, PWL was measured in both the ipsilateral and contralateral paw. Results are presented as PWL at 30 min after s.c. injection of morphine or vehicle (veh). *, p < 0.05 compared with sham-vehicle; #, p < 0.05 compared with injured-vehicle. D through F, dose-response curves of s.c., i.t., and i.c.v. morphine in sham-operated and nerve-injured mice at 7 days following nerve ligation. The data are presented as AUC analgesia (see Materials and Methods for details). Each data point represents the mean ± S.E.M. from six mice. *, p < 0.05.
Loss of Peripheral Morphine Analgesia in Partial Sciatic Nerve-Injured Mice. With the thermal paw withdrawal test, i.pl. injection of morphine produced dose-dependent analgesic action in sham-operated mice from 3 to 30 nmol (Fig. 2, A and C). The peak analgesia produced by 10 and 30 nmol of morphine (at 10 min after injection) was almost of the same level, and the analgesic action persisted for about 40 min after the i.pl. injections. On the other hand, in nerve-injured mice, even 30 nmol of morphine could not produce analgesic action (Fig. 2, B and D), suggesting a loss of morphine analgesia at the periphery after nerve injury. We also performed experiments with additional higher doses of i.pl. morphine (100 nmol) to see if there was any analgesia. As shown in Fig. 2C, ipsilateral i.pl. injection of 100 nmol of morphine only slightly increased the withdrawal latency in the injured paw, whereas it considerably increased the withdrawal latency in the contralateral side. This result suggests that 100 nmol of i.pl. morphine produced some systemic effects; thus, this dose was not used in subsequent studies at 14 and 21 days. However, 30 nmol of i.pl. morphine did not increase the withdrawal latency in the contralateral paw (Fig. 2C), suggesting only local action at this dose. We further examined the morphine analgesia following i.pl. injection of 30 nmol of morphine into the contralateral side of the nerve-injured mice at different time points. As shown in Fig. 2E, i.pl. injection of 30 nmol of morphine into the uninjured contralateral paw produced significant analgesia at 7, 14, and 21 days after nerve injury. However, injection of morphine into the ipsilateral paw failed to produce significant analgesia at any of these time points, suggesting that the loss of peripheral morphine analgesia was not time-specific.

Effects of i.pl. injection of morphine in sham-operated (sham) and nerve-injured (injured) mice with the Hargreaves thermal paw withdrawal test. A and B, time course of the effects of i.pl. morphine in sham and injured mice at 7 days following nerve ligation. Results are presented as PWL in seconds. C, effects of ipsilateral i.pl. morphine on contralateral paw withdrawal latency. Morphine (30 or 100 nmol i.pl.) was injected into the ipsilateral paw, and the PWL was measured in both the ipsi- and contralateral paw at 10 min following injection. The dotted lines indicate the approximate PWL in untreated sham and injured mice. D, dose-response curve of i.pl. morphine in sham-operated and nerve-injured mice at 7 days after nerve ligation. The data are presented as AUC analgesia (see Materials and Methods for details). E, effects of 30 nmol of i.pl. morphine following injection into either the ipsi- or contralateral paw of the nerve-injured mice at 7, 14, and 21 days after surgery. Results are presented as PWL in either the ipsi- or contralateral paw at 10 min after i.pl. injection of morphine (M) or vehicle (V). Each data point represents the mean ± S.E.M. from six mice. *, p < 0.05 compared with vehicle.
Effects of Subcutaneous and Intraplantar Morphine against the Intraplantar BK-Induced Nociception with the ANF test. Next, we examined the analgesic effects of morphine with the more sensitive ANF test (Inoue et al., 2003). The use of a single pain-producing ligand such as bradykinin in the ANF test would be helpful in identifying the immunohistochemical colocalization between that pain-producing ligand receptor and the receptor for the applied analgesics. We found that the analgesic potency of systemic morphine (s.c.) against BK nociception in the ANF test was greatly reduced in nerve-injured mice (Fig. 3A). In sham-operated mice, 1.5 mg/kg morphine decreased the BK-induced flexion responses to 24.6 ± 10.3% of the control response, whereas in nerve-injured mice 5 mg/kg morphine blocked the BK nociception to only 61.8 ± 10.5% of the control response. Analgesic effect of peripheral morphine, however, completely disappeared in nerve-injured mice in the ANF test. The i.pl. pretreatment with 1 nmol of morphine completely blocked the BK nociception in sham-operated mice at 30 min after injection (Fig. 3B). However, in nerve-injured mice, i.pl. pretreatment with even 10 nmol of morphine could not block the BK nociception (Fig. 3C).
