

Abstract
The release of dopamine (DA) in the nucleus accumbens (NAc) is thought to be critical for mediating natural rewards as well as for the reinforcing actions of drugs of abuse. DA and amphetamine depress both excitatory and inhibitory synaptic transmission in the NAc by a presynaptic D1-like DA receptor. However, the mechanisms of depression of excitatory and inhibitory synaptic transmission appear to be different. DA depressed the frequency of spontaneous miniature EPSCs, but the frequency of miniature IPSCs was depressed only when spontaneous release was made dependent on Ca2+influx through voltage-dependent Ca2+ channels. Furthermore, the K+ channel blocker Ba2+ attenuated the effects of DA on evoked IPSPs, but not on EPSPs. Thus, DA appears to depress inhibitory synaptic transmission in the NAc by reducing Ca2+ influx into the presynaptic terminal, but depresses excitatory transmission by a distinct mechanism that is independent of the entry of Ca2+.
- amphetamine
- cocaine
- dopamine
- miniature inhibitory postsynaptic currents
- minature excitatory postsynaptic currents
- nucleus accumbens
- presynaptic
The nucleus accumbens (NAc) is a critical component of reward circuitry and, as such, is an important site of action of drugs of abuse. It has been implicated in both the self-administration of psychostimulants (Zito et al., 1985;Koob, 1992) and psychostimulant-induced sensitization, a phenomenon that may mimic aspects of the development of addiction as well as of psychosis (Kalivas and Stewart, 1991; Robinson and Berridge, 1993). Psychostimulants interfere with the dopamine (DA) transporter (Ritz et al., 1987), which is located on the terminals of the profuse dopaminergic projection from the ventral tegmental area (VTA) to the NAc. The resulting increase in synaptic levels of DA in the NAc is thought to be a critical mechanism by which psychostimulants produce reward (Koob and Bloom, 1988; Nestler, 1992; Hyman, 1996).
Although the molecular targets of DA (i.e., DA receptors) and the psychostimulants have been characterized, development of comprehensive hypotheses of the neurobiological mechanisms of reward and drug abuse will require a thorough understanding of the actions of DA on synaptic transmission in the NAc. The major cell type in the NAc is the GABAergic medium spiny neuron (DeFrance et al., 1985a; Pennartz et al., 1991; O’Donnell and Grace, 1993). These cells receive excitatory input from a variety of brain regions (DeFrance et al., 1985a,b;Christie et al., 1987; Yim and Mogenson, 1988; Sesack and Pickel, 1990,1992; Johnson et al., 1994; O’Donnell and Grace, 1995) and exhibit extensive networks of recurrent axon collaterals that form GABAergic synapses with other NAc cells (Chang and Kitai, 1985; Pennartz and Kitai, 1991; Kawaguchi et al., 1995). Ultrastructural studies of the NAc and striatum have found dopaminergic terminals apposed to both symmetrical and asymmetrical synapses (Bouyer et al., 1984; Sesack and Pickel, 1992), as well as a significant proportion unapposed to any postsynaptic structures (Bouyer et al., 1984; Descarries et al., 1996). Thus, dopaminergic afferents to the NAc may modulate synaptic transmission either by releasing DA directly onto synapses or by causing a more diffuse increase in extracellular DA levels (Garris et al., 1994; Descarries et al., 1996).
Surprisingly few studies have examined the synaptic effects of DA in the NAc. Early in vivo studies reported that VTA stimulation or DA iontophoresis reduced stimulus-evoked excitatory responses (Yang and Mogenson, 1984; DeFrance et al., 1985b; Yim and Mogenson, 1988). Later work using slice preparations of the NAc was consistent with the hypothesis that DA causes a reduction in synaptic transmission (Higashi et al., 1989; Pennartz et al., 1992a,b; O’Donnell and Grace, 1994). Recently, detailed studies from our lab (Nicola et al., 1996) and others (Harvey and Lacey, 1996b) have demonstrated that DA and psychostimulants depress excitatory synaptic transmission, most likely by activation of a presynaptic D1-like DA receptor. Here, we present evidence that inhibitory synaptic transmission in the NAc is also modulated by DA, but by a mechanism different from that responsible for the modulation of excitatory transmission.
