


Abstract
The effects of riluzole, a neuroprotective drug, on high voltage-activated (HVA) calcium channels of rat dorsal root ganglion neurons were studied using the whole-cell patch-clamp technique. Riluzole inhibited HVA calcium channel currents in a dose-dependent, time-dependent and reversible manner. The apparent dissociation constants for riluzole inhibition of the transient and sustained components of the current were 42.6 and 39.5 μM, respectively. Riluzole accelerated the activation kinetics of calcium channels without affecting the voltage dependence of activation. It accelerated the fast component of deactivation kinetics without affecting the slow component. It also accelerated fast and slow inactivation kinetics of the HVA channels. However, only one of the two components in the steady-state inactivation curve for the HVA channels was shifted in the hyperpolarizing direction by riluzole, which indicates differential block of the multiple-type HVA channels. By use of the specific blockers nimodipine, ω-conotoxin GVIA and ω-agatoxin IVA, the HVA calcium channels were found to comprise L-type (10%), N-type (63%), P/Q-type (23%) and R-type (9%). Riluzole blocked N- and P/Q-type channels, but not L-type channel, with the order of efficacy of P/Q- > N- ≫ L-type channels. Riluzole inhibition of N- and P/Q-type calcium channels may result in reduced calcium influx at presynaptic terminals, which thereby decreases excessive excitatory neurotransmitter release, especially glutamate, a mechanism known to cause neuronal death in ischemic conditions.
Riluzole, a novel neuroprotective drug with anticonvulsant, sedative and anti-ischemic properties (Malgouris et al., 1989; Stutzmannet al., 1991; Romettino et al., 1991; Prattet al., 1992; Bryson et al., 1996), has been used to prolong the survival of patients with amyotrophic lateral sclerosis (Bensimon et al., 1994; Bryson et al., 1996;Lacomblez et al., 1996).
Ischemic damage is known to be caused by the neurotoxic effects of excitatory amino acids released as a result of hyperexcitation of presynaptic nerves (Choi and Rothman, 1990; Rothman, 1984; Meldrum, 1985; Rothman and Olney, 1986). The excess activation of the glutamatergic receptors, especially the NMDA receptors, causes a massive influx of Ca++ through the open channels leading to cell death. Riluzole blocked kainate and NMDA receptors (Debono et al., 1993); this would lessen the cell damage caused by ischemia. Several lines of evidence suggested that riluzole inhibited synaptic transmission by reducing the release of glutamate from the presynaptic nerve terminals (Martin et al., 1993;Boireau et al., 1995; Rothstein and Kuncl, 1995).
The neuroprotective effect of riluzole is also known to rely in part on inhibition of sodium channels (Hebert et al., 1994;Benoit and Escande, 1991), because the inhibition of sodium channels was correlated with postischemic neuronal protection (Prenen et al., 1988; Boening et al., 1989; Yamasaki et al., 1991). Riluzole had a higher affinity for the inactivated state than the resting state of sodium channels, which resulted in a selective block of damaged or depolarized nerve thereby preventing excess stimulation of the glutamatergic receptors (Song et al., 1996).
Because transmitter release from the nerve terminals is mediated by voltage-gated calcium channels, neuroprotective drugs such as riluzole could also block these channels, thereby preventing excess release of transmitter. The present study was undertaken to test this hypothesis. It was indeed demonstrated that riluzole blocked the HVA calcium channels, with much higher affinities for N-type and P/Q-type calcium channels than L-type channels.
Methods
Cell culture
DRG were dissected from the lumbosacral area of newborn Sprague-Dawley rats (1–5 days old) under methoxyflurane anesthesia, and were transferred into Ca++-/Mg++-free phosphate-buffered saline solution supplemented with 6 g/ld-glucose. The ganglia were digested in this solution added with 2.5 mg/ml trypsin (type XI, Sigma Chemical Co., St. Louis, MO) for 25 min at 37°C and then washed with DMEM containing 10% neonatal calf serum and 0.08 mg/ml gentamicin. The ganglia were dissociated acutely by repeated triturations with use of a fire-polished Pasteur pipette in 2.0 ml DMEM. The dissociated neurons were plated onto coverslips coated with poly-l-lysine (0.1 mg/ml, Sigma). Cells were maintained in DMEM containing neonatal calf serum and gentamicin in a 90% air/10% CO2 atmosphere controlled at 36°C. Neurons cultured for less than 2 days were used for experiments. In most experiments, recording began 4 hr after the plating. Cells cultured more than 1 day usually develop processes and may result in poor space clamp condition. Any recording with inadequate space clamp, such as that with slow tail current and slow settling of capacity transient, was discarded.
Electrophysiology.
