

Abstract
A brief exposure to light can shift the phase of mammalian circadian rhythms by 1 hr or more. Neuropeptide Y (NPY) administration to the hypothalamic suprachiasmatic nucleus, the circadian clock in the brain, also causes a phase shift in circadian rhythms. After a phase shift, the neural clock responds differently to light, suggesting that learning has occurred in neural circuits related to clock function. Thus, certain stimuli can produce effects that last for an extended period, but possible mechanisms of this long-term effect have not been previously examined at the cellular level. Here, we report that NPY caused a long-term depression in both electrical activity and intracellular calcium levels of neurons, as studied with whole-cell patch-clamp recording and Fura-2 digital imaging. In contrast to the immediate (1 sec) recovery after relief from glutamate receptor blockade, a brief single application of NPY (100 nm) depressed cytosolic Ca2+ for >1 hr. The mechanism of this long-term calcium depression, a form of cellular learning, is dependent on the simultaneous release of glutamate and activation of NPY receptors, because both the extended response to NPY and any aftereffect were blocked by coapplication of glutamate receptor antagonists. Postsynaptic actions of NPY, mediated by both Y1- and Y2-like receptors, were short term and recovered rapidly. The primary site of long-term NPY actions may be on presynaptic glutamatergic axons, because the frequency of miniature excitatory postsynaptic currents in the presence of tetrodotoxin was reduced by transient exposure to NPY in both cultures and slices.
- suprachiasmatic nucleus
- mammalian circadian clock
- glutamate
- NPY
- LTD
- learning biological clock
- phase-shift
- neuroendocrine
- hypothalamus
In the absence of environmental cues, the brain’s circadian clock in the suprachiasmatic nucleus (SCN) generates a daily rhythm that is remarkably accurate in its timing (for review, see van den Pol and Dudek, 1993). The phase of this rhythm can be shifted by photic stimulation of the retina via glutamatergic pathways that project directly to the SCN (Meijer et al., 1988; Cahill and Menaker, 1989; Colwell et al., 1991; Kim and Dudek, 1991) or to the intergeniculate leaflet (IGL) and then to the SCN (Harrington et al., 1985; Card and Moore, 1989). Stimulation or damage to the neuropeptide Y (NPY)-containing cells of the IGL or direct stimulation of the SCN with NPY causes a significant phase shift of circadian rhythms (Harrington and Rusak, 1986; Johnson et al., 1989; Rusak et al., 1989), as does altering glutamate transmission to the SCN (Meijer et al., 1988; Colwell et al., 1991).
Neurons of the circadian clock respond to light differently depending on the phase of the circadian clock. A pulse of light at one part of the circadian cycle will advance the phase, whereas a similar pulse at a different part of the cycle will delay the phase (Meijer et al., 1988; Nelson and Takahashi, 1991). Furthermore, photic stimulation at certain times of day alters the response of the clock neurons to photic stimulation at later times in the same 24 hr period. This change in the response to light can be considered a novel form of learning and is consistent with Thompson’s (1967) definition of learning as a response modification resulting from experience.
A single pulse of light (Nelson and Takahashi, 1991) or a single application of NPY (Albers and Ferris, 1984; Medanic and Gillette, 1993; Huhman and Albers, 1994) can generate a phase shift of 1 hr or more, suggesting a change in neuronal behavior may occur during this period. This hypothetically could be achieved by a temporally extended change in neuronal activity, followed by its resumption at a new phase. Because brief application of NPY produces dramatic and long-term shifts in clock-regulated physiology and behavior, we studied the ability of NPY, found in a high density of axons innervating the SCN (Chronwall et al., 1985) to induce long-term changes in neuronal Ca2+ levels and electrical activity by a mechanism involving modulation of glutamate neurotransmission. Because NPY also may exert long-lasting effects on hypothalamic regulation of food intake (Stanley and Leibowitz, 1985) and endocrine secretion (McDonald et al., 1985, 1989) via a similar mechanism, we also studied medial hypothalamic neurons in parallel experiments.
NPY, one of the most widespread neuroactive peptides in the brain, can act at several different Y receptors, of which Y1 and Y2 are the most completely characterized. In other regions of the brain, such as the CA1 region of the hippocampus where NPY has been extensively studied, the majority of effects are mediated through presynaptic Y2-like receptors (Colmers et al., 1991; Bleakman et al., 1993). Using selective agonists, we found that hypothalamic neurons were quite different from hippocampal pyramidal neurons in the subcellular location and type of receptors that mediated NPY effects. Some of our data have been presented in abstract form (van den Pol et al., 1995a).
MATERIALS AND METHODS
Whole-cell patch-clamp recordings: culture.Whole-cell recordings were made with an Axoclamp-2B amplifier and a List EPC-7 amplifier. Glass pipettes were filled with (in mm): 145 potassium methyl sulfate, 10 HEPES, 5 MgCl2, 1.1 EGTA, 4 Na-ATP, 0.5 Na-GTP, pH 7.2, 310 mOsm. In most cases, neither negative nor positive current was applied to the cells during current-clamp recording. Cells were recorded in an external solution containing (in mm): 158.5 NaCl, 2.5 KCl, 2 CaCl2, 10 HEPES, 1 × 10−3 glycine, and 10 glucose, pH 7.3, 325 mOsm. Glutamate receptor block containedd,l-2-amino-5-phosphonovalerate (AP-5) (100 μm) and 6-cyano-7-nitroquinoxaline (CNQX) (10 μm). A multibarrel flow-pipe perfusion system was used to stimulate the cells. Neurons were continuously perfused (2 ml/min) in a chamber with 1 ml volume. The flow of AP-5/CNQX solution was stopped during the application period of both control buffer and buffer containing NPY. Agonists could be applied or completely washed away from the recorded neuron in 1–2 sec with flow-pipe application. Only one cell was recorded from each coverslip. Recordings were done at 20–22°C. Cytosine arabinofuranoside and glutamate were from Sigma (St. Louis, MO); AP-5, CNQX, t-aminocyclopentane-1,3-dicarboxylic acid (t-ACPD), ω-conotoxin-GVIA, bicuculline methiodide, and TTX from Research Biochemicals (Natick, MA); Fura-2 AM ester, Fura-2 acid, and Ca2+ standards from Molecular Probes (Eugene, OR); papain from Worthington Biochemicals (Freehold, NJ); NPY agonists from Sigma, Peninsula (Hubbell, MI), and gift of Dr. L. Cornfield.
