

Abstract
Nitric oxide and cGMP influence plasticity of nociceptive processing in spinal cord. However, effectors for cGMP have not been identified in sensory pathways. We now demonstrate that cGMP-dependent protein kinase I (cGKI) occurs in the DRGs at levels comparable to that in cerebellum, the richest source of cGKI in the body. Immunohistochemical studies reveal that cGKI is concentrated in a subpopulation of small- and medium-diameter DRG neurons that partially overlap with substance P and calcitonin gene-related polypeptide containing cells. During development, cGKI expression throughout the embryo is essentially restricted to sensory neurons and to the spinal floor and roof plates. Neuronal nitric oxide synthase (nNOS) is coexpressed with cGKI in sensory neurons during embryonic development and after peripheral nerve axotomy. The primary target for cGKI in cerebellum, G-substrate, is not present in developing, mature, or regenerating sensory neurons, indicating that other proteins serve as effectors for cGKI in sensory processing. These data establish sensory neurons as a primary locus for cGMP actions during development and suggest a role for cGKI in plasticity of nociception.
Nitric oxide (NO) serves as a cellular mediator for diverse developmental and physiological processes (Marletta, 1993;Moncada and Higgs, 1993; Bredt and Snyder, 1994a; Nathan and Xie, 1994;Garthwaite and Boulton, 1995). Neuronal type NO synthase (nNOS, or type I) is localized to a discrete population of neurons in the embryonic and mature nervous system (Bredt et al., 1991; Bredt and Snyder, 1994b). nNOS is a calcium/calmodulin-dependent enzyme that is stimulated by activation of NMDA type glutamate receptors (Garthwaite et al., 1988; Knowles et al., 1989; Bredt and Snyder, 1990). NO formed during excitatory transmission participates in synaptic plasticity mediated by NMDA receptor activity (Schuman and Madison, 1994).
A role for NO in sensory signaling was initially suggested based on the specific localization of nNOS in sensory pathways (Bredt et al., 1990;Aimi et al., 1991). In normal adult mammals, nNOS is expressed in a few small- and medium-diameter DRG neurons, and in most of these cells, nNOS coexists with calcitonin gene-related peptide (CGRP) and rarely with substance P (Zhang et al., 1993). By contrast a large percentage of DRG neurons contain nNOS transiently during embryonic development (Bredt and Snyder, 1994b) and after peripheral nerve damage (Zhang et al., 1993). Direct evidence for involvement of NO in plasticity of sensory processing derives from pharmacological studies. NOS antagonists do not alter baseline responses indicating that NO does not function as a fast transmitter at primary afferent synapses. NOS antagonists, however, do prevent spinal sensitization of nociceptive reflexes (Moore et al., 1991; Kitto et al., 1992; Meller et al., 1992;Meller and Gebhart, 1993). This NO-dependent sensitization contributes to hyperalgesia after peripheral injury and is also blocked by NMDA receptor antagonists (Dickenson, 1990).
NO signaling occurs primarily through activation of soluble isoforms of guanylyl cyclase. In the vascular and nervous systems, cGMP-dependent protein kinases (cGKs) serve as major effectors for NO and cGMP (Lincoln et al., 1994). Endothelial-derived NO stimulates guanylyl cyclase and subsequently cGK in vascular smooth muscle leading to blood vessel relaxation (Ignarro, 1993; Moncada and Higgs, 1993; Warner et al., 1994). Similarly, physiological actions of NO in sensory pathways appear to involve cGMP (Meller and Gebhart, 1993). NO-mediated hyperalgesia is blocked by extracellular hemoglobin (Kitto et al., 1992), indicating a role for intercellular NO signaling. Furthermore, membrane permeable cGMP analogs mimic the effects of NO on sensory neuron responsiveness (McGehee et al., 1992; Niedbala et al., 1995). Downstream effectors for cGMP, however, have not previously been characterized in sensory neurons.