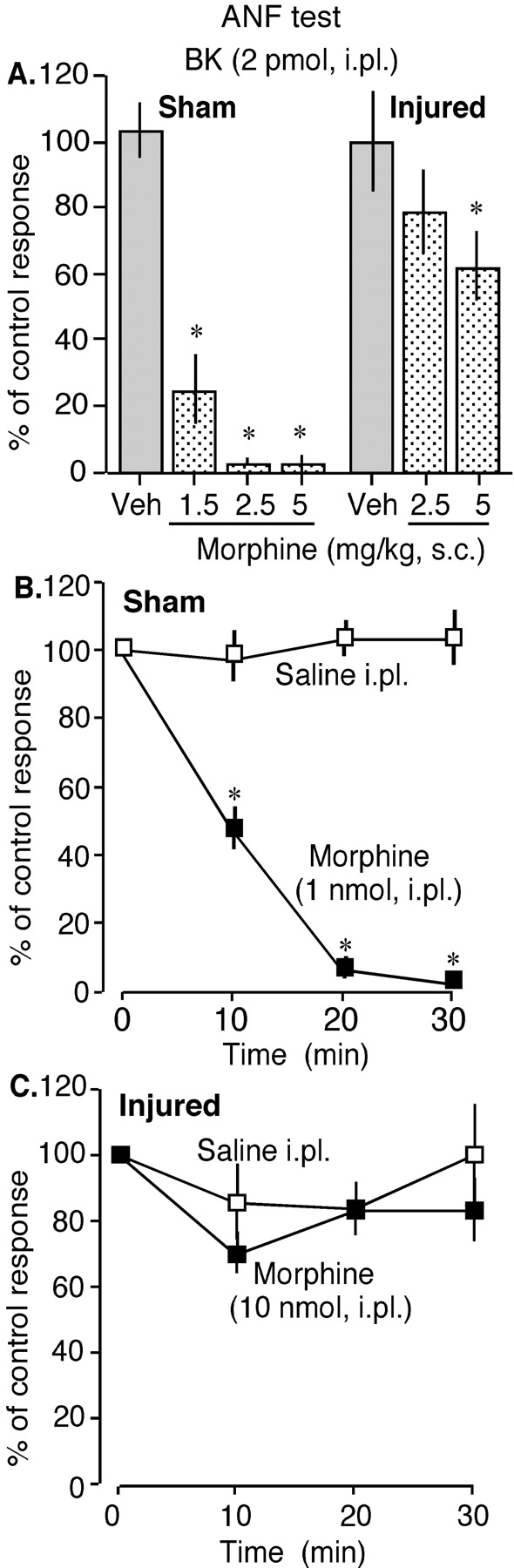
Effects of s.c. and i.pl. morphine in sham-operated (sham) and nerve-injured (injured) mice with the ANF test at 7 days following surgery. A, effects of different doses of s.c. morphine on 2 pmol of BK-induced nociceptive flexion responses (i.pl.) in sham-operated and nerve-injured mice. B and C, effects of i.pl. morphine on 2 pmol of BK-induced nociceptive flexion responses (i.pl.) in sham-operated and nerve-injured mice. Bradykinin was given at 10, 20, and 30 min after morphine injection. The results are presented as a percentage of control BK responses (see Materials and Methods for details). The vertical columns in Fig. 3A are the percentage of control responses at 30 min after s.c. morphine injection. Each data point represents the mean ± S.E.M. from six mice. *, p < 0.05.
Expression Pattern of the μ-Opioid Receptor in the DRG Neurons of Sham-Operated and Partial Sciatic Nerve-Injured Mice. We examined the expression pattern of MOP in the DRG neurons of control sham-operated and nerve-injured mice by using an immunohistochemical technique. As shown in Fig. 4A, in the DRG of control sham-operated mice, many MOP-immunoreactive neurons were observed (38.3 ± 4.4 neurons were MOP-immunoreactive out of 99.0 ± 10.7 total neurons; n = 3). Moreover, most of the MOP-immunoreactive neurons were not colabeled with the myelinated fiber marker N52 (only 3.7 ± 1.0% of total DRG neurons), indicating their presence on small diameter unmyelinated DRG neurons under naive condition (Fig. 4, C and H). However, in DRG of partial sciatic nerve-injured mice, the total number of MOP-immunoreactive cells in the ipsilateral DRG was drastically reduced (Fig. 4, B and G; 5.7 ± 0.7 neurons were MOP-immunoreactive out of a total 142.0 ± 11.4 neurons; n = 3), correlating our behavioral data of the loss of peripheral morphine analgesia in such a condition (Figs. 2B and 3C). An approximately 90% decrease in MOP expression was observed in the DRG of nerve-injured mice at 7 days after injury. The decrease in MOP expression in the ipsilateral DRG was also similar at later time points of 14 and 21 days following surgery (data not shown). However, MOP expression in the sham mice and in the contralateral side of the nerve-injured mice was unaffected at all these time points (data not shown). We also observed that in control sham-operated mice, bradykinin B2 receptors were expressed in the small-diameter unmyelinated neurons that coexpressed the MOP (Fig. 4, D, E, F, and I). Of 95.3 ± 4.9 total DRG neurons from three separate mice, 37.6 ± 2.3 neurons were MOP-positive, 38.33 ± 1.2 neurons were B2-positive, and 31.0 ± 3.4 neurons were positive for both MOP and B2. The high incidence (∼90%) colocalization between MOP and B2 receptor in sham mice was also consistent with our behavioral data of complete blockade of BK nociception by i.pl. morphine in sham-operated mice (Fig. 3B).