Whole-cell patch-clamp recordings: slice. Sprague Dawley rats (4 weeks old) were used to prepare SCN slices. After decapitation under full halothane anesthesia, the brain was removed rapidly and placed in ice-cold, aerated buffer (described below). The hypothalamus was dissected out, and 400-μm-thick coronal SCN slices were cut with a vibroslicer (Technical Products International).
After preparation, the slices were kept in artificial CSF (ACSF) at room temperature for at least 1 hr before using. ACSF contained (in mm): 124 NaCl, 3.0 KCl, 2.0 CaCl2, 2.0 MgCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, continuously aerated with 95% O2/5% CO2, pH kept at 7.4. A slice then was adhered to a piece of lens paper and transferred to the experimental interface chamber with constant flow of the oxygenated medium (4 ml/min). Glass pipettes pulled from borosilicate glass capillaries of 2 mm diameter and 0.2 mm wall thickness were filled with an internal solution containing (in mm): 145 potassium methyl sulfate, 2 MgCl2, 0.1 CaCl2, 1.1 EGTA, 10 HEPES, 2 Na-ATP, 0.3 Na-GTP, pH 7.2, 290 mOsm. After filling, the patch electrodes had resistances of 5–7 MΩ. They were inserted into the SCN during observation through a stereomicroscope. With positive pressure applied to the recording pipette to eject a small fluid stream, the recording pipette was advanced into the brain slices until a partial seal was obtained. Whole-cell access then was obtained by applying negative pressure to the pipette. The recordings were made at room temperature (20–22°C). Bicuculline (50 μm) was added to the ACSF to block GABAA-mediated neurotransmission. To stimulate excitatory axons projecting to the SCN, bipolar electrodes were placed at the optic nerve. Both retinal and nonretinal axons were stimulated. The electrical impulses (0.2 msec, 0.5 Hz) were delivered through an isolation unit from a Grass 44 stimulator. The stimulating current was 50–400 μA. Drugs were delivered by bath application in the slice chamber. Complete bath exchange took 90–120 sec.
Cultures. Cells were cultured from Sprague Dawley rats on embryonic days 18–21 (E18–E21). Cultures were made either from the 500-μm-diameter punches of the SCN area or were made from a more extensive area that included the medial hypothalamus. Tissue was enzymatically treated (Obrietan and van den Pol, 1995; van den Pol et al., 1995b), and plated onto poly-lysine-treated glass coverslips. Cultures were maintained in glutamate and glutamine-free DMEM (Life Technologies, Gaithersburg, MD) with 10% fetal bovine serum, 100 U/ml penicillin/streptomycin, 6 gm/l glucose, and cytosine arabinofuranoside (1 μm). Cells were kept in a Napco incubator at 37°C and 5% CO2 for 20–41 d before use. CNQX (10 μm) and AP-5 (100 μm) were added to the tissue culture medium 4–7 d after the initial plating to inhibit glutamate-mediated excitotoxicity.
Fura-2 Ca2+ digital imaging. During long periods of whole-cell electrical recording, cells may be dialyzed through the recording pipette. Digital imaging eliminates the potential damage to the cell membrane during patch-clamp recording that may interfere with long-term experiments. Cells were loaded with Fura-2 AM (5 μm) and studied in a perfusion solution containing (in mm): 137 NaCl, 25 glucose, 10 HEPES, 5 KCl, 3 CaCl2, 1 × 10−3 glycine, pH 7.4. The coverslip was held in a laminar flow, 180 μl perfusion chamber (Forscher et al., 1987) that allowed the rapid (5 sec) and complete change in solutions. Cells were imaged on a Nikon Diaphot 300 inverted microscope with an Olympus DApo 40× objective with high UV light transmittance. Sequential switching between 340/380 nm excitation filters was performed by a Sutter filter wheel. Emitted light was first passed through a 480 nm filter and then directed at a Hamamatsu 2400 silicon-intensified target video camera. Excitation light from a 150 W xenon lamp was attenuated by 90% using neutral density filters to allow for continuous recording over long durations without significant photobleaching or phototoxicity. Data were collected every 2 sec in short experiments and every 10 sec in long experiments. Video background was subtracted, and ratiometric data calculated. In some of the figures, several neurons recorded from the same video field are depicted, allowing analysis of temporal and amplitude variability in response to different NPY agonists. Most experiments were performed in at least 3 cultures, and some used up to 14 cultures. Peripheral devices were controlled by a 486 computer using Fluor software (Universal Imaging, West Chester, PA). Ca2+ calibrations were performed with Ca2+ standards and Fura-2 acid (Grynkiewicz et al., 1985).
RESULTS
Electrical activity: slice
Whole-cell recordings were made from the SCN in coronal slices. Of 51 cells recorded, 8 showed good Na+ and K+currents, generated action potentials, and maintained a stable resting membrane potential of 58 mV ± 3.1 mV (SD). Electrical stimulation of glutamatergic axons innervating the SCN evoked stable EPSPs in SCN neurons that could be blocked with the glutamate receptor antagonists AP-5 (100 μm) and CNQX (10 μm) (Fig.1C). NPY (500 nm) added to the bath for 8 min caused a substantial decrease in the amplitude of the evoked EPSP (Fig. 1A,B). When the time integral of the EPSP (Fig. 1B) (time × ΔV) was compared, NPY evoked a large decrease (>60%). When NPY was washed out, the EPSP amplitude remained depressed for long recording periods > 90 min. During that time, the amplitude slowly returned toward its pre-NPY level, but did not reach it.