Ionic currents were recorded under voltage-clamp conditions by the whole-cell patch clamp technique (Hamill et al., 1981). Pipette electrodes were made from 1.5-mm (outside diameter) borosilicate glass capillary tubes and had a resistance of 2 megohm when filled with standard internal solution. The transmembrane voltage was clamped at −80 mV. Unless otherwise indicated, a period of 10 min was allowed after rupture of the membrane to ensure adequate equilibration between the internal pipette solution and the cell interior. Cells showing a rundown rate of calcium currents more than 1%/min were excluded. Membrane currents passing through the electrode were recorded with an Axopatch amplifier (Axopatch-1B, Axon Instruments, Foster City, CA), and currents were stored in an SX 386 computer (DELL Computer Company, Austin, TX) using pCLAMP 6 software (Axon Instruments).
The external and internal solutions for whole-cell current recording were designed to eliminate sodium and potassium channel currents. The standard external solution contained (in mM): BaCl2, 10; tetraethylammonium chloride, 130;d-glucose, 25; HEPES, 5; and tetrodotoxin, 200 nM. The standard internal solution contained (in mM): CsCl, 105; HEPES, 40;d-glucose, 5; MgCl2, 2.5; EGTA 10; Mg-ATP, 5; and Li3-GTP, 0.5. Unless otherwise indicated, the pH of all solutions was adjusted to 7.3 with 1 M CsOH, and the osmolarity was raised to 300 mOsm with sucrose.
Drug application.
The recording chamber was perfused continuously with the normal external solution by gravity at a rate of 1 ml/min. Unless otherwise described, all the drugs were applied to the bath by gravity. The total volume of the chamber was only 1 ml facilitating the rate of application and washout of the drug. All experiments were carried out at a room temperature of 20–23°C.
Calcium channel toxins were dissolved in distilled water to make a stock solution which was divided into 100-μl aliquots and stored at −20°C. The test solution was pipetted directly to the bath solution to achieve the desired concentration, while the bath perfusion was stopped. Stopping the bath perfusion for less than 15 min did not accelerate the rundown of the currents. The final solution also contained 1% cytochrome C to saturate the nonspecific peptide binding to the chamber. Because of the diffusion time of the toxin in the bath, sometime the onset of the toxin action was slow. However, we found that a few more pipettings of the bath solution after addition of the toxin facilitated its diffusion and shortened its onset of the effect.
Nimodipine and Bay K 8644 were dissolved in 100% ethanol as 10 mM stock solution, and kept in light-proof containers. Experiments were performed under restricted light conditions in the presence of dihydropyridines. Riluzole was dissolved in DMSO at a concentration of 100 mM and diluted to external solution at the desired concentrations on the day of experiment. The final concentration of ethanol or DMSO was less than 0.3% (v/v), which did not affect calcium channel currents.
ω-CTx was purchased from Bachem California (Torrence, CA), nimodipine from Tocris Cookson (St. Louis, MO), and Bay K 8644 from Calbiochem (La Jolla, CA). ω-Aga was a gift from Pfizer Inc. (Groton, CT) and riluzole from Mitsui Pharmaceuticals (Tokyo, Japan). All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Data analysis.
The dose-response relationship of riluzole inhibition of the transient and sustained calcium channel currents was fitted to a sigmoid curve as calculated by the four-parametric logistic function equation (Tandel Scientific, Corte Madera, CA):
Statistical analysis was done by use of the Student’s ttest at a significant level of P = .05. Data are given as mean ± S.E.M. with the number of experiments (n).
Results
Time- and Dose-Dependent Inhibition of HVA Ca++ Channel Currents by Riluzole
With 10 mM barium as charge carrier, the T-type calcium channel current was activated by a test pulse to −40 mV from a holding potential of −80 mV. The current had fast inactivation kinetics and was found in only 15% of the cells tested. A test pulse to −20 mV from the same holding potential activated the HVA calcium channel currents which reached the maximal amplitude at a test pulse to 0 mV or 10 mV. Cells generating T-type calcium channel currents were discarded to avoid the contamination of HVA calcium channel currents.
Riluzole was applied to the bath while HVA calcium channel currents were evoked by 200-msec test pulses from −80 mV to +20 mV at 20-sec intervals. Riluzole at 300 μM inhibited the calcium channel currents, and the effect reached the maximum taking more than 3 min after bath perfusion (fig. 1). Both peak and steady-state amplitudes were suppressed. After washout, the riluzole inhibitory effect persisted for a while, and a complete recovery usually took more than 3 min. In 20% of the cells studied, a rebound to 118% of the control current amplitude was observed after washout (fig. 1). The slow onset and offset of the riluzole inhibitory effect and the rebound phenomenon suggest that intracellular components may be involved in the riluzole action.

Riluzole inhibition of the HVA calcium channel current. Currents are evoked by a step pulse to +20 mV from a holding potential of −80 mV at an interval of 20 sec. (A) Current traces a to e correspond to currents at times a to e in panel B. (B) Riluzole 300 μM inhibits the HVA channel current with a slow onset and offset. A rebound of the current amplitude occurs after washout of riluzole.