Molecular studies have identified two genetic loci encoding mammalian cGKs. The first gene product characterized, cGKI, was purified and sequenced from soluble extracts of vascular smooth muscle from bovine lung (Lincoln et al., 1977; Flockerzi et al., 1978; Takio et al., 1984). Molecular cloning of the cDNA demonstrated N-terminal alternative splicing yielding cGKIα and cGKIβ isoforms (Wernet et al., 1989). More recently, a membrane-associated cGKII has been cloned from brain (Uhler, 1993) and from the epithelium of intestinal mucosa (Jarchau et al., 1994). In the CNS cGK enzyme activity is selectively enriched in the cerebellum where high levels of cGKI protein are histochemically apparent in Purkinje neurons (Lohmann et al., 1981). Low levels of cGK activity found in forebrain tissues appear to primarily represent cGKI from vascular smooth muscle (Lohmann et al., 1981). A prominent target for cGK, G-substrate, is exclusively found in cerebellum (Detre et al., 1984).
The prominent functions for NO and cGMP in sensory plasticity led us to investigate the possible expression of cGKs in sensory neurons. Strikingly, we demonstrate that cGKI occurs in DRG at levels equivalent to that in cerebellum. cGKI is found in small- and medium-diameter neurons of DRG and is concentrated in axonal processes in laminae I and II of spinal cord, consistent with known roles for NO and cGMP in nociceptive processing. During embryogenesis, cGKI is found only in DRG neurons and cells of the spinal floor and roof plates. Whereas cerebellar Purkinje cell soma contain both cGKI and G-substrate, sensory neurons lack G-substrate, suggesting that novel targets mediate actions of cGKI in sensory neuron development and plasticity.
MATERIALS AND METHODS
Antisera. The following primary polyclonal rabbit antibodies were used: cGKI (Lohmann et al., 1981; Wyatt et al., 1991), nNOS (Bredt et al., 1990), substance P (Peninsula Laboratories), CGRP (Peninsula Laboratories). For double-immunofluorescence labeling, a polyclonal guinea pig antiserum to substance P was used (Too and Maggio, 1991).
Affinity purification of G-substrate antibody. Polyclonal antibody to G-substrate (Dolphin et al., 1983) was further affinity purified by presorbing nonspecific antibodies with a crude cerebral cortical extract. Rat cerebral cortex was homogenized in 10 volumes of 25 mm Tris-HCl buffer, pH 7.4, 1 mm EDTA, 100 μm PMSF and diluted to 5 mg protein/ml. Ten micrograms of antibody to G-substrate (Dolphin et al., 1983) were incubated overnight with 5 mg of cortical extract. Soluble antibodies were recovered by filtration through a 0.44 μm mesh.
Western blotting. Tissues were dissected and homogenized in 10 volumes of Tris-HCl buffer, pH 7.4, containing 1 mm EDTA and 100 μm PMSF. Acid soluble extracts were prepared using the method of Aswad and Greengard (1981). Proteins were separated by SDS-PAGE and transferred to PVDF membranes, and blots were incubated in blocking solution containing 3% bovine serum albumin in PBS and 0.1% Tween 20. Antibodies were diluted in this same buffer and hybridized to blots overnight. Immunoreactive bands were visualized by ECL according to the manufacturer’s specifications (Amersham, Arlington Heights, IL).
Immunohistochemistry. Adult and embryonic rats were anesthetized with pentobarbital and perfused with 4% freshly depolymerized paraformaldehyde in 0.1 m phosphate buffer. Tissues were removed and postfixed in paraformaldehyde for 1 hr at 4°C. Tissues were cryoprotected overnight in 20% sucrose. Thin sections were cut on a cryostat (6 μm), and free-floating sections (40 μm) were cut on a sliding microtome. Endogenous peroxidase activity was inactivated by incubating sections in 0.5% H2O2 for 15 min. Sections were blocked for 1 hr in PBS containing 1.5% NGS and then incubated overnight in the same buffer containing diluted antiserum. Immunohistochemical staining used an avidin/biotin/peroxidase system (ABC Elite; Vector Laboratories, Burlingame, CA) according to the manufacturer’s instructions. Peroxidase staining was developed using 3,3′-DAB as the chromogen. For indirect immunofluorescence, secondary goat anti-rabbit Cy-3, and donkey anti-guinea pig FITC conjugated antibodies were used according to the manufacturer’s specifications (Jackson Laboratories, 1:200).