Whole-cell patch-clamp recording: slice.A, Evoked EPSP amplitude was decreased substantially by NPY in the bath solution. The amplitude remained depressed for ∼1.5 hr after NPY introduction and had not fully returned to normal by the end of the trace shown. At the end of the experiment, the amplitude showed a second decrease; because this second decrease (after 90 min) (data not shown) was accompanied by an increase in access resistance (+50%), we only show the time when access resistance was stable, between 12 and 15 MΩ. Access resistance was determined from voltage pulses at different stages of the recording. B, Representative traces from the neuron shown in A. Control (pre-NPY) EPSP is shown (−10 min), together with 5 min after NPY introduction, and 85 min after NPY introduction (75 min after NPY washout). C, AP-5 (100 μm) and CNQX (10 μm) completely blocked the evoked EPSP. Before NPY exposure, the input resistance was 450 MΩ. In the period after NPY exposure, the input resistance decreased by 10% and remained within 10% of the pre-NPY input resistance for the entire experiment.
These data from SCN slices suggested NPY exerted long-term depressing actions on glutamate-mediated EPSPs. Because the long duration of the effect could be attributable to the slow washout of NPY, a potentially sticky peptide, from the slice, we turned to monolayer tissue culture in which complete washout can be more quickly and effectively achieved. Experiments below were performed on cultured neurons, except for the series of experiments on the presynaptic site of action, which were performed on neurons both in slice and in culture.
Electrical activity: culture
With whole-cell recording of cultured neurons, we found that NPY (100 nm–1 μm) caused a depression in the frequency of EPSPs and in the membrane potential of 13 of 15 cultured neurons (Fig.2A,D,F). In 10 current-clamped neurons recorded for at least 40–50 min, a 2 min exposure to NPY (100 nm–1 μm) caused a hyperpolarization of −5 to −23 mV that had not returned to baseline membrane potential even by 30 min after NPY washout (Fig.2A,D,G,H). Of the 13 neurons that responded to NPY, the mean spike frequency was reduced from a pre-NPY level of 2.1 ± 0.4 to 0.4 ± 0.1 Hz during NPY treatment, and the I–V relationship was shifted (Fig. 2F). Five minutes after complete NPY washout, spike frequency still was depressed to 0.7 ± 0.1 Hz (p < 0.01). EPSPs were reduced in amplitude by NPY. After NPY washout, the hyperpolarized membrane potential continued to recover to a more depolarized potential, and the spike frequency also showed continued recovery with an increase in spikes. That neurons continued to show recovery during the time course of the experiments suggests that the long-duration depression exerted by NPY on electrical activity was not just the result of neuronal rundown. In contrast to cells stimulated with NPY, control neurons (n = 5) not stimulated by NPY showed little change in activity or membrane potential (Fig. 2B).

Whole-cell patch-clamp recording: culture.A, Typical change in SCN neuron spike frequency and membrane potential (mV) after 2 min application of 100 nm NPY and subsequent recovery period in the absence of NPY. B, Representative control cell showing a constant membrane potential and spike frequency. C, In the presence of glutamate receptor blockers AP-5 (100 μm) and CNQX (10 μm), this SCN neuron (solid line) showed a 5 mV hyperpolarizing response to NPY (1 μm) that returned to a pre-NPY membrane potential rapidly at NPY washout, typical of neurons responding to NPY in the presence of AP-5/CNQX.Dotted line shows second neuron that did not respond to NPY. No spikes were seen in the presence of AP-5/CNQX. D, Record from a single SCN neuron before, during, and after flow-pipe application of 1 μm NPY. No excitatory activity was seen in the presence of AP-5/CNQX, and activity was depressed for almost 1 hr after a single exposure to NPY. E, No change in activity or membrane potential was found in this SCN neuron treated with NPY (1 μm) in the presence of the glutamate receptor blockers AP-5 (100 μm) and CNQX (10 μm).F, I–V curve of a typical voltage-clamped neuron in three conditions: AP-5/CNQX, control with no AP-5/CNQX, and NPY in control buffer. G, In current-clamp experiments, NPY (1 μm) caused a mean hyperpolarization (ΔV) (n = 8) of 12–13 mV after a 2 min application. Even after a 30 min recovery period, the membrane potential had only made a partial recovery. Asterisksindicate groups statistically different from controls; ***p < 0.001. H, Mean spike frequency after 2 min exposure to 1 μm NPY (n = 6) was reduced sixfold. Recovery was not complete even after extended recovery times. Asterisksdenote groups significantly different from pre-NPY control data (two-tailed t test); **p < 0.01, *p < 0.05. All cells in this group responded to NPY, some with long-term and some with short-term responses. Error bars indicate SEM.
If glutamate receptors were blocked with AP-5 (100 μm) and CNQX (10 μm), 6 of 12 neurons responded to NPY with a transient hyperpolarization of −4 to −18 mV lasting only for the duration of peptide presence, and showing full and rapid recovery at NPY washout (Fig. 2C). In 6 of 12 neurons, no response to NPY was seen in the presence of AP-5/CNQX (Fig. 2C). NPY had no long-term effect on membrane potential or electrical activity in any neuron if endogenous glutamate activity was blocked with AP-5 and CNQX (Fig. 2E), suggesting that glutamate neurotransmission was necessary for NPY to produce effects with long durations. This was tested in additional experiments below.
Ca2+ responses
Intracellular Ca2+ can play an important role as a second messenger in a number of neuronal functions, including enzyme activation, gene expression, transmitter release, and gating ion channels (Tsien, 1987), all factors that might affect clock function. Furthermore, Ca2+ digital imaging is an excellent noninvasive approach to studying long-term effects on neurons that might be sensitive to perfusion of the intracellular milieu. To determine whether NPY influenced Ca2+ levels, we used digital imaging with Fura-2. We defined baseline Ca2+ as the cytosolic Ca2+ level for each cell in the presence of glutamate receptor blockers AP-5 (100 μm) and CNQX (10 μm), a condition that blocks all excitatory synaptic activity in these neurons (van den Pol and Trombley, 1993; Obrietan and van den Pol, 1995). Baseline Ca2+ levels in the absence of AP-5 and CNQX were between 40 and 100 nm. In the absence of AP-5 and CNQX, the Ca2+ levels were higher (100–200 nm) because of the actions of synaptically released glutamate. Of the neurons tested (n > 600 in 14 experiments), 95% showed a Ca2+ depression (mean decrease, 76%; minimum decrease, 15%) in response to NPY (100 nm in most experiments) in the absence of AP-5 and CNQX. Exposure to NPY caused an extended Ca2+ depression in 39% of 190 spontaneously active neurons (by at least 50% amplitude for > 10 min) (Fig. 3A–C). Some neurons (6% of 190) showed a striking Ca2+ depression to a level equivalent to blocking glutamate receptors with AP-5 and CNQX.