To study the dose-response relationship for the riluzole block of the HVA calcium channels, different concentrations of riluzole were perfused cumulatively. Riluzole inhibited both the peak current and sustained currents measured at the end of 200 msec pulse in a dose-dependent manner, and a complete block occurred at 300 μM riluzole. Using equation 1, the dose-response relationships for riluzole inhibition of peak and sustained calcium channel currents were fitted to sigmoid curves, which yielded apparentKd values of 42.6 ± 7.4 μM (n = 10–16) and 39.5 ± 17.26 μM (n = 7–16), and the Hill coefficients of 1.1 ± 0.160 and 0.83 ± 0.212, respectively (fig.2).

The dose-response relationship for riluzole inhibition of the peak transient (•) and sustained (○) components of HVA calcium currents with apparent K dvalues of 42.6 ± 7.4 μM (n = 10–16) and 39.5 ± 17.26 μM (n = 7–16), respectively.
Effects of Riluzole on the Kinetics of HVA Calcium Channels
Activation.
To study the effect of riluzole on the activation of calcium channel currents, current-voltage (I-V) relationships for the transient calcium channel currents were plotted by 5-mV incremental step pulses from −80 mV to +75 mV in the absence and presence of riluzole (fig. 3A). Because the I-V relationship was nonlinear in the positive potential range, the constant field theory (Goldman, 1943; Hodgkin and Katz, 1949) was used to calculate the maximal current amplitude at its full activation (Ohmori and Yoshii, 1977, Yoshii et al., 1988). The constant field equation for divalent cations is:

Effect of riluzole on the activation of HVA channels. (A) Current-voltage relationships for HVA channels in the absence (•) and presence of 30 μM (▪) and 100 μM (▴) riluzole. (B) The voltage dependence of activation curve for HVA channels is fitted by the constant field equation (see text). Riluzole at 30 and 100 μM reduces the mid-point potential from the control value of −1.6 ± 0.34 mV to −2.7 ± 0.23 mV (n = 7) and −1.2 ± 0.21 mV, respectively (P > .05). (C) The time to peak of HVA channel currents in the presence and absence of riluzole is measured as shown in the inset, and the data are plotted as a function of riluzole concentration. (D) The time to peak for HVA channel currents in the absence (○) or presence (•) of 30 μM riluzole is plotted against the step pulse potential. Riluzole reduction of the time to peak for HVA channel currents is not voltage dependent (n = 5).
The time to peak of the calcium channel current was measured in a time-expanded record as shown in the inset of figure 3C, and was normalized to that of the control current. Riluzole shortened the time to peak in a dose-dependent manner (fig. 3C) with a maximal reduction to 70 ± 3.12% of control (n = 7–18) at a concentration of 300 μM. Figure 3D shows the time to peak of calcium channel currents at different test pulse potentials in the presence and absence of 30 μM riluzole. There was no voltage dependence in riluzole reduction of the time to peak (n = 5). Thus, riluzole shortens the time to peak in a dose-dependent and voltage-independent manner.
Inactivation.
To study the kinetics of channel inactivation, calcium channel currents were generated at a test pulse to +20 mV preceded by a 5-sec conditioning prepulse to various potentials and were normalized to the current associated with −100 mV prepulse. Plot of the normalized current amplitude against the prepulse potential yielded a steady-state inactivation curve for the calcium channel, which could be fitted by two exponential functions, which indicates the existence of at least two groups of HVA calcium channels with distinct inactivation kinetics (fig. 4A). Riluzole at 30 μM shifted the half-inactivation potentials for one group of calcium channels from 48.2 ± 1.31 mV to 55.4 ± 1.70 mV (P < .05), and for the other group from −8.1 ± 1.98 mV to −9.0 ± 2.53 mV (P > .05) (n = 7). The slope factor was changed by riluzole from 9.6 ± 0.81 mV to 11.6 ± 1.29 mV for the former (P < .05), and from 7.6 ± 1.09 mV to 7.91 ± 1.49 mV for the latter (P > .05) (n = 7). Thus, riluzole selectively shifted the steady-state inactivation curve for one group of HVA calcium channels in the hyperpolarizing direction.

(A) The steady-state inactivation curve for HVA channels in the absence (○) and presence (•) of 30 μM riluzole showing two components. Riluzole shifts the half-inactivation potential from −48.2 ± 1.31 mV to −55.4 ± 1.7 mV for one component (P < .05), and from −8.1 ± 1.98 mV to −9.0 ± 2.53 mV for the other component (P > .05) (n = 7). (B) Riluzole accelerates the inactivation kinetics. The time course of inactivation of HVA channel currents generated by step pulses from −80 mV to +20 mV for a duration of 300 msec can be fitted to a double-exponential curve. The time constants of the HVA channel inactivation are reduced by rulizole to 47.5 ± 5.49% and 41.4 ± 5.71% of control for the fast and slow inactivation, respectively.
The time course of inactivation of calcium channel currents was fitted to two exponential functions. Riluzole reduced both the fast and slow time constants of calcium channel inactivation to 69.7 ± 8.99% (n = 11) and 79.8 ± 8.43% (n = 8) of controls, respectively, at 3 μM, and to 16.3 ± 8.09% (n = 16) and 24.7 ± 7.64% (n = 13) of controls, respectively, at 300 μM (fig. 4B).