MATERIALS AND METHODS
Slices were prepared and recordings obtained as described (Kombian and Malenka, 1994; Nicola et al., 1996). Sprague Dawley rats (16- to 20-d-old) were anesthetized with halothane, and 400 μm saggital slices of the NAc were cut using a vibratome. At least 1 hr after cutting, slices were transferred to a submersion-type recording chamber and perfused with external solution at 2 ml per minute at room temperature (21–23°C). Except where noted, the external bathing solution contained (in mm): 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2, 18 NaHCO3, and 11 glucose. Solutions without NaH2PO4 were used for experiments involving CoCl2 or CdCl2 (see Figs. 10, 11). For high-K+ experiments (see Fig.10), the bathing solution was prepared by equimolar substitution of NaCl with the indicated concentration of KCl, and CaCl2 was increased to 3.8 mm. When spontaneous miniature events were recorded, 1.5 μm tetrodotoxin (TTX) was included in the external solution throughout the experiment. Antagonists of excitatory synaptic transmission (75 μmd,l-APV and 10 μm DNQX) were included in most experiments, except as noted.

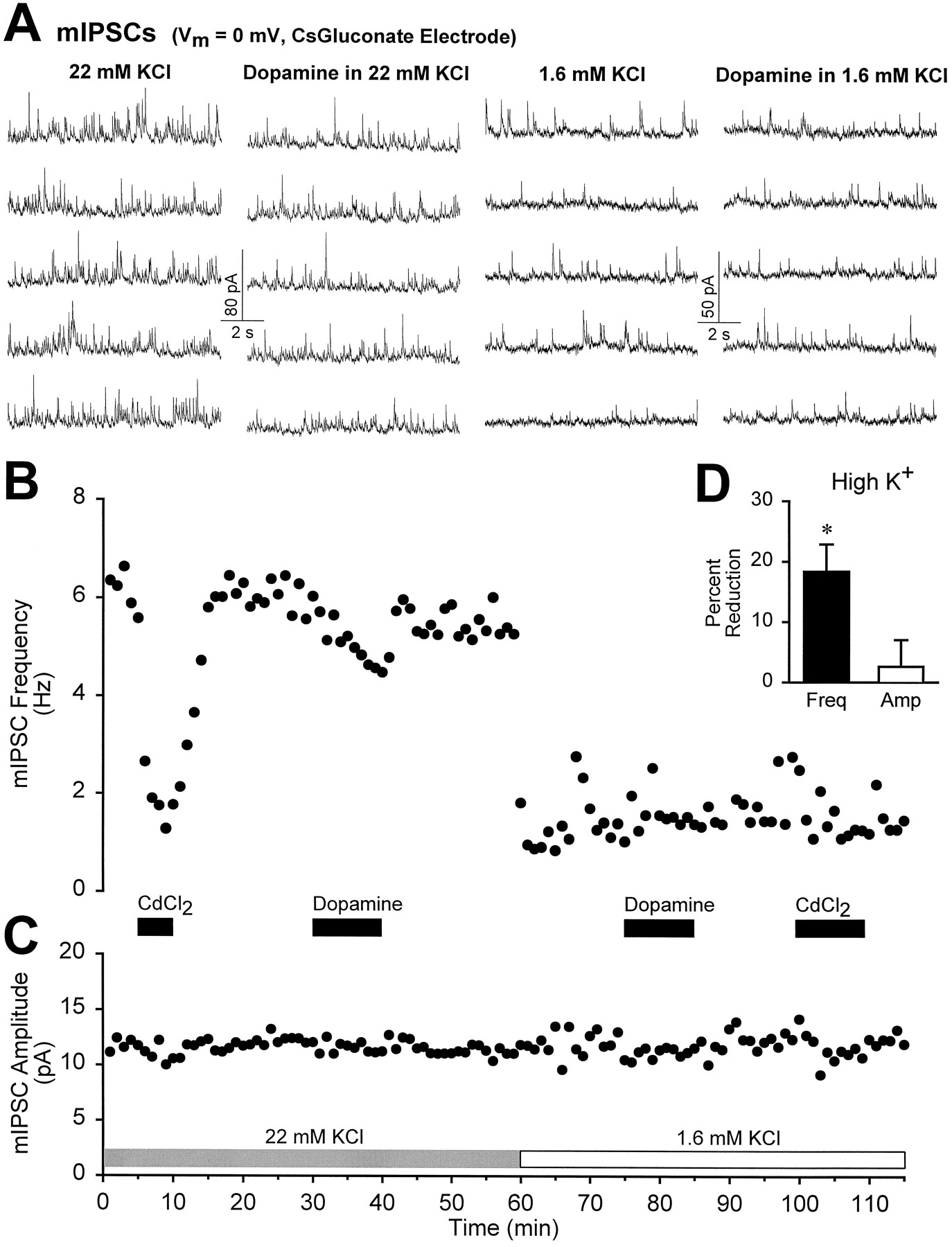
Dopamine depresses Ca2+-dependent mIPSCs. A, Consecutive traces taken from the experiment in B show that the frequency of mIPSCs (holding potential = 0 mV, cesium gluconate-based electrode solution) was greatly increased by raising extracellular KCl from 1.6 to 22 mm. B, The time course of the frequency of mIPSCs is shown for one experiment. In 22 mm KCl, the Ca2+ channel antagonist CdCl2 reduced the frequency of mIPSCs, whereas in 1.6 mmKCl, Cd2+ was without effect. Dopamine (100 μm) decreased the frequency of Ca2+-dependent mIPSCs recorded in high KCl but not the frequency of Ca2+-independent mIPSCs recorded in low KCl. C, Neither Cd2+ nor dopamine influenced the )amplitude of mIPSCs in either condition.D, Summary graph (n = 4) demonstrating the effects of dopamine (100 μm) on the frequency and amplitude of mIPSCs in high KCl (22–25 mm). The asterisk indicates a statistical difference from baseline (p < 0.01). In high K+, the mean mIPSC frequency was 6.5 ± 1.5 Hz, and the mean amplitude was 11.1 ± 0.8 pA.