Surgery. Lesions of the rat sciatic nerve were performed by exposing the right nerve in the mid thigh and crushing it for 10 sec with a number 6 jeweler’s forceps. After the lesion, the skin was sutured and the animals were housed in isolation for 18 d. Affected DRGs were isolated by tracing the sciatic nerve back to the spinal cord (L4 or L5). Isolated DRGs were dissected, fixed in paraformaldehyde, and stained as described above.
RESULTS
cGKI is expressed at high levels in DRG extracts
We first characterized the specificity of antisera to cGKI and nNOS by Western blotting. Crude protein extracts from lumbar DRG, cerebellum, and cerebral cortex of an adult rat were resolved by SDS-PAGE and probed with antisera to cGKI and nNOS (Fig.1A,B). Each antiserum reacts specifically with a single band of appropriate molecular weight. cGKI (MW ∼ 74,000) occurs at comparable levels in DRG extracts as in cerebellum, the richest known source of cGKI in the body (Fig. 1A). As previously reported (Lohmann et al., 1981), cGKI is present at very low levels in forebrain extracts. nNOS (MW ∼ 160,000) occurs most prominently in cerebellar extracts and is only faintly detectable in mature DRG tissue (Fig. 1B). To ensure specificity of immunohistochemical staining (see below) we used two different affinity purified antisera to cGKI. Both of these antisera were raised against intact purified cGKI protein and have been well characterized in previous immunohistochemical studies (Lohmann et al., 1981; Wyatt et al., 1991). Both antisera give essentially identical patterns of staining in all tissues examined. For consistency, all cGKI histochemistry presented here used the affinity purified antiserum described by Wyatt et al. (1991). Immunoreactivity was not observed in sections labeled with nonimmune serum or in incubations lacking primary antibody (data not shown).

cGMP-dependent protein kinase but not nNOS or G-substrate is enriched in dorsal root ganglion extracts. A, Western blotting shows that cGKI occurs at similar levels in crude extracts of DRGs as in cerebellum (Cb) but is only weakly detectable in cerebrocortical (Cx) tissue. B, nNOS is most enriched in cerebellum, is moderately expressed in cerebral cortex, and is only faintly detectable in DRG. C, G-substrate is only detectable in cerebellar extracts and does not occur in cortex or DRG. A, B, Crude tissues were homogenized in 25 mm Tris-HCl buffer, pH 7.4, containing 1 mm EDTA, 100 μm PMSF, and 0.5% Triton X-100. Soluble proteins were resolved by 10% SDS-PAGE (80 μg/lane). ForC, acid-soluble extracts were separated on a 15% SDS-PAGE gel (50 μg/lane). Immunoreactive bands were detected by ECL. Positions of molecular weight standards (in kDa) are indicated on theleft.
cGKI occurs in small- and medium-diameter sensory neurons
Immunohistochemical analysis reveals that cGKI is discretely localized to specific neurons in lumbar DRG (Fig.2A). cGKI staining of DRG cell bodies is heterogeneous; some cells are intensely stained, others stain weakly and some not at all. DRG neurons intensely stained by cGKI were all of small (<32 μm) or medium (32–50 μm) diameter, the class of neurons involved in nociceptive processing. Weakly stained neurons were of both medium and large (>50 μm) cell diameters. Because many small- and medium-diameter sensory neurons contain substance P or CGRP, neuropeptides that participate in nociceptive transmission (Hokfelt, 1991), we stained adjacent thin (6 μm) sections for cGKI and substance P or CGRP. We found that neurons intensely stained for cGKI partially overlap with neurons containing substance P (Fig.3A,A′) or CGRP (Fig. 3B,B′). We also conducted double-immunofluorescent labeling studies of L5 DRG using antibodies to cGKI and substance P (Fig. 3C,C′). We found that cGKI immunoreactivity was found in essentially all substance P positive cells, whereas substance P occurred in 64/356 cGKI containing neurons.

cGKI is enriched in small- and medium-diameter DRG neurons, and expression does not change after axotomy. A, cGKI is heterogeneously expressed in specific neurons of the rat L5 DRG. Examples of densely stained small- and medium-diameter neurons are indicated with a solid arrow. Unstained large cells are identified by an open arrow (magnification 200×).B, C, Expression of cGKI does not dramatically change 18 d after unilateral sciatic nerve axotomy as a similar pattern and density of staining of DRG cells is noted contralateral (CN) and ipsilateral (IP) to the lesion (100×).D, E, nNOS expression is dramatically induced in ipsilateral L5 DRG neurons 18 d after sciatic nerve axotomy (100×). Ten micrometer cryostat sections of DRG were processed for cGKI or nNOS immunohistochemistry and photographed with light-field microscopy.