Deactivation.
The tail current evoked upon step hyperpolarization of the membrane after a depolarizing test pulse reflects the fraction of calcium channels remaining open at the end of the test pulse. The decay component of this tail current represents the deactivation or closure of the activation gate of the channel. With an increase in the duration of depolarizing pulse, the tail current amplitude reached a maximum and gradually decreased (fig.5, A and B). To ensure the full activation of the channel and to minimize the effect of inactivation, the subsequent tail current experiments were performed with a 10-msec test pulse. The rate of decay of the tail current varied with the level of repolarization after a depolarizing pulse to a fixed potential level (fig. 5Ca), and could be fitted by a double-exponential curve (fig.5Cb). Figure 5D plots the fast time constant against various repolarizing potentials after the same depolarizing pulse in the presence and absence of 30 μM riluzole. Riluzole decreased the fast time constant (fig. 5D), but not the slow time constant (data not shown) for the decay of tail current. At repolarizing potentials more positive then −40 mV, riluzole clearly reduced the fast time constant (n = 7, P < .05), whereas at the more negative repolarizing potentials, this effect was less pronounced because of the fast deactivation kinetics. As shown in figure 5E, the deactivation kinetics did not change significantly by varying the depolarizing potential preceding the same repolarizing potential (○,n = 5). However, riluzole reduction of the fast time constant for the decay of tail current was absent if preceded by large positive depolarizing test pulses (•, n = 5). Thus, riluzole accelerates the deactivation kinetics of HVA calcium channels by reducing the fast time constant for the decay phase in a voltage-dependent manner.

Effects of riluzole on the deactivation of HVA channels. (A) The tail current amplitude is time dependent. The currents are evoked by step pulses from −80 mV to +20 mV with 1- to 14-ms durations. (B) Normalized tail current amplitude as a function of pulse duration. (C) HVA channel currents are generated by the same test pulse which is followed by repolarization to various potentials. Decay of the calcium tail current is slowed with repolarization to less negative potentials (Ca) and can be fitted to a double-exponential curve (Cb). (D) Riluzole at 30 μM reduces the fast time constant for the decay of HVA channel tail current, and the effect is more pronounced at more positive repolarizing potentials. (E) HVA channel currents are evoked by the protocol shown in the inset. Plot of the fast time constant for the decay of HVA tail current against the test pulse potential. Riluzole reduces the fast time constant in a voltage-dependent manner.
Pharmacological Dissection of HVA Calcium Channels
Effects of dihydropyridines.
Nimodipine was used as a dihydropyridine antagonist to block the L-type calcium channel current because of its less voltage-dependent action. Nimodipine at 10 μM suppressed HVA channel currents reversibly (fig.6A). Bay K 8644 (5 μM), a dihydropyridine agonist known to prolong the opening of dihydropyridine-sensitive calcium channels (Fox et al., 1987; Nowycky et al., 1985), potentiated HVA calcium channel currents (fig. 6, A and C). Current traces a to d in figure 6B correspond to the currents at times a to d in figure 6A. Subtracting the current in nimodipine (trace b) from the control current (trace a) yields the L-type current blocked by 10 μM nimodipine, whereas subtracting current trace c from current trace d yields the L-type current in the present of 5 μM Bay K 8644 (fig. 6B2). Thus, 5 μM Bay K 8644 potentiated the L-type current 3.1 ± 0.53 times (n = 5). Note that the decay of tail current was prolonged by Bay K 8644 (fig. 6B2). To test if 10 μM nimodipine can completely block the L-type current, 5 μM Bay K 8644 was applied in the absence and presence of 10 μM nimodipine (fig. 6C). Bay K 8644 potentiation of the L-type current was observed in the control and was completely abolished in the presence of 10 μM nimodipine. Thus, nimodipine, at a concentration of 10 μM, was sufficient to block all the L-type current.

Effects of dihydropyridines on HVA channel currents. (A) Nimodipine at 10 μM inhibits the HVA channel current and Bay K 8644 at 5 μM potentiates it. (B) Current traces a to d were recorded at times a to d of panel A. (C) Nimodipine at 10 μM inhibits 5 μM Bay K 8644 potentiation of HVA channel currents. Thus, 10 μM nimodipine inhibits the dihydropyridine-sensitive L-type current completely.
Various types of calcium channels.