Fura-2 Ca2+ digital imaging. In these experiments, the effect of NPY receptor agonists (100 nm) was examined on spontaneously active synaptically coupled neurons. Glutamate receptor antagonists AP-5 (100 μm) and CNQX (10 μm) blocked excitatory activity and were used in some experiments to allow a comparison of the relative efficacy of NPY in reducing activity. A, A1, andA2 are two SCN neurons recorded simultaneously.A1 shows an NPY-mediated LTDCa that lasted for the duration of the experiment. A2 showed recovery as soon as NPY was washed out and was depressed by a second application of NPY. A3 is a control neuron showing a typical maintenance of Ca2+ levels over the course of recording. NPY was not applied to this neuron. B, A 2 min application of NPY led to an LTDCa that lasted >90 min.B1, Partial recovery of Ca2+ spikes after 45 min is seen, but the level of activity and elevated Ca2+baseline did not recover. B2, The baseline shows an LTD after NPY exposure, but Ca2+ spikes continued.C1, An 8 sec application of NPY depressed cytosolic Ca2+ for the length of the experiment. C2shows partial recovery after 10 min. D, Application of Y1 agonist [Leu31,Pro34]-NPY designated NPY-Pro34 (D1) and Y2 agonist NPY13-36 (D2) caused substantial reductions in Ca2+baseline and transient activity. E, Relative efficacy of NPY agonists in reducing Ca2+. Although not tested for relative effectiveness, examples of LTDCa were found after each of the NPY agonists. Error bars indicate SEM. Number of cells tested for each agonist, in same sequence as bars, leftto right: n = 103 (NPY tested in general medial hypothalamic cultures), 42 ([Leu31,Pro34]-NPY), 69 (NPY13-36), 35 (PYY3-36), 47 (PYY13-36), 40 (tested in SCN cultures). The Ca2+ level in each group before NPY agonist application served as the control level (100%). Both selective SCN and general medial hypothalamic cultures showed very similar amplitudes of responses.
Long-term Ca2+ depression (LTDCa)
In longer experiments, a brief application of NPY generated a Ca2+ depression in spontaneously active neurons that lasted for the duration of the recording (>1 hr) (Fig. 3B). Even a very brief (8 sec) application of NPY (1 μm) caused a Ca2+ depression that lasted > 30 min, or 225 times longer than the exposure to the peptide (Fig. 3C1). Some neurons began returning to their pre-NPY Ca2+ level as soon as NPY was washed off (Fig. 3A2), whereas others in the same culture showed a long-lasting depression (Fig. 3A1). That some neurons showed a rapid recovery after NPY washout indicates that the long-term effect was not attributable to partial washout of the NPY or to a general adhesion of the peptide to cells. Control neurons not stimulated with NPY maintained constant mean Ca2+ levels (Fig. 3A3).
Receptor-selective agonists
To determine which NPY receptor types might be involved in the depression of cytosolic Ca2+, we used Y1 ([Leu31,Pro34]-NPY) (Fig. 3D1) and Y2 [NPY13-36 (Fig. 3D2) and PYY13-36] preferring agonists (Wahlestedt et al., 1990) and examined the effects of these agonists on spontaneously active neurons. Figure 3E shows the relative Ca2+ depression elicited by different Y1 and Y2 agonists. NPY had the greatest effect, but all agonists evoked a strong Ca2+ depression, suggesting that both Y1- and Y2-like receptors were involved in reducing ongoing activity.
Postsynaptic effects
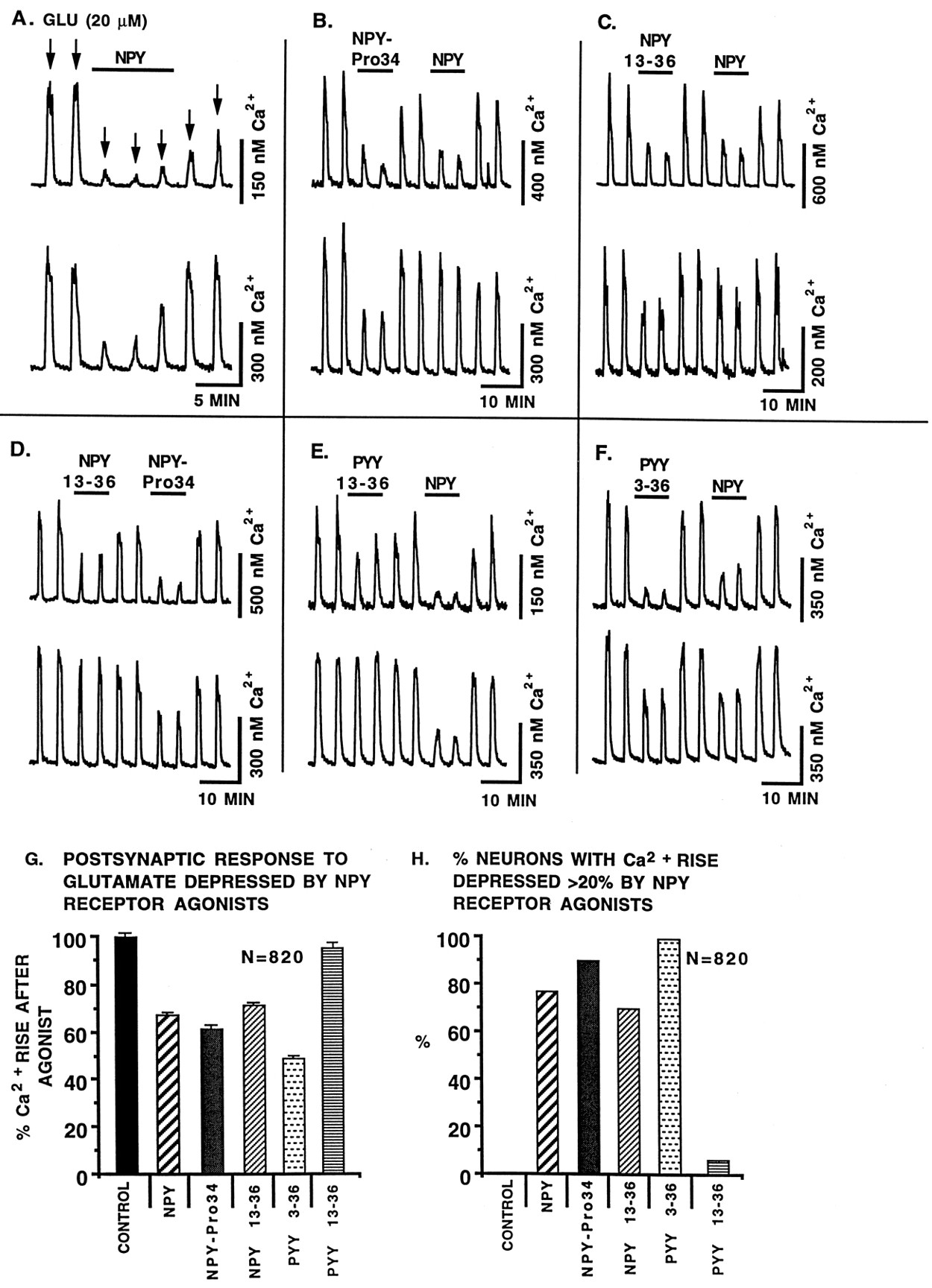
In the experiments above showing NPY effects on spontaneously active neurons, the Ca2+ depression could be attributable to either presynaptic or postsynaptic NPY receptors, or both. Previous work in the hippocampus (Colmers et al., 1991; Bleakman et al., 1992,1993) and raphe (Kombian and Colmers, 1992) suggested that NPY acted by a presynaptic mechanism; postsynaptic effects were not detected, except in a recent report in hippocampal granule cells (McQuiston et al., 1996). In contrast, in the present study, SCN neurons and hypothalamic neurons in general showed striking NPY-mediated depressions of Ca2+ rises evoked by coapplication