Amphetamine reduces mEPSC frequency in the presence of the Ca2+ channel blockers Co2+ and Cd2+. A, Consecutive traces taken from an experiment in which pharmacologically isolated mEPSCs were recorded in the presence of the Ca2+ channel antagonist Co2+ at a concentration (5 mm) sufficient to abolish evoked EPSCs. Compared with baseline (left), 10 μmamphetamine (right) reduced the frequency but not the amplitude of mEPSCs. B, A summary graph (n = 8) showing the effects of amphetamine (10 μm) on the frequency and amplitude of mEPSCs recorded either in 5 mm Co2+(n = 4) or in 100 μmCd2+ (n = 4). Theasterisk indicates a statistical difference from baseline (p < 0.005). Mean baseline mEPSC frequency was 3.4 ± 0.5 Hz, and mean amplitude was 12 ± 0.7 pA.
Whole-cell recordings (Blanton et al., 1989) were made using an Axopatch 2D amplifier (Axon Instruments, Foster City, CA). For most experiments, electrodes (8–12 MΩ) contained (in mm): 117.5 cesium gluconate, 17.5 CsCl, 8 NaCl, 10 HEPES, 0.2 EGTA, 2.5 MgATP, and 0.1 GTP, pH 7.2. With this electrode solution, large IPSPs could be recorded when the cell was held at 0 mV. In some miniature IPSC (mIPSC) experiments (see Fig. 7A–C), the cesium gluconate was replaced with CsCl. For the experiments in Figure1A,B, the electrode solution consisted of (in mm): 134.5 potassium methylsulphate, 3 NaCl, 5 QX314 chloride, 10 HEPES, 0.2 EGTA, 2.5 MgATP, and 0.1 GTP, pH 7.2. For experiments in Figure 12 the electrode contained 137.5 cesium gluconate, 10 HEPES, 0.2 EGTA, 5 QX314 chloride, 2.5 MgATP, and 0.1 GTP. All IPSPs were evoked at 0.1 Hz with bipolar stimulating electrodes that were placed in the NAc near the cortex. Data were filtered at 1 kHz, digitized at 3–10 kHz and collected on-line using acquisition software developed in this laboratory by D. Selig. The amplitudes of IPSPs/IPSCs were calculated by taking the mean of a 2–4 msec window around the peak and comparing this with the mean of a window immediately before the stimulation artifact. For paired-pulse experiments, single pulses were given alternately with paired pulses. Because the second IPSC was usually superimposed on the decay phase of the first IPSC, the amplitude of the second pulse was adjusted by subtraction of the amplitude of a running average of single-pulse IPSCs measured at the time point of the maximal amplitude of the second pulse in paired-pulse sweeps. Synaptic potential amplitudes were displayed on-line during the course of each experiment, along with the input and (in voltage-clamp experiments) access resistance calculated by voltage or current pulses given at 0.1 Hz.