ω-Aga is known to block the P-type calcium channel (Mintz et al., 1992a, b; Mintz and Bean, 1993). It also blocks the Q-type channel in certain preparations (Randall et al., 1993; Sather et al., 1993; Wheeler et al., 1994; Randall and Tsien, 1995; Rusin and Moises, 1995). ω-CTx blocks the N-type calcium channel selectively (McCleskey et al., 1987). The residual current insensitive to dihydropyridine antagonists, ω-Aga and ω-CTx, is designated as the R-type current, which can be blocked by the nonspecific calcium channel blocker CdCl2(Ellinor et al., 1993; Zhang et al., 1993;Randall and Tsien, 1995). To separate various types of calcium channels present in the same cell, 10 μM nimodipine, 1 μM ω-CTx, 300 nM ω-Aga and 150 μM CdCl2 were applied to the bath in sequence (fig. 7A). Current traces a to e in figure 7B represent the currents indicated at times a to e in figure 7A. Similar to figure 6, L-, N-, P- and R-type calcium channel currents were isolated by subtracting the current after application of respective blocker from that before application (fig.7B). Thus, the precentages of L-, N-, P/Q- and R-type calcium channel currents out of the total current are 10.5 ± 1.10, 62.8 ± 2.35, 22.8 ± 1.81 and 9.1 ± 1.62%, respectively (n = 4–20). To test whether ω-CTx and ω-Aga block part of the L-type current, 10 μM nimodipine was applied in the presence of 1 μM ω-CTx and 300 nM ω-Aga (fig. 7C). Nimodipine was able to block a small portion of the HVA calcium channel current still remaining in the presence of ω-CTx and ω-Aga. Thus ω-CTx and ω-Aga do not seem to block the L-type current.

(A) Sequential applications of various calcium channel blockers including 10 μM nimodipine, 1 μM ω-CTx, 300 nM ω-Aga and 150 μM CdCl2. (B) Currents subtracted before and after the administration of respective HVA channel blockers yield L-, N-, P/Q- and R-type currents. (C) Nimodipine 10 μM, applied after 1 μM ω-GTx and 300 nM ω-Aga, still blocks the HVA channel current. L-, N-, P/Q- and R-type currents represent 10.5 ± 1.10%, 62.8 ± 2.35%, 22.8 ± 1.81%, and 9.1 ± 1.62% of the total current, respectively (n = 4–20).
Riluzole Inhibition of Various Types of HVA Calcium Channel Currents
Because riluzole affects only one of the two groups contained in the steady-state inactivation curve (fig. 4A), riluzole may have selective actions on some of the HVA calcium channels. To prove this hypothesis, riluzole inhibition of HVA calcium channel currents was studied in the presence of various calcium channel blockers. As shown in figure 8, 30 μM riluzole blocked HVA calcium channel currents by 17.1 ± 0.83%, 17.1 ± 1.49%, 3.9 ± 0.58% and 9.3 ± 1.45% in the control (n = 30), in the presence of 10 μM nimodipine (n = 11), 1 μM ω-CTx (n = 11) and 300 nM ω-Aga (n = 10), respectively. Thus, ω-CTx-GIVA and ω-Aga-IVA, but not nimodipine, partially block riluzole inhibition of HVA calcium channel currents.

Riluzole inhibition of various HVA channel currents. Riluzole at 30 μM inhibits 17.1 ± 0.83, 17.1 ± 1.49, 3.9 ± 0.58 and 9.3 ± 1.45% in the control (n = 30), in the presence of 10 μM nimodipine (n = 11), 1 μM ω-CTx (n = 11), and 300 nM ω-Aga (n = 10), respectively.
To determine the efficacy of riluzole on various HVA calcium channels, riluzole inhibitory effect was studied further in the absence and presence of different calcium channel blockers in the same cell. Nimodipine did not affect riluzole inhibition of HVA calcium channel currents (fig. 9A), which indicates that the L-type channel is not the target of riluzole. However, riluzole reduced the fast time constant for the decay of tail current in Bay K 8644-modified calcium channel from 0.33 ± 0.039 ms to 0.25 ± 0.034 ms (n = 4, fig. 9, B and C). Thus, riluzole does not affect the native L-type current, but accelerates the deactivation of the Bay K 8644-modified L-type calcium channel.