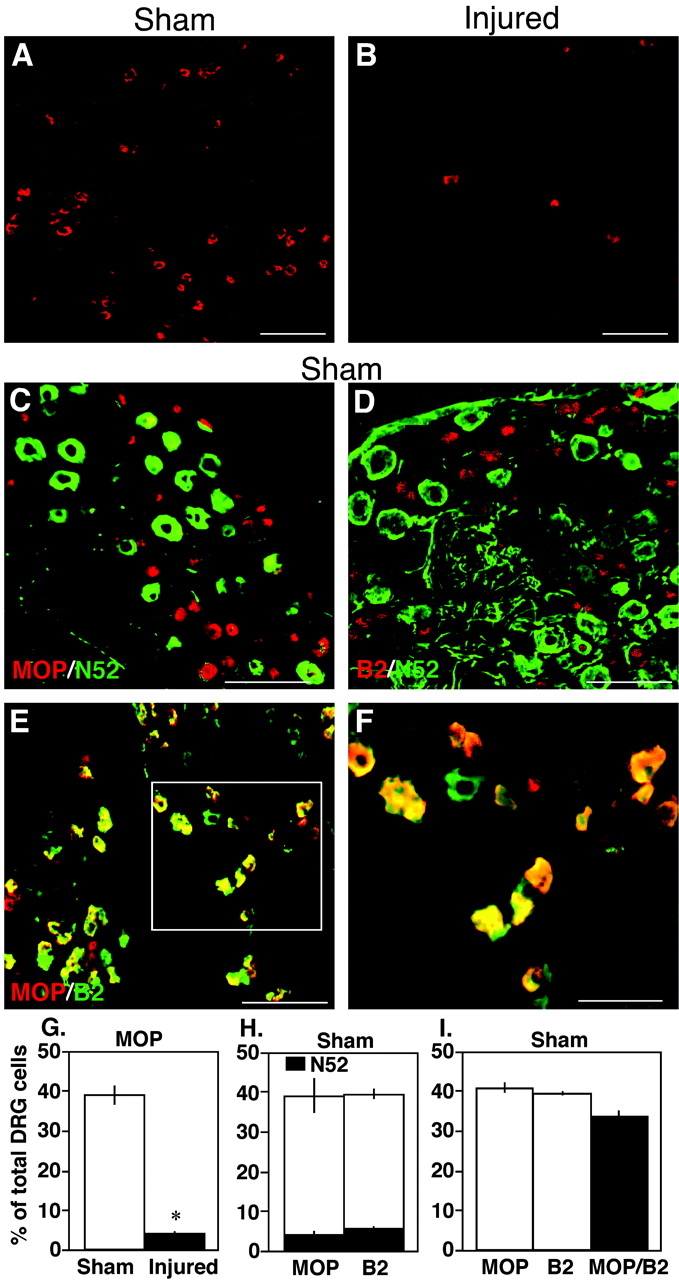
Immunohistochemical localization of MOP in the DRG neurons of sham-operated (sham) and nerve-injured (injured) mice at 7 days after surgery. A and B, single immunolabeling of the DRG sections of sham (A) and injured (B) mice with an antibody specific for the MOP. MOP-immunoreactive (IR) neurons were drastically reduced in the DRG of nerve-injured mice. C, immunohistochemical double labeling between MOP and N52, a marker of myelinated fibers, in sham mice. Most of the MOP immunoreactivities (red) were located in unmyelinated DRG neurons. D, immunohistochemical double labeling between the bradykinin B2 receptor and N52 in sham mice. Similar to MOP, most B2 receptors (red) were located in unmyelinated DRG neurons in sham mice. E and F, immunohistochemical double labeling between MOP (red) and B2 receptors (green) in the DRG of sham mice. Most of the MOP-IR neurons were colocalized with the B2-IR neurons (yellow). Panel F is the high-magnification image of the marked box in panel E. G, percentage of total cells expressing MOP in the DRG of sham-operated and nerve-injured mice from three separate animals. H and I, percentage of total cells expressing MOP, bradykinin B2 receptor (B2), or both (MOP/B2) in the DRG of sham-operated mice from three separate animals. *, p < 0.05. Scale bars: A through E, 100 μm; F, 20 μm.