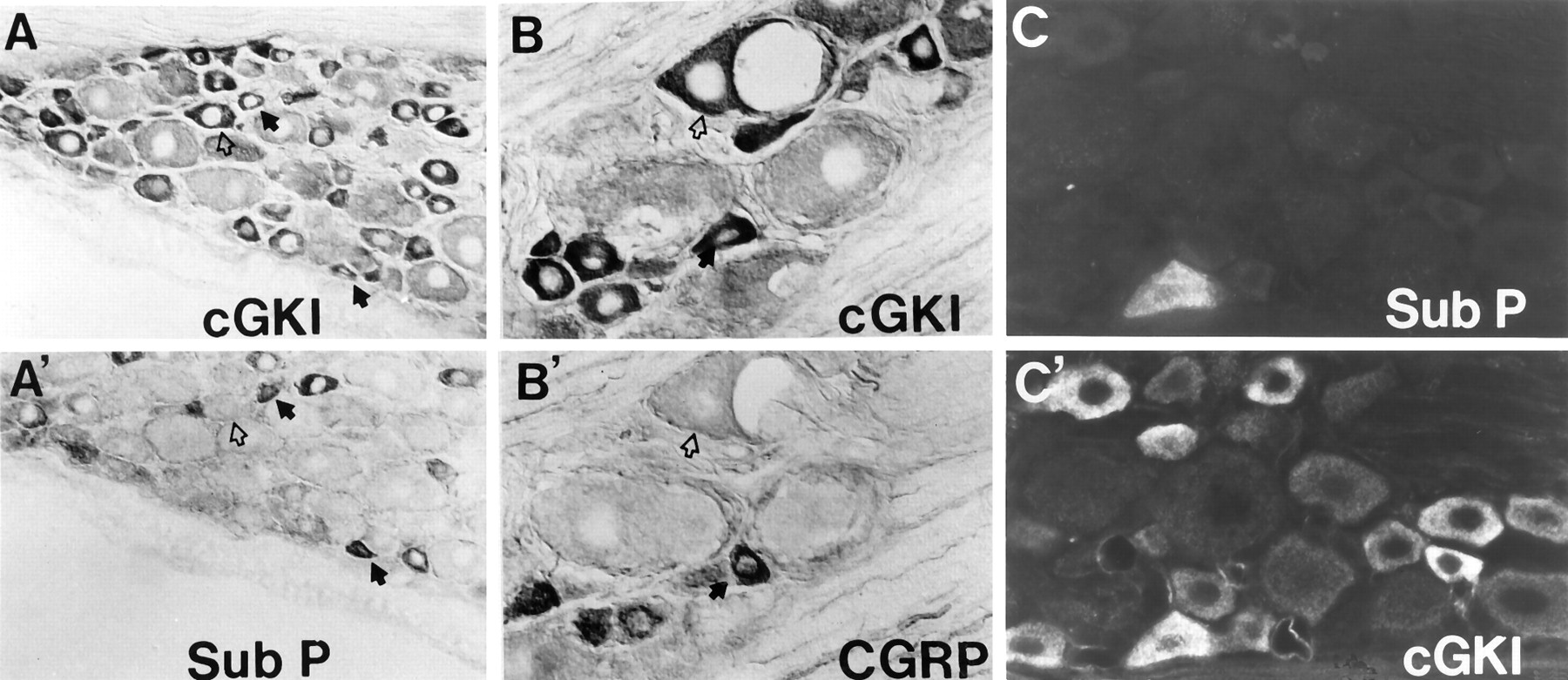
cGKI partially overlaps with substance P and CGRP in DRG neurons. Staining of adjacent 6 μm sections shows that cGKI (A) occurs in a greater fraction of DRG cells than substance P (A’). In certain cells, cGKI coexists with substance P (solid arrow), whereas others contain cGKI but not substance P (open arrow) (200×). A cell labeled for cGKI and CGRP (solid arrow) is shown in the middle panels(B, B’). These panels also show a medium-diameter neuron stained for cGKI but not CGRP (open arrow) (400×). Double-immunofluorescent labeling shows colocalization of cGKI (C) and substance P (C’) in a single neuron.