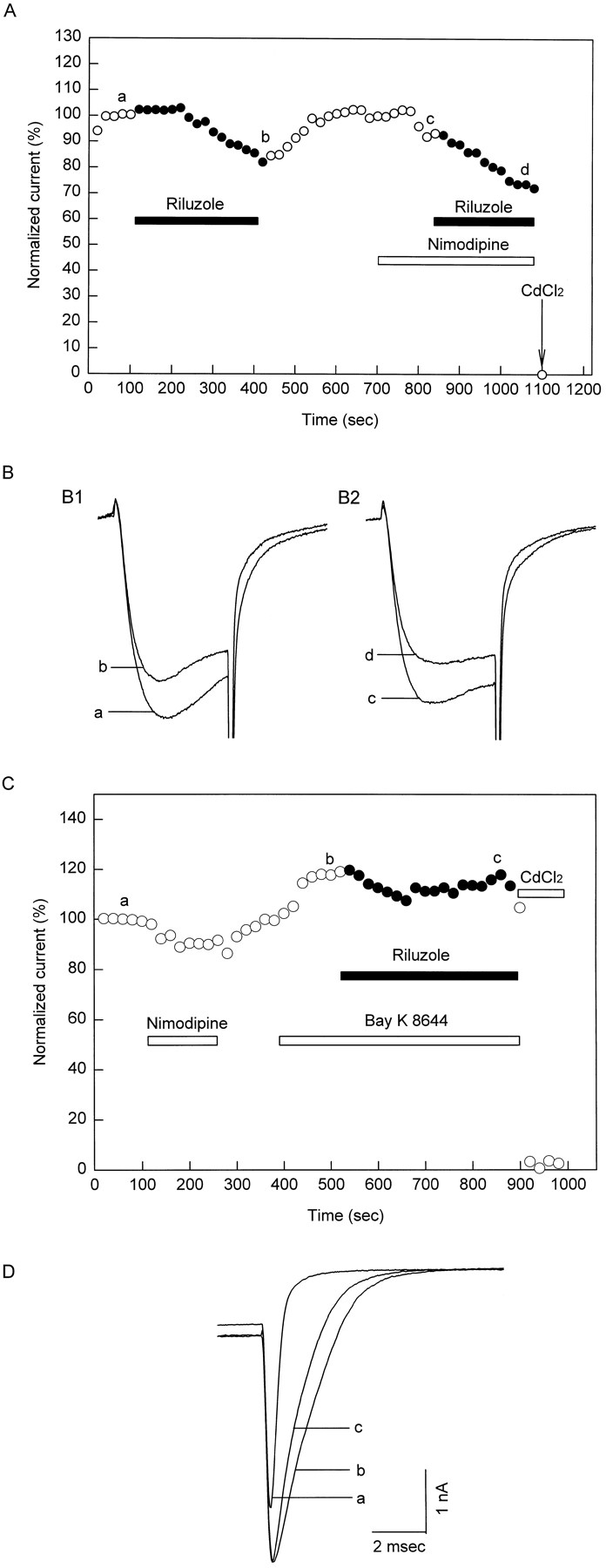
Riluzole inhibitory effect on the HVA channel current is tested in the presence and absence of dihydropyridines in the same cell. (A) Riluzole 30 μM inhibition of HVA channel currents is not affected by 10 μM nimodipine. (B) Riluzole inhibitory effect on HVA channel currents in the presence of 5 μM Bay K 8644. (C) Riluzole still accelerates the decay of the Bay K 8644-modified HVA channel tail current. Thus, 30 μM riluzole does not affect unmodified L-type current, but accelerates the decay of Bay K 8644-modified L-type tail current.
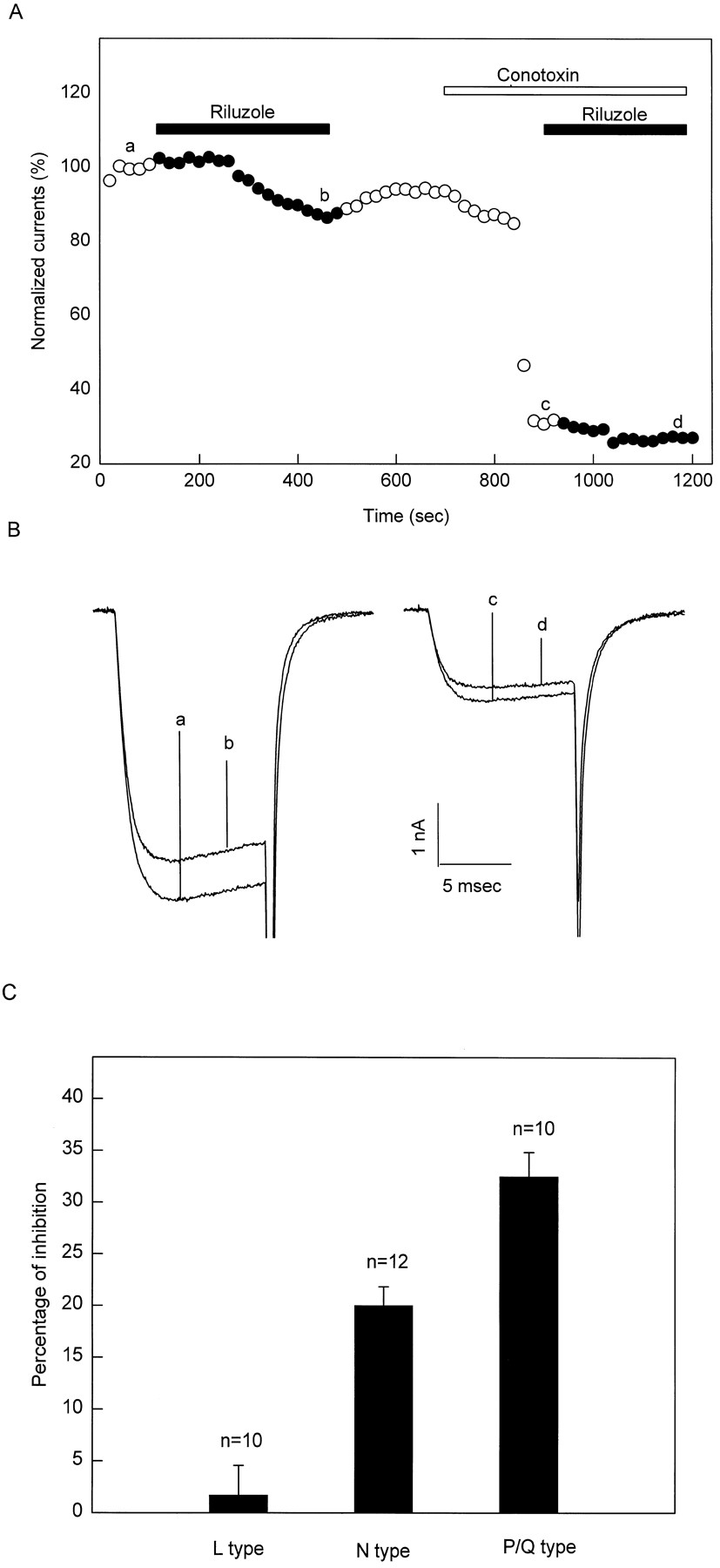
Figure 10A shows the riluzole inhibitory effect on the HVA calcium channel current in the presence and absence of 1 μM ω-CTx in a representative cell. Current traces a to d in figure 10B represent the currents at times a to d in figure10A. Current trace a minus current trace b (a − b), 13.42% of the total HVA current, represents the portion of the total HVA calcium channel current blocked by 30 μM riluzole. Because ω-CTx blocks the N-type current, current trace c minus current d (c − d), 4.6% of the total HVA calcium channel current, equals the portion of the current other than the N-type current blocked by 30 μM riluzole. Thus, (a − b) minus (c − d), 8.8%, equals the N-type current that was blocked by riluzole. The total N-type current equals (a − c) 62.8%. Therefore, the percentage of the total N-type current blocked by 30 μM riluzole is 14.2% (8.8 ÷ 62.8) in this cell. Through similar calculation, 30 μM riluzole blocks 1.7 ± 2.88%, 20 ± 1.84%, 32.5 ± 2.35% of the total L- (n = 10), N- (n = 12) and P/Q (n = 10)-type currents, respectively (fig. 10C). Thus, 30 μM riluzole blocks P/Q- and N-type calcium currents selectively.

(A) Riluzole at 30 μM inhibits the current to a lesser extent in the presence of 1 μM ω-CTx than in the absence. (B) Current traces a to d were recorded to at times a to d in A. (C) Riluzole at 30 μM inhibits L-, N- and P/Q-type currents 1.7 ± 2.88%, 20 ± 1.84% and 32.5 ± 2.35%, respectively.
Discussion
The HVA channels in neonatal rat DRG neurons contained various types with relative fractions of N- ≫ P/Q- > L- ∼R-type. Riluzole inhibited these channel types with relative efficacies of P/Q- > N- ≫ L-type.
HVA calcium channels in rat DRG neurons.