Discussion
In clinic, neuropathic pain caused by peripheral nerve injury is often reported to be less sensitive to morphine (Arnér and Meyerson 1988; Kupers et al., 1991; Bleeker et al., 2001). Similar results were also observed with experimental animal models of neuropathic pain (Ossipov et al., 1995; Idanpaan-Heikkila et al., 1997). Consistent with these lines of evidence, a marked decrease in the analgesic potency of both s.c and i.t. morphine was observed in partial sciatic nerve injury-induced neuropathy model mice in the present study (Fig. 1, A-E). In contrast, effects of morphine after i.c.v. injection did not differ in nerve-injured mice compared with control sham-operated mice (Fig. 1F), indicating that the analgesic potency of morphine remained unchanged at the supraspinal level. Local injection of morphine (like i.t. or i.c.v.) is expected to act on the local μ-opioid receptors while systemic morphine (like s.c.) is distributed throughout the body and acts at different locations, including peripheral, spinal, and supraspinal sites. Since the supraspinal component of morphine analgesia remained intact after nerve injury, the decreased effectiveness of systemic morphine in nerve-injured mice might be caused by its reduced potency in the periphery or at the spinal level.
As such, we examined the analgesic effect of morphine after i.pl. administration in sham-operated and nerve-injured mice. Local intraplantar injection of morphine produced dose-dependent (3-30 nmol) analgesia in sham-operated mice (Fig. 2, A and D). Moreover, consistent with our previous report (Ueda et al., 2000), i.pl. morphine (1 nmol) completely blocked the i.pl. BK-induced nociception in sham mice (Fig. 3B). These results were also confirmed by our immunohistochemical data. In the DRG of control sham-operated mice, many small diameter unmyelinated neurons expressed the μ-opioid receptor (Fig. 4, A, C, and G). In control sham mice, approximately 40% of the total DRG cells expressed MOP, and approximately 90% of these were located in unmyelinated DRG neurons. These results are in agreement with many previous reports (Arvidsson et al., 1995; Zhang et al., 1998). The presence of MOP in unmyelinated afferent C-fiber neurons, most of which are nociceptive, indicates that morphine produced its peripheral analgesia in sham mice through these μ-opioid receptors. The coexistence of MOP and bradykinin B2 receptor in the DRG of sham-operated mice (Fig. 4, E, F, and I) also strengthens our results of complete blockade of BK nociception by i.pl. morphine in the ANF test (Fig. 3B). Bradykinin is a well known mediator of pain, which acts through the constitutively expressed bradykinin B2 receptors. The presence of MOP on bradykinin B2 receptor-containing DRG neurons further indicates the pain-modulatory effects of morphine at the periphery under naive condition.