of glutamate (20 μm) applied by rapid bath perfusion. Neurons did not respond to NPY with a change in cytosolic Ca2+ in the absence of glutamate. These experiments were performed in the presence of tetrodotoxin (TTX) (1 μm) to block action potential-dependent synaptic release of transmitters and, therefore, a mechanism involving secondary release of unidentified transmitters is unlikely. Because these effects cannot be explained by modulation of presynaptic release, the results suggest that cytosolic Ca2+ levels in hypothalamic neurons can be regulated by NPY modulating a postsynaptic response to glutamate. Only a very small number of cells (1% of 167) showed a delayed recovery (LTDCa) after NPY stimulation (Fig.4A, top), whereas most recovered by the subsequent transmitter stimulation (Fig.4A, bottom), suggesting that long-term actions of NPY probably were not mediated postsynaptically.

NPY receptor agonists modulate postsynaptic glutamate responses: Fura-2. In these experiments with 820 tests, glutamate (20 μm) was added to each neuron several times (30 sec application duration). Each spike represents the addition of glutamate. NPY agonists were coapplied with glutamate as indicated by the horizontal lines. All agonists were 100 nm. TTX (1 μm) was used in the buffer to prevent release of endogenous transmitters that might complicate data interpretation. Two representative neurons recorded simultaneously are shown for each condition. Because we found no difference in the responses of selective SCN cultures and medial hypothalamic cultures that included the SCN, data are pooled here. In A, NPY (horizontal bar) was applied 30 sec before the third glutamate (arrows) application. In the atypical upper neuron, even after 10 min, the cell did not recover to its pre-NPY glutamate-evoked Ca2+ amplitude. The lower cell recovered as soon as NPY was removed. In B–F, the relative efficacy of two NPY agonists is compared. G andH show the relative efficacy of different NPY agonists.G shows the mean glutamate-evoked Ca2+ rise in the presence of NPY agonists, and H shows the percent neurons within each group that showed a NPY-mediated depression of glutamate-evoked Ca2+ rises. Control is the response to glutamate in the absence of NPY agonists. Number of cells tested with each agonist, n = 412 (NPY), 181 (NPY13-36), 114 [Leu31,Pro34]-NPY called NPY-Pro34, 53 (PYY13-36), and 60 (PYY3-36). PYY3-36 was the most effective (p < 0.05), whereas the shorter C-terminal fragment PYY13-36 was least effective.
To determine whether Y1- or Y2-like receptors were responsible for the postsynaptic Ca2+ depression, we tested a number of NPY agonists. Both Y1 and Y2 receptor agonists (100 nm) including NPY (Fig. 4A), [Leu31,Pro34]-NPY (Fig. 4B), NPY13-36 (Fig. 4C,D), PYY13-36 (Fig.4E), and PYY3-36 (Fig. 4F) reduced the amplitude of the Ca2+ rise evoked by glutamate (20 μm). Interestingly, the most effective agonist was PYY3-36, which depressed glutamate-evoked Ca2+ rises even more than NPY itself (Fig.4G,H). That PYY3-36 has a modulatory effect greater than NPY raises the question of whether it may be acting on a novel hypothalamic Y-type receptor or whether it is activating a Y2 or Y5 receptor (Gerald et al., 1996). PYY is a gut hormone with a 69% amino acid sequence homology to NPY (Tatemoto et al., 1982), and its presence in the brain has been suggested (Broome et al., 1985). PYY has been suggested to be equipotent with NPY in its actions on Y1 and Y2 receptors (Walker and Miller, 1988; Wahlestedt et al., 1992; Foucart et al., 1993). PYY3-36 is not working through a putative Y3 receptor, because that receptor is thought to be insensitive to PYY activation (Grundemar et al., 1993). The sensitivity of the postsynaptic effect to PYY3-36 is in contrast to the experiments with spontaneously active cells that found NPY itself was the most effective agonist in reducing Ca2+. This difference may be attributable to receptors with greater PYY sensitivity postsynaptically and a greater NPY sensitivity presynaptically.