There are at least four pharmacologically distinguishable HVA calcium channels in rat DRG neurons, i.e., L-, N-, P/Q- and R-type. Without separating the Q-type current from the P-type current, our results show that 62.8% of the HVA channel currents are a ω-CTx-sensitive N-type current, 22.8% are a ω-Aga-sensitive P/Q-type current, and 10.5% are a dihydropyridine-sensitive L-type current. The remaining 9.1% of the HVA channel currents are insensitive to the these blockers but sensitive to Cd++; they are termed as R-type currents.
The HVA calcium channel current of rat DRG neurons contains a strikingly higher percentage of the ω-CTx-sensitive N-type current. This may have several causes. First, calcium channel blockers may have overlapping effects. In chick sensory neurons, ω-CTx reversibly blocks part of the L-type current in addition to the irreversible inhibition of N-type currents (Aosaki and Kasai, 1989). In contrast, dihydropyridines can partially block the N-type current in DRG neurons (Regan et al., 1991). Therefore, it is possible that 1 μM ω-CTx in our study may have blocked the N-type current completely and the L-type current partially, resulting in a larger inhibition of HVA channel currents. However, the administration of ω-CTx was preceded by nimodipine (fig. 7A) at a concentration high enough to block the L-type current completely, and 10 μM nimodipine, preceded by the application of 1 μM ω-CTx, still blocked the HVA channel current (fig. 7C). Thus, this is not likely the case.
Second, rundown of the calcium channel current may have contributed to the calculated percentage of inhibition of HVA channel currents by various inhibitors. However, only cells with a rundown rate less than 1%/min within 10 min after rupture of the membrane were used in our experiments. Furthermore, experiments with calcium channel blockers were completed within 15 min. Thus, rundown of the calcium channel current may have played only a minor role in the calculation of the fraction of various components of HVA channel currents.
Third, developmental changes are known to occur in the HVA calcium channel density in DRG neurons (Bickmeyer et al., 1993;Kostyuk et al., 1993). The HVA channel current responds to calcium channel blockers in an age-dependent manner (Disterhoftet al., 1993), which indicates that the proportion of different HVA channels may also change with age. Therefore, a higher proportion of the N-type current found in pups with an age of 1 to 5 days in our study may be because of the early age of development. Because the N-type calcium channels are located mostly at the nerve terminals controlling neurotransmitter release (Dutar et al., 1989; Leveque et al., 1994), their high expression in DRG neurons at the early age of development may play a major role in the synaptic plasticity and transmission.