nNOS, the upstream activator cGKI, is present in only 1–2% of rat lumbar DRG neurons (Aimi et al., 1991). After peripheral nerve axotomy, nNOS expression is induced in specific DRG neurons, that correspond to small- and medium-diameter cells (Zhang et al., 1993). To determine the relationship between nNOS and cGKI, we performed unilateral sciatic nerve axotomy and evaluated cGKI and nNOS labeling of DRG cells ipsilateral and contralateral to the lesion. We confirmed dramatic induction of nNOS after axotomy (Fig. 2D,E) but found no significant change in cGKI staining up to 18 d after the lesion (Figs.2B,C).
Central projections of primary nociceptors terminate in laminae I and II of the spinal cord. Immunohistochemical staining of L5 spinal cord segments demonstrated cGKI in nerve terminals of lamina I and II (Fig.4). Spinal neurons and white matter tracks are devoid of cGKI staining. The distribution of cGKI in dorsal spinal cord is similar to that of substance P (Fig. 4) (Chung et al., 1988). nNOS containing fibers, by contrast, are found in all layers of the spinal cord, but are most concentrated in the lamina II, confirming previous studies (Fig. 4) (Bredt et al., 1991; Zhang et al., 1993). There are also many nNOS containing small interneurons that are most numerous in lamina II.

cGKI is colocalized with substance P in dorsal spinal cord. A, cGKI is restricted to neuronal fibers in laminae I and II of the spinal cord. cGKI is not present in white matter tracks of the dorsal columns (DC) or in spinal neuron cell bodies (50×). B, cGKI also stains the vascular smooth muscle of blood vessels (BV, 100×). C, nNOS occurs in neuronal processes and throughout the dorsal horn (DH) and is present in occasional spinal interneurons (100×). D, Substance P is colocalized with cGKI in laminae I and II of the dorsal horn (100×).
cGKI is concentrated in embryonic sensory neurons and spinal floor plate
Although present in only few mature DRG neurons, nNOS is transiently expressed in many sensory neurons during embryonic development (Bredt and Snyder, 1994b). We therefore evaluated expression of cGKI during development. Western blotting indicated that cGKI is present in extracts of DRG from embryonic day 15 (E15) rat, but that cGKI is barely detectable in brain extracts at this stage (Fig.5). nNOS is found at similar levels in E15 DRG and brain extracts. Immunohistochemistry of rat embryos demonstrated that cGKI is restricted to developing sensory pathways (Fig. 6). In sagittal sections of E15 embryos, cGKI is concentrated in cell bodies of the DRG, as well as fine processes in dorsal spinal cord and periphery. cGKI is not apparent in any other cell type or tissue in E15 embryos. Micrographs of horizontal sections revealed that cGKI occurs in sensory ganglia as early as E12 (Fig. 7A). Interestingly, cGKI is also prominently expressed in the spinal floor and roof plate at E12 (Fig. 7B). Spinal neurons themselves do not express cGKI in embryonic animals (Figs. 7B,E). By E15 labeling of the floor plate is nearly absent, whereas cGKI staining remains prominent in a subset of sensory neurons (Figs.7C–E). Immunostaining occurs both in cell bodies and in central and peripheral axons of embryonic sensory neurons. cGKI labeling of sensory projections to the dorsal spinal cord (Figs.7A,C,E) accounts for staining seen in dorsal cord (Fig. 6). cGKI also labels cutaneous branches of sensory axons throughout the embryo (Fig. 7G), which accounts for staining of fine processes in the periphery seen at low magnification (Fig. 6).

cGKI is uniquely enriched in DRG tissue extracts during embryonic development. A, Western blotting shows that cGKI is present in DRG, but not brain tissue, in E15 rat extracts. cGKI from E15 DRG comigrates with a similar 74 kDa band from adult cerebellum (Cb). B, Probing a duplicate blot shows that nNOS is expressed at comparable levels in E15 brain and DRG extracts. Solubilized tissue extracts were separated by 10% SDS-PAGE. Five times more protein was loaded from embryonic tissues (100 μg/lane) than from cerebellum (20 μg/lane). Note that E15 DRG extracts were unavoidably contaminated with adjacent spinal tissues and may therefore underestimate true enrichment of cGKI.