The relative efficacies of NPY agonists in modulating postsynaptic glutamate responses in 820 trials are shown in Figure 4, Gand H. Each neuron was stimulated with glutamate and two NPY agonists in the presence of TTX (1 μm). Two neurons are shown for each experiment, revealing different relative sensitivities to NPY agonists. The heterogeneous response of single neurons to application of different NPY agonists suggests that a single cell may express several different types of NPY receptors on the cell soma.
Mechanisms of NPY action
We have suggested previously that most of the fast synaptic activity in the hypothalamus was mediated by glutamate and GABA, and that a principal role of hypothalamic peptides was to modulate these fast amino acid transmitters (van den Pol et al., 1990). To test further this hypothesis, we undertook a series of experiments to examine the mechanism of NPY action. These experiments were also designed so that we could compare possible mechanisms of LTD in the hypothalamus with that reported previously by other laboratories examining LTD in cortical brain regions.
Because an NPY-mediated reduction of activity could be attributable to an NPY enhancement of GABA inhibitory activity, we blocked the GABAA receptor with bicuculline (20 μm). In contrast to previous reports of GABAergic involvement in some other models of LTD (Thiels et al., 1994; Yang et al., 1994; Obrietan and van den Pol, 1996), we found that blocking the GABA receptors did not interfere with NPY-mediated long-term (Fig.5A1) or short-term (Fig. 5A2) Ca2+ depression in mature neurons. Previous work on hippocampal LTD has suggested that the NMDA receptor is a necessary component of many forms of LTD (Dudek and Bear, 1992; Mulkey and Malenka, 1992). However, blocking the hypothalamic NMDA receptor with AP-5 (100 μm) did not block LTDCa (Fig.5C). Other reports have demonstrated the crucial importance of voltage-gated Ca2+ channels in the induction of hippocampal LTD (Bolshakov and Siegelbaum, 1994). In contrast, selective blockade of the L-type (Fig. 5B) or N-type (Fig.5D) voltage-activated Ca2+ channel in hypothalamic neurons with nimodipine (1 μm) or conotoxin (1 μm) did not block the NPY extended reduction of Ca2+ in hypothalamic neurons, but did reduce the amplitude of the NPY effect, as shown in Figure 5. Involvement of the N channel is potentially interesting in light of the finding that NPY acted through an N-type Ca2+ channel in its action on terminals of sympathetic neurons (Toth et al., 1993). The possibility remains that one or more voltage-gated Ca2+ channels may mediate part of the long-term effect of NPY, but that a single type may not account for the entire response.

Mechanisms of NPY action. A, The GABAA antagonist bicuculline (20 μm) did not block either NPY-mediated long- or short-term Ca2+depression (n = 28). B, Blocking the L-type voltage-activated Ca2+ channel with nimodipine (1 μm) did not block NPY-mediated long- or short-term Ca2+ depression (n = 50).C, Blocking the NMDA receptor with AP-5 (100 μm) caused a reduction in general Ca2+levels, but did not block NPY-mediated long- or short-term Ca2+ depressions (n = 39).D, Blocking the N-type voltage-activated Ca2+ channel with ω-conotoxin (1 μm) did not block NPY-mediated long- or short-term Ca2+ depression (n = 58). E, Preincubation with PTX (150 ng/ml for 17 hr) completely blocked the NPY depression (compare lack of depression with the NPY-mediated depression in the second error bar in Fig. 3E), indicating the involvement of Gi/Go proteins (n = 26). In contrast, control application of AP-5 (100 μm) and CNQX (10 μm) generated a significant drop in cytosolic Ca2+.
To test the hypothesis that Gi or Go G-proteins were necessary for the long-term effect of NPY, we treated the cells with pertussis toxin (PTX) (Colmers et al., 1991; Bleakman et al., 1992, 1993). Neurons treated with PTX showed no response to NPY (Fig.5E), whereas control neurons from sister cultures did show strong depressions. AP-5 and CNQX decreased Ca2+ in PTX-treated neurons, indicating that the cells were healthy and capable of regulating cytosolic Ca2+. These results demonstrate that NPY action may be dependent on a PTX-sensitive G-protein-coupled mechanism.
Presynaptic site of NPY action: culture and slice
To test further the hypothesis that NPY acted at presynaptic axons, we used TTX (1 μm) to block Na+-dependent action potentials in cultured neurons, and examined the frequency of miniature EPSCs (mEPSCs) in cultures of medial hypothalamus that contained the SCN. Neurons were voltage clamped at −60 mV in the presence of bicuculline (20 μm) to block the actions of synaptically released GABA. Eight of 13 neurons tested responded to NPY, and all 8 showed a decrease in the frequency of mEPSCs. Three of the eight showed a depression in mEPSC frequency lasting for the duration of the recording period (mean, 1 hr). Figure6 shows an example of one neuron in which NPY application (4 min duration) reduced mEPSC frequency by 80%. Figure6A shows representative EPSCs during different stages of the recording. The EPSCs were fully blocked by the addition of AP-5 (100 μm) and CNQX (10 μm), indicating that they were caused by glutamate release (Fig. 6B). The time course is shown in Figure 6C. Even 1 hr after NPY washout, the mEPSC frequency still was significantly lower (p < 0.05) than pre-NPY levels. That the frequency was starting to return to pre-NPY levels at the end of the experiment suggests that the frequency reduction was not simply attributable to a compromised cell showing rundown. The magnitude of the mEPSCs showed no decrease over the course of the experiments, suggesting that the results were not attributable to increases in access resistance.

Presynaptic NPY action: LTD of mEPSCs.A, In this example, the frequency of EPSCs was reduced by a 4 min exposure to NPY (200 nm) in a voltage-clamped neuron (−60 mV) and remained depressed long after NPY washout. Two miniature events are shown in greater detail at thebottom of A, indicating that the time course and amplitude of the EPSC did not change from the beginning of the experiment to the end (54 min). All buffers contained TTX (1 μm) to block spike-dependent transmitter release.B, AP-5 (100 μm) and CNQX (100 μm) completely blocked EPSCs, indicating glutamate as the transmitter. Washout of AP-5/CNQX resulted in recovery of EPCSs (data not shown). C, The time course of the raw data inA are shown over the entire recording period. The SEMs are shown by the small error bars at each time interval, based on seven to eight consecutive samples of 30 sec duration. All points from the first one after NPY introduction to the last one at the end of the experiment (asterisk) were significantly different from pre-NPY EPSC frequency.