HVA calcium channel currents other than the N-type current may also play an important role in neurotransmission (Lundy et al., 1991; Hillyard et al., 1992; Mintz et al., 1992b). P-type currents are inhibited by γ-aminobutyric acid in rat Purkinje neurons (Mintz and Bean, 1993) and potentiated by adenosine in guinea pig hippocampal neurons (Mogul et al., 1993). Furthermore, several ligands including carbachol and (−)-baclofen inhibit the Q-type currents in rat hippocampal neurons. A single type of receptor can be coupled to several types of calcium channels. Activation of the mu opioid receptor in rat DRG neurons modulates multiple components of HVA calcium channel currents, including P-, Q- and N-type calcium currents (Rusin and Moises, 1995).
Differential block of HVA channel currents by riluzole.
Riluzole differentially inhibits HVA channel currents. First, riluzole affects only one of the two components of steady-state inactivation curve for the HVA channel current (fig. 4). Second, ω-CTx and ω-Aga, but not nimodipine, partially abolish riluzole suppression of HVA channel currents, which indicates the involvement of N- and P/Q-type currents and the exclusion of L-type current in riluzole’s inhibitory effect (fig. 8). Third, the inhibitory action of riluzole on the various types of HVA channel currents is in the order of P/Q- > N- ≫ L-type currents (fig. 10C). Because P- and Q-type currents are different in the inactivation kinetics and the sensitivity to ω-Aga (Randall et al., 1993; Randall and Tsien, 1995; Rusin and Moises, 1995), riluzole may not block P- and Q-type currents equally.
Although nimodipine does not block riluzole inhibition of HVA channel currents, thereby excluding its effect on the L-type current, riluzole accelerates the decay of the BAY K 8644-modified HVA channel tail current (fig. 9C). This indicates that the Bay K 8644-modified L-type current and unmodified L-type current may have different affinities for riluzole. This is in line with the evidence that the BAY K 8644-modified L-type channel was different from the unmodified L-type channel in their activation and deactivation kinetics (Grabner et al., 1996).
Mechanism of riluzole block of HVA calcium channel currents.
The kinetic values of deactivation and inactivation of HVA channel currents are altered by riluzole, but the activation kinetics remains unchanged. Riluzole selectively blocks the sodium channel in the inactivated state without affecting the activation kinetics (Hebertet al., 1994; Benoit and Escande, 1991; Song et al., 1996). However, there are differences between sodium and calcium block by riluzole. Riluzole blocks sodium channels rapidly and the effect is reversed rapidly after washout, which suggests a direct action on the channel (Benoit and Escande, 1991; Song et al., 1996). In contrast, the slow time course of the onset and offset of riluzole inhibitory effect on HVA channel currents suggests an indirect mechanism involving riluzole interactions with some intracellular components. This idea is further supported by the rebound phenomenon after washout of riluzole, reminiscent of the rebound of baclofen effect on the calcium channel (Matsushima et al., 1993).
Several lines of evidence indicate that G proteins may be involved in riluzole neuroprotective effects. Riluzole inhibition of glutamate-induced aspartate release from cerebellar granule cells is abolished by pretreatment of the cells with pertussis toxin (Dobleet al., 1992). Furthermore, riluzole inhibition of NMDA-, but not veratridine-induced neurotoxicity, is abolished by pertussis toxin pretreatment (Malgouris et al., 1994; Hubert et al., 1994). Riluzole inhibition of HVA channel currents was eliminated by GDP-β-S or N-ethylmaleimide in the internal solution, whereas pretreatment of DRG neurons with pertussis toxin failed to attenuate riluzole’s inhibitory effect, which suggests the involvement of the pertussis toxin-insensitive G proteins in riluzole modulation of HVA channels (Huang et al., in press, 1997).
Because calcium channels control the presynaptic neurotransmitter release, riluzole inhibition of P/Q- and N-type calcium channels may reduce glutamate release in presynaptic terminals and thus avoid excess activation of NMDA receptors, which would cause massive calcium influx leading to cell death in ischemic brain insult.
Acknowledgments
We thank Julia Irizarry for secretarial assistance.
Footnotes
-
Send reprint requests to: Dr. Toshio Narahashi, Department of Molecular Pharmacology and Biological Chemistry, Northwestern University Medical School, 303 E. Chicago Avenue, Chicago, IL 60611-3008.
-
↵1 This work was supported by National Institutes of Health Grant NS14144.
- Abbreviations:
- HVA
- high voltage-activated
- DRG
- dorsal root ganglion
- DMEM
- Delbecco’s Modified Eagle Medium
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- DMSO
- dimethylsulfoxide
- ω-CTx
- ω-conotoxin GVIA
- ω-Aga
- ω-agatoxin IVA
- I-V
- current-voltage
- NMDA
- N-methyl-d-aspartate, EGTA, [ethylenebis(oxyethylenenitrilo)]tetraacetic acid,
- Received November 14, 1996.
- Accepted May 9, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}