cGKI and nNOS are colocalized in embryonic sensory neurons. At E15, cGKI is apparent only in cells and axonal processes of sensory neurons. Intense staining of the DRG, dorsal cord (DC), and peripheral cutaneous axons (Ax) is noted. nNOS is present in DRG but also occurs in olfactory epithelium (O), cerebrocortical plate (CP), kidney (K), adrenal gland (A), and intestine (In), all of which lack cGKI. Twenty micrometer cryostat sections of E15 rat embryos were stained with antisera to cGKI or nNOS. In these dark-field images, positive staining appears white. Note that fixation conditions for cGKI and nNOS immunohistochemistry are somewhat different and do not allow staining of adjacent sections from the same embryo.

cGKI expression in embryonic spinal ganglia and floor plate. A, At E12, cGKI is specifically enriched in DRG neurons (100×) and cells of the floor plate (FP) and roof plate (B; 400×). Spinal neurons themselves are unstained, and weak labeling of blood vessels (BV) is detected at this stage. C, D, Parasagittal sections of E15 embryos show that cGKI is enriched in a subset of DRG neurons and is present in both peripheral axons (AX) and central projections to the dorsal spinal cord (DSC). E, F, At E15, nNOS is colocalized with cGKI in DRG and in axonal projections to the dorsal spinal cord (50×). Note that nNOS occurs in certain spinal neurons such as those of the intermediolateral column (IL) that lack cGKI. G, H, Both nNOS cGKI also colocalize in cutaneous branches of sensory axons such as those depicted here near the snout that derive from the trigeminal ganglia.
To evaluate possible regulation of cGKI by NO during development, we stained appropriate sections with antibodies to nNOS. Low-power dark field images show that nNOS occurs together with cGKI in E15 DRG cells and sensory projections (Fig. 6). As previously reported, nNOS also occurs in other embryonic sensory cells such as primary olfactory neurons (Bredt and Snyder, 1994b; Roskams et al., 1994), which lack cGKI (Fig. 6). nNOS immunoreactivity is also apparent in cerebral cortical plate, neurons of the midbrain, myenteric neurons, epithelial cells of the gastrointestinal tract, and certain epithelial cells of the embryonic kidney all of which lack cGKI (Fig. 6) (Bredt and Snyder, 1994b). High-power micrographs show that nNOS is colocalized with cGKI in E15 DRG cells as well as the central and peripheral projections of these sensory neurons (Fig. 7F,H). As previously reported (Bredt and Snyder, 1994b), nNOS is also present in embryonic spinal neurons of the intermediolateral cell column that lack cGKI (Fig.7E,F).
G-substrate is enriched in Purkinje cell soma but not in sensory neurons
Purkinje neurons of the cerebellum, the only other neuronal cell type to express cGKI at high density (Lohmann et al., 1981), are also enriched with G-substrate (Detre et al., 1984), a potent protein target for cGKI. This localization was based on studies showing decreased G-substrate levels in the cerebellum of mutant mice lacking Purkinje cells (Detre et al., 1984). To directly evaluate the distribution of G-substrate in cerebellum and DRG, we generated an affinity purified antiserum by taking advantage of the absence of G-substrate in cerebral cortex (Detre et al., 1984). A G-substrate antiserum (Nairn et al., 1982; Dolphin et al., 1983) was preabsorbed with crude protein extracts from rat cerebral cortex to remove cross-reacting antibodies. Western blotting of cerebellar extracts indicates that this affinity purified antiserum reacts with a major 23,000 Da band, corresponding to G-substrate (Detre et al., 1984). By contrast, no bands were detected in extracts of cerebral cortex or DRG (Fig. 1C). We also failed to detect G-substrate in brain or DRG extracts from embryonic rat (data not shown).