Parallel experiments were performed in SCN slices under voltage clamp (−75 mV). These slice experiments have the advantage that the responses of the existing circuitry and axonal innervation of identified SCN neurons could be studied. In the example shown in Figure7, the frequency of spontaneous mEPSCs in TTX (1 μm) showed a significant decrease after NPY (500 nm) bath application. The frequency remained depressed for long recording periods (1 hr). During that time, current injections were used to show that the membrane input resistance remained stable (445 ± 20 MΩ). Bicuculline (50 μm) was used in all solutions to block GABA-mediated IPSCs. mEPSCs could be fully blocked with AP-5 (100 μm) and CNQX (10 μm) (data not shown), indicating that glutamate release was responsible for the EPSCs. All mPSCs were effectively blocked with the combination of GABA and glutamate receptor antagonists bicuculline and AP-5/CNQX. Thus, the slice work is consistent with and fully supports the conclusions based on experiments with cultured neurons.

mEPSCs in SCN slice. mEPSCs were recorded in an SCN neuron in the presence of TTX (1 μm), and bicuculline (50 μm). A, A decrease in the frequency of mEPSCs was seen when NPY (500 nm) was bath applied (horizontal line). After NPY was washed out, the mEPSC frequency remained depressed for the duration of the recording period. B, Representative traces during the course of the experiment are shown with the time in minutes on theright of each trace. NPY was added at 0 time.C, mEPSCs from the record above (B) are shown in the boxed area at the bottomwith a faster time sweep. Holding potential, −75 mV.
Long-term effects require coincident activation of NPY and glutamate receptors
In a particularly interesting set of experiments, we tested the hypothesis that NPY would exert a long-lasting effect on cytosolic Ca2+ only if glutamate activity were ongoing at the time of NPY application. We therefore added NPY (100 nm) in the presence of glutamate receptor antagonists, and 2 min later removed NPY and then washed out the glutamate receptor antagonists. In these experiments (n = 102) we found no cases in which NPY would exert either an immediate or a latent effect on glutamate-mediated Ca2+ activity (Fig. 8). This is in striking contrast to our previous observation of LTDCa lasting 90 min (e.g., Fig. 3B) if NPY was applied during ongoing glutamate activity. To demonstrate that the cells would respond to NPY, we removed the AP-5/CNQX glutamate receptor block from the same cells and found that NPY then did exert a long-lasting depression of activity on one third of the group of neurons (n = 102) that showed no immediate or latent response to NPY in the presence of glutamate receptor antagonists (Fig.8). These results indicate that glutamate-dependent activity was necessary for NPY to influence intracellular Ca2+.

NPY in the presence of glutamate receptor antagonists AP-5 and CNQX had no Ca2+ effect, either at the time of NPY application or after the rapid removal of the glutamate receptor antagonists (n = 102). For control purposes, AP-5 and CNQX were washed off twice, and each time a large increase in cytosolic Ca2+ was found. NPY (100 nm) then was added in the presence of AP-5 and CNQX, and NPY then was washed out. Thirty seconds later, AP-5/CNQX was washed out, and no effect of NPY was detected. However, on a subsequent trial, in the absence of AP-5 and CNQX, NPY evoked a large decrease in Ca2+ that persisted for the duration of the recording session.
DISCUSSION
Taken together, our experiments suggest that both Y1- and Y2-like NPY receptors are functionally expressed at both presynaptic and postsynaptic sites; this has not been reported in other regions of the CNS. NPY depressed both the intracellular Ca2+ and the electrical activity of neurons, in part by a presynaptic mechanism that reduced glutamate release from axons bearing NPY receptors. Both the induction and the expression of a long-term depression of excitatory activity, found with whole-cell recording in cultures and slices and with noninvasive digital Ca2+ imaging, was dependent on the simultaneous release of glutamate and NPY receptor activation.
Widespread NPY receptor expression
In contrast to studies based on radioactive ligand binding suggesting that NPY receptor expression is relatively low in the hypothalamus (Lynch et al., 1989), our data, based on both patch-clamp recording and digital imaging, indicate that almost all hypothalamic neurons in vitro are influenced by presynaptic or postsynaptic functional NPY receptors. Acting both presynaptically and postsynaptically, NPY can cause a substantial depression of cytosolic Ca2+ and glutamate-mediated excitatory activity, not only in SCN neurons but also in other mediobasal hypothalamic neurons.
The magnitude of the Ca2+ depression elicited by NPY in spontaneously active neurons, considered as a percentage decrease, was two to three times greater than the depression elicited during responses evoked by glutamate application. This was true over a range of glutamate-regulated Ca2+ levels. Because the evoked responses were done in the presence of TTX, and would demonstrate postsynaptic actions, the greater effects of NPY on spontaneously active cells were probably attributable to additional presynaptic effects of the peptide. This is consistent with the relative absence of long-term action of NPY on glutamate-evoked postsynaptic responses, but a sizable LTDCa in 39% of spontaneously active neurons. In contrast to pyramidal neurons of the hippocampus, where NPY exerts its effects presynaptically through NPY Y2 receptors (Colmers et al., 1991), our data indicate that both presynaptic and postsynaptic NPY receptors participate in the SCN and surrounding hypothalamus, via Y1, Y2, and perhaps additional Y-type receptors. NPY actions have been divided into two groups based on similarity to Y1- and Y2-like responses (Grundemar et al., 1993). Recent cloning studies, however, have revealed novel NPY Y4 and two Y5 receptors that have some pharmacological similarities to Y1 and Y2 receptors (Bardt et al., 1995; Gerald et al., 1996; Weinberg et al., 1996). These recently isolated receptors are expressed in the hypothalamus, including the SCN. Thus, some of the physiological responses to NPY described in the present paper may be mediated by these NPY receptors for which subtype-selective agonists are not yet available. The heterogeneity of NPY receptor expression presynaptically and postsynaptically may, in part, explain why some neurons show LTDCa, whereas other neurons show only transient depressions. Another factor that may explain why some neurons show LTDCa and others show only a transient response to NPY may relate to the different ionotropic and metabotropic glutamate receptors expressed by subsets of these neurons (van den Pol, 1994; van den Pol et al., 1994). In the case of SCN neurons, different circadian phases of neurons in the same culture may contribute to the response heterogeneity.