Dopamine does not affect mIPSC frequency or amplitude. A, Consecutive 10 sec traces before (left) and during (right) application of dopamine (100 μm). Spontaneous mIPSCs were recorded with a CsCl-based electrode solution at −80 mV, and with DNQX and APV in the external bathing solution. B, Cumulative probabilityhistograms of mIPSC amplitudes taken from the cell shown in A showing the lack of effect of dopamine on mIPSC amplitude. C, Normalized averages (1 min bins) of the mIPSC frequency (top) and amplitude (bottom) from 10 experiments demonstrate that dopamine (75–100 μm) does not affect mIPSC frequency or amplitude. Mean baseline frequency was 0.8 ± 0.1 Hz, and mean amplitude was 14.3 ± 1.8 pA (n = 10).

Monosynaptic IPSPs are depressed by amphetamine and dopamine. A, B, In DNQX (10 μm) and APV (75 μm), IPSPs recorded with a potassium methylsulphate-based electrode solution exhibited a linear dependence on membrane potential and reversed at the estimated Cl− reversal potential of −58 mV.C, The traces depict the effects of picrotoxin (50 μm) on IPSPs recorded at 0 mV. In this, and subsequent, figures, the negative voltage deflection after the synaptic potential is the result of a −0.03 nA current pulse given through the recording electrode. The traces are averages of consecutive sweeps taken at the times indicated by the numbers in the bottom graph, which demonstrates the time course of experiment. D, Representative IPSP traces (top) taken from one experiment before application of amphetamine, in the presence of bath-applied amphetamine (10 μm) and after recovery of the response. The bottom graph is a normalized average (n = 10) of the time course of the effects of amphetamine on IPSPs. Error bars (this and subsequent graphs) indicate SEM. E, The traces are taken from one experiment during the baseline, before application of dopamine (75 μm), and during recovery. The bottom graph is an average (n = 28) demonstrating that dopamine reversibly depressed the IPSP.

Dopamine depresses compound synaptic potentials in response to lower stimulus strengths to a greater extent than those in response to higher stimulus strengths. A, Traces taken from the experiment shown in B. B, In this example experiment, low (top) and high (bottom) stimulus strengths were delivered alternately (5 sec between stimuli). DA (100 μm) was applied first in the absence and then in the presence of picrotoxin (200 μm). Note that picrotoxin causes a greater increase in the synaptic potential that results from high stimulation strengths than in that resulting from low stimulation strengths, despite their similar initial amplitudes. C, Average graphs demonstrate that in the absence of picrotoxin (left), DA depresses the compound potential for smaller stimulus strengths more than for larger stimulus strengths. In the presence of picrotoxin (right), however, DA is equally effective in depressing the EPSP no matter the stimulus strength.
Each point on the illustrated graphs represents the mean of all data points in a 1 min bin. Each representative data trace is the mean of all traces in a 1.5 min bin. For mIPSC and miniature EPSC (mEPSC) experiments, spontaneous synaptic events were detected using software (generously provided by J.H. Steinbach, Washington University, St. Louis, MO) that detected the fast rise time of synaptic events. In most experiments, each putative event was then accepted or rejected manually. For high-K+ experiments, the frequency of mIPSCs was too high for manual analysis and, therefore, stretches of data were analyzed by eye to ensure that the software was detecting genuine synaptic events adequately. Statistics were calculated as described previously (Nicola et al., 1996). Briefly, to determine whether an agonist applied alone had a significant effect, pairedt tests compared the average of all points in the 10 min baseline with the average of all points in a 3 min window at the point at which agonist washout was begun. When antagonists were applied, two-way repeated-measures ANOVAs were computed. For all analyses,p ≤ 0.05 was considered statistically significant.
Drug stock solutions were made daily and diluted with external solution just before bath application to the slice. Antagonists were applied for at least 10 min before application of agonists. Dopamine hydrochloride, (+)-SKF38393 hydrochloride, dihydrexidine, (−)-quinpirole hydrochloride, and (±)-2-amino-6,7-dihydroxy-1,2,3,4-tetrahydronaphthalene hydrobromide (6,7-ADTN) stocks were made in water containing sodium metabisulfite such that the final solution applied to the slice contained 50 μm metabisulfite. (+)-Amphetamine sulfate, (+)-SCH23390,d-2-amino-5-phosphonovaleric acid (d-APV),d,l-APV and picrotoxin stock solutions were made in water. TTX, (−)-sulpiride, 6,7-dinitroquinoxaline-2,3-dione (DNQX) and 8-cyclopentyltheophylline (8-CPT) stock solutions were made in dimethylsulfoxide at 1000–10,000 times their final concentrations. CGP35348 was dissolved directly into the final solution. LidocaineN-ethyl chloride (QX314 chloride) was synthesized for us by Precision Biochemicals (Colton, CA). Other drugs were obtained from RBI (Natick, MA), Sigma (St. Louis, MO), or Tocris-Cookson (St. Louis, MO).