On the other hand, peripheral morphine analgesia observed in sham mice was almost completely lost in partial sciatic nerve-injured mice. Ipsilateral i.pl. injection of morphine (30 nmol in the thermal paw withdrawal test and 10 nmol in the ANF test) was unable to block the thermal hyperalgesia or the BK nociception in nerve-injured mice (Figs. 2B and 3C). Consistent with these behavioral data, we found that partial sciatic nerve injury in mice caused a drastic decrease in μ-opioid receptor expression in the injured DRG neurons (Fig. 4, B and G). An approximately 90% decrease in μ-opioid receptor expression was observed in the DRG of nerve-injured mice compared with sham-operated mice. These immunohistochemical data confirm our behavioral results of an almost complete loss of peripheral morphine analgesia in nerve-injured mice. However, MOP expression and morphine analgesia in the contralateral side of nerve-injured mice was unaffected (Fig. 2E and data not shown), suggesting that the loss of peripheral morphine analgesia was only injury-specific. Recently, Pertovaara and Wei (2001) reported that i.pl. morphine was more effective in the ipsilateral paw than in the contralateral paw in the spinal nerve ligation model of neuropathic pain in the rat. However, the i.pl. dose of morphine that produced such an effect (200 μg i.pl.) in the study of Pertovaara and Wei (2001) was much higher than our i.pl. doses (30 nmol or approximately 1 μg i.pl.). Systemic effects of such higher doses cannot be excluded. Indeed, in our experiments, ipsilateral injection of 100 nmol of i.pl. morphine produced analgesia in the contralateral paw, indicating the presence of systemic effects at this higher dose (Fig. 2C). Nevertheless, other factors, such as the use of different neuropathy models, different modality of nociception tests, and different species of animals might also underlie the discrepancies between our results and that of Pertovaara and Wei (2001). On the other hand, Truong et al. (2003) reported an increase in MOP expression in DRG neurons of the chronic constriction injury model rat by immunohistochemistry; however, the same report could not detect an increase in MOP expression by Western blot or reverse transcription-polymerase chain reaction experiments. Our result of a drastic decrease in MOP expression in DRG neurons following peripheral nerve injury is consistent with numerous previous reports. For instance, Li et al. (1996) reported a drastic decrease in MOP expression in DRG and nodose ganglion 7 days following unilateral ligation of rat sciatic or vagus nerve, and Zhang et al. (1998) reported down-regulation of MOP in rat and monkey DRG and spinal cord following peripheral axotomy. Most recently, a microarray study on DRG neurons in a rat neuropathy model demonstrates approximately 5 times down-regulation of μ-opioid receptor gene (Xiao et al., 2002). The decrease in MOP expression in the DRG of our partial sciatic nerve injury model mice was, however, higher (∼90%) than that observed by Zhang et al. (1998) in the rat axotomy model (∼30%); however, in the same report, there was considerable species difference for the reduction of MOP expression following axotomy. Whereas axotomy caused an approximately 30% reduction in MOP expression in the rat, it caused more than a 99.5% decrease in MOP expression in the monkey DRG (Zhang et al., 1998). We speculate similar species difference for the higher reduction in MOP expression in our study with mice compared with the axotomy experiments in rat by Zhang et al. (1998). Moreover, use of different neuropathy models might also underlie such differences. For the decreased effectiveness of i.t. morphine in partial sciatic nerve-injured mice (Fig. 1E), we speculate a reduction of MOP expression in the spinal dorsal horn presynaptic afferent terminals as a result of a drastic decrease in MOP expression in the DRG neurons (Zhang et al., 1998). The reduction in analgesic potency of i.t. morphine against nerve injury-induced thermal hyperalgesia (Wegert et al., 1997) and a spinal segment-specific reduction in MOP expression (Porreca et al., 1998) had already been reported elsewhere. On the other hand, although we did not perform B2 receptor expression experiments in nerve-injured mice in the present study, we found that the flexion responses induced by 2 pmol of bradykinin (i.pl.) remained almost similar in sham-operated and nerve-injured mice (72.3 ± 6.6% of maximal reflex in sham versus 78.9 ± 4.9% in injured). In a recent study (Rashid et al., 2004), we found decreased expression of B2 receptors with some novel expression of B1 receptors in the DRG of nerve-injured mice. However, the loss of peripheral morphine analgesia observed in the present study should be caused by the drastic reduction in MOP expression, but not by the changes in B2 receptor expression since bradykinin still produced potent nociceptive flexion responses in the nerve-injured mice. Nevertheless, the use of a single pain-producing ligand such as bradykinin in the ANF test was helpful in identifying the immunohistochemical colocalization between that pain-producing ligand receptor (i.e., B2) and the receptor for the applied analgesics (i.e., MOP). The high incidence of colocalization between B2 receptor and MOP in the DRG of control sham mice confirmed that morphine could modulate the nociceptive mechanisms at the periphery under normal condition.
In conclusion, our results indicate that the lower analgesic potency of systemic morphine in neuropathic pain in clinic could be at least partly caused by the loss of morphine analgesia at the periphery. The findings of the present study could help in clinical management of neuropathic pain, with possible modifications in the use of morphine.
Acknowledgments
We thank Fumiko Fujiwara, Saori Kondo, and Toshiko Kawashima for technical assistance.
Footnotes
-
This study was supported in part by grants-in-aid and special coordination funds from the Ministry of Education, Science, Culture and Sports of Japan and a grant from the Human Frontier Science Program.
-
DOI: 10.1124/jpet.103.060582.
-
ABBREVIATIONS: ANF, algogenic-induced nociceptive flexion; BK, bradykinin; DRG, dorsal root ganglion; MOP, μ-opioid receptor; PBS, phosphate-buffered saline; AUC, area under the curve; ANOVA, analysis of variance; RT, room temperature; FITC, fluorescein isothiocyanate; PWL, paw withdrawal latency; IR, immunoreactive.
- Received September 26, 2003.
- Accepted December 15, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}