LTD in hypothalamic neurons
Our experiments support the hypothesis that NPY requires the simultaneous release of glutamate for the generation of LTDCa, and suggest that NPY binding to its receptor for an extended period is not the cause of the long-lasting effect. The LTD effects are specific to NPY, because parallel experiments with the modulator adenosine showed only short-term reductions of glutamate-mediated excitatory activity (Obrietan et al., 1995). We showed that the NMDA receptor is not necessary for the LTDCa; therefore, AMPA/kainate or metabotropic glutamate receptors are sufficient to achieve LTDCa in combination with NPY stimulation in hypothalamic neurons. Furthermore, because we found little LTDCa with experiments in which we focused on postsynaptic responses to glutamate + NPY, the presynaptic axon may be critical for LTDCa.
NPY reduced the frequency of mEPSCs in the presence of TTX. Because TTX effectively isolated the axon terminals from their parent perikarya, any NPY actions on the cell body would not be expected to influence the cell’s axon terminals or transmitter release. These data support the hypothesis that NPY can act at a presynaptic site on glutamate-secreting axons to reduce transmitter release in both SCN area cultures and SCN slices. This is consistent with previous observations that NPY acts on presynaptic axons in several brain areas (Colmers et al., 1991; Bleakman et al., 1992, 1993) and in peripheral neurons (Walker et al., 1988). An extended reduction of the mEPSC frequency was found with NPY application in some cells even under conditions of relatively low activity typically found in the presence of TTX. The long-lasting effect of NPY on Ca2+ and action potentials, therefore, may depend on local microsignaling or feedback at the synaptic level between the excitatory presynaptic axon and the postsynaptic cell. Another consideration is that the reduction of the dynamic electrical activity and cytosolic Ca2+ levels, at least in part, may be dependent on cellular feedback in a network of synaptically coupled neurons, and may require a combination of presynaptic and postsynaptic NPY actions at glutamatergic synapses. A final possibility that merits exploration is that the presynaptic axons may bear glutamate receptors, and these need to be activated for NPY to evoke a maximal effect.
NPY exerts a depressing action on developing GABAergic neurons that is not dependent on coactivation of GABAergic axons (Obrietan and van den Pol, 1996). In contrast, in the present paper we demonstrate that robust LTD of excitatory activity, membrane potential, and cytoplasmic Ca2+ requires the coincident release of glutamate and activation of NPY receptors. The importance of glutamate may be to increase transmitter release, and NPY may act to induce postsynaptic LTDCa by reducing transmitter release from active presynaptic axons. Because we found little latent effect of NPY on LTDCa if administered in the transient absence of glutamate activity, NPY may be more effective on active glutamate-secreting axons than on inactive ones in mediating LTDCa. The long-lasting actions of NPY may be state dependent, requiring the initial glutamate-mediated activation or elevation of some substrate, perhaps Ca2+. Previous work has suggested that an increase in Ca2+ may be necessary for LTD in a different model (Christofi et al., 1993). This is consistent with our finding that at the time of induction of LTDCa, cytosolic Ca2+ was raised by glutamate. Our data are unique in showing LTDCa in these neurons.
An extended depression of intracellular Ca2+ would have the capacity to alter many aspects of neuronal function, including transmitter release and electrical activity, Ca2+ gated ion channels, Ca2+ binding proteins, and Ca2+modulated gene expression. This extended change in the behavioral state of a neuron would have a significant influence on its functional output, and may represent a cellular substrate for the long-term effects of NPY on phase shifts in circadian rhythms controlled by the suprachiasmatic nucleus. Similar long-term changes in cellular behavior in the form of depression or potentiation may explain other paradigms of phase shifts of the circadian clock involving other peptides that may modulate glutamate activity.
Functional considerations
A long-term depression in cytosolic Ca2+, hypothetically, may be a cellular substrate in SCN neurons for NPY-induced phase shifts (Albers and Ferris, 1984; Medanic and Gillette, 1993; Huhman and Albers, 1994) in circadian rhythms. A decrease in intracellular Ca2+ would have widespread effects on cellular function and transmitter release. In addition to its long-term effects on the circadian clock, NPY application also causes an extended bout of vigorous feeding that may last hours (Stanley and Leibowitz, 1985) and long-term alterations in the timing and release of gonadal hormones (McDonald et al., 1985, 1989). The long-lasting depression of Ca2+ responses generated by NPY in cultured mediobasal hypothalamic neurons was similar to that of selective cultures of the suprachiasmatic nucleus, suggesting that NPY-mediated LTD could be involved in a number of hypothalamic homeostatic functions requiring long lasting changes in the activity of neurons.
Hypothalamic regulatory mechanisms involved in caloric maintenance, hormone control, and circadian time keeping depend on comparisons among different internal states over time, and that requires some form of long-term information processing of afferent signaling and cellular memory. Taking together the findings that glutamate appears to be the primary fast excitatory transmitter in the hypothalamus (van den Pol et al., 1990), that NPY-containing terminals are found throughout the hypothalamus (Chronwall et al., 1985) and that the majority of hypothalamic neurons respond physiologically to NPY (present paper), then NPY-mediated LTD via a mechanism involving presynaptic modulation of glutamate transmission merits additional exploration as to whether it may explain other long-term effects of NPY on hypothalamic homeostatic regulation.
Footnotes
This research was supported by National Institutes of Health Grants NS 34887 and NS 10174, the National Science Foundation, and the Air Force Office of Scientific Research. We thank Dr. Fred Sigworth and Dr. Craig Heller for helpful suggestions.
Correspondence should be addressed to Anthony N. van den Pol, Section of Neurosurgery, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}