Immunohistochemical studies demonstrate that G-substrate is most concentrated in Purkinje neurons of the cerebellum (Fig.8). Neuronal fibers in thalamus and pyramidal layer of the hippocampus also contain G-substrate immunoreactivity. cGKI is concentrated together with G-substrate in Purkinje cells of the cerebellum as previously reported (Fig. 8; Lohmann et al., 1981). cGKI is also enriched in pyramidal cells and dentate granule cells of the hippocampus. By comparison, nNOS displays a more general neuronal distribution, as described previously (Bredt et al., 1990). High power micrographs of the cerebellum demonstrate that G-substrate is specifically concentrated in Purkinje cell soma (Fig.9). By contrast, cGKI is present throughout the Purkinje cell, in the apical dendrites, cell soma and axon as reported previously (Lohmann et al., 1981). Specific G-substrate or cGKI immunoreactivity is not apparent in cells in the granular layer or molecular layers of cerebellum. nNOS in cerebellum occurs in a complementary distribution, in basket cells of the molecular layer and granule cell neurons as reported previously (Bredt et al., 1990).

G-substrate and cGKI are colocalized in cerebellum and hippocampus. Immunohistochemistry demonstrates that (A) cGKI is expressed at highest levels in Purkinje cell processes of the molecular layer of the cerebellum (Cb). Lower levels of cGKI are apparent in dentate neurons and pyramidal cells of the hippocampus.B, G-substrate is also enriched in the cerebellum but is restricted to a linear layer of cells that correspond to Purkinje cell soma. The thalamus (T) contains G-substrate immunoreactivity but lacks cGKI. The cerebral cortex and the rest of the forebrain are essentially devoid of cGKI or G-substrate. C, nNOS is also most enriched in cerebellum but displays a broader forebrain distribution than cGKI or G-substrate. Forty micrometer free-floating rat brain sections were stained as described above. In these dark-field images, positive labeling appears white.

G-substrate and cGKI are coexpressed in Purkinje cell soma. C, D, Immunoperoxidase staining of sagittal sections of rat cerebellum shows that G-substrate is specifically restricted to Purkinje (P) cell bodies and does not stain dendritic processes in the molecular layer (M) or axons in the granule cell layer (G). A,B, cGKI is also uniquely expressed in Purkinje cells but stains dendrites and axons as well as the cell body. E,F, nNOS shows a complimentary distribution to that of G-substrate and cGKI and is uniquely absent from Purkinje cells but, instead, stains granule cells in the granule cell layer and basket cells in the molecular layer.
DISCUSSION
A principle finding of this work is that a subset of sensory neurons is dramatically enriched in cGKI. Previous biochemical and histochemical studies suggested that Purkinje neurons of the cerebellum were the only cells to be enriched with cGKI at levels comparable to those in vascular smooth muscle (Lohmann et al., 1981). Much lower levels of cGKI have been detected in other cell types including macrophages, neutrophils, osteoclasts, and vascular endothelial cells (for review, see Lincoln, 1995). In these tissues cGKI is associated with cytoskeletal proteins such as vimentin (MacMillan-Crow and Lincoln, 1994). Similarly, we have recently noted that cGKI is present at low levels in skeletal muscle cells and is selectively associated with neuromuscular endplates (D. Chao, F. Silvagnano, T. Cornwell, T. Lincoln, and D. Bredt, unpublished observations).
The abundance of cGKI in small- and medium-diameter neurons of the DRG has important implications for mechanisms of NO-mediated nociceptive processing. In several animal models NOS inhibitors have been found to block hyperalgesia subsequent to peripheral tissue injury (Moore et al., 1991; Niedbala et al., 1995). This sensitization is mediated by NMDA receptors and occurs at the level of the spinal cord. In support of this conclusion, intrathecal administration of NOS inhibitors or NMDA receptor antagonists prevents hyperalgesia produced by a variety of experimental stimuli without affecting normal reflex function (Dickenson, 1990; Meller and Gebhart, 1993). Furthermore, intrathecal administration of either an NO donor or NMDA itself produces a marked hyperalgesia in nociceptive tests (Aanonsen and Wilcox, 1987; Coderre and Yashpal, 1994). These hyperalgesic actions of NO and NMDA appear to be mediated by activation of guanylyl cyclase as they are mimicked by membrane permeant cGMP analogs and are blocked by guanylyl cyclase antagonists (McGehee et al., 1992; Niedbala et al., 1995).
Consistent with our proposed role for cGKI in spinal sensitization, pharmacological studies suggest that cGKI mediates synaptic plasticity in several other systems. In Purkinje neurons of the cerebellum, cGKI activity is required for expression of long term depression, a model of motor learning (Shibuki and Okada, 1991; but see Linden and Connor, 1995). Similarly, effects of NO and cGMP on hippocampal long term potentiation are believed to be mediated by cGK (Zhuo et al., 1994). Both of these models involve a Hebbian mechanism (Hebb, 1949); that is, coincident activity of the pre- and postsynaptic neuron yields long lasting changes in synaptic efficacy. NO functions as an anterograde messenger in cerebellar long term depression, transmitting information from granule cell axons to Purkinje cell dendrites (Lev-Ram et al., 1995). By contrast, a retrograde function for NO in hippocampal plasticity is suggested by experiments showing that injection of cGK activators into the presynaptic neuron can substitute for postsynaptic neuron activity or NO synthesis (Arancio et al., 1995). Our studies demonstrating high densities of cGKI in laminae I and II of the spinal cord suggest that similar retrograde signaling by NO may participate in spinal sensitization. In this model, neuron-derived NO from the spinal cord would serve as a retrograde messenger to increase cGMP levels and activate cGKI in primary nociceptors. In support of this, high densities of nNOS are found in neuronal fibers and interneurons in superficial layers of the dorsal spinal cord (Fig. 4; Bredt et al., 1991; Zhang et al., 1993).
Presynaptic targets for cGKI that participate in synaptic plasticity are uncertain. In vitro phosphorylation studies identified G-substrate as a prominent target for cGKI in cerebellum (Detre et al., 1984). G-substrate is a small heat-stable protein that shares biophysical properties with certain protein phosphatase inhibitors, such as DARPP-32 and inhibitor 1. Our immunohistochemical studies demonstrate that G-substrate is restricted to cell soma of cerebellar Purkinje cells and is entirely absent from sensory neurons. This restricted distribution makes G-substrate an unlikely mediator of synaptic plasticity. On the other hand, recent studies demonstrate that rabphilin-3A is an efficient substrate for phosphorylation by endogenous cGK in vitro (Fykse et al., 1995). Rabphilin-3A is a Ca-binding synaptic vesicle protein that is implicated in exocytosis or vesicular transport (Shirataki et al., 1993). It will now be important to determine whether rabphilin 3A or other synaptic vesicle proteins serve as the physiological substrates for cGKI that mediate presynaptic plasticity.
Our developmental studies yield insight into mechanisms for NO- and cGMP-mediated actions during embryonic development. nNOS is transiently expressed in developing sensory neurons (Bredt and Snyder, 1994b), and NO has been implicated in neuronal differentiation (Peunova and Enikolopov, 1995) and axon outgrowth (Hess et al., 1993). High levels of cGKI expression restricted to sensory neurons during embryonic development suggest possible functions for this kinase in axonal outgrowth or cellular differentiation of these cells. Further support for this idea derives from our observation of a transient expression of cGKI in the spinal floor plate, a tissue the inductive signals of which specify certain neuronal cell types (Yamada et al., 1991). Interestingly, protein kinase A has recently been noted to play a critical role in specifying neuronal fate during Drosophiladevelopment (Li et al., 1995; Pan and Rubin, 1995). Disregulation of protein kinase A also prevents floor plate-mediated differentiation of dopaminergic neurons of the substantia nigra in mammals (Hynes et al., 1995). It will now be important to evaluate whether modulation of cGKI activity influences development and differentiation of sensory neurons.
Footnotes
This work was supported by grants (to D.S.B.) from the National Science Foundation, the Lucille P. Markey Charitable Trust, and the National Association for Research on Schizophrenia and Depression, the McKnight Endowment Fund for Neuroscience, the Esther A. and Joseph Klingenstein Fund, and National Institutes of Health Grants HL 34646 (T.M.L.) and MH-40899 (P.G.). We thank Allan Basbaum for helpful discussions and for review of this manuscript.
Correspondence should be addressed to David S. Bredt, Department of Physiology, School of Medicine, University of California at San Francisco, 513 Parnassus Avenue, San Francisco, CA 94143-0444.
Dr. Qian’s present address: Department of Psychiatry, San Mateo County General Hospital, San Mateo, CA 94113.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}