RESULTS
Monosynaptic IPSPs or IPSCs were recorded from cells in the core region of the NAc by blocking excitatory synaptic transmission with the glutamate receptor antagonists DNQX (10 μm) andd,l-APV (75 μm) and directly stimulating within the NAc. IPSPs or IPSCs were observed in all cells recorded under these conditions (n = 138) and were found to have a reversal potential of approximately −58 mV (Fig.1A,B; n = 3), in close agreement with the calculated reversal potential for Cl− under our recording conditions (potassium methylsulphate-based pipette solution with the Na+channel blocker QX314). Picrotoxin (50–200 μm) completely abolished the IPSPs (Fig. 1C, n = 7), confirming that they were mediated by activation of GABAA receptors. Because IPSPs were often small when cells were held near their resting membrane potentials, in most experiments, cells were held at 0 mV using a cesium gluconate-based pipette solution.
Pharmacology of DA and amphetamine modulation of IPSPs
Bath application of amphetamine (10 μm) reversibly reduced IPSPs by 19 ± 4% (n = 10,p < 0.005) without affecting the input resistance of the cell as measured by the amplitude of a constant current pulse applied after each stimulus (Fig. 1D). Consistent with the ability of amphetamine to reverse the DA transporter and cause efflux of DA into the extracellular space (Ritz et al., 1987; Seiden et al., 1993; Sulzer et al., 1995), DA (75 μm) also reversibly depressed IPSPs (by 28 ± 4%, n = 28,p < 0.001), again without changing the cells’ input resistance (Fig. 1E). As with their actions on excitatory synaptic transmission (Nicola et al., 1996), amphetamine and DA reversibly depress inhibitory synaptic transmission.
Based on their pharmacology, DA receptors can be divided into two general classes, the D1-like and D2-like receptors (Civelli et al., 1993; Sibley, 1995). To determine which receptor subtype is responsible for the depressant effects of DA on IPSPs, we first examined the effects of the D2 antagonist sulpiride. As shown in Figure2A, sulpiride (20 μm) had no significant effect on the depression elicited by DA (60 μm). DA caused a depression of 20 ± 5% when initially applied and a depression of 16 ± 4% when reapplied to the same cells (n = 8) in the presence of sulpiride (p > 0.34). In contrast, SCH23390, a D1 antagonist, was very effective in blocking the effects of DA on IPSPs (Fig. 2B). In these experiments, DA (75 μm) initially caused a depression of 18 ± 1% but no significant depression (3 ± 2%) when reapplied to the same cells in the presence of SCH23390 (10 μm,n = 7, p < 0.001). SCH23390 (10 μm) also antagonized the depressant effect of amphetamine (10 μm), which reduced the IPSPs by 28 ± 7% in control conditions but by only 9 ± 4% in the same cells (n = 8) in the presence of SCH23390 (p < 0.02) (Fig. 3).

D1 but not D2 antagonists reduce the effects of dopamine on IPSPs. A, The IPSP traces (top) were taken at the times indicated in the example experiment (middle) in which dopamine (60 μm) was applied first in the absence and then in the presence of the D2 antagonist sulpiride (20 μm). Thebottom graph is a summary (n = 8) comparing the effects of dopamine on IPSPs in the presence and absence of sulpiride. B, Traces (top) taken from an experiment (middle) in which dopamine (75 μm) was applied first in the absence and then in the presence of the D1 antagonist SCH23390 (10 μm). Thebottom graph is a summary (n = 7) comparing the effects of dopamine in the presence and absence of SCH23390.

SCH23390 antagonizes the effects of amphetamine on IPSPS. A, Traces from the experiment inB. B, A representative experiment in which amphetamine (10 μm) was first applied in the absence and then in the presence of SCH23390 (10 μm).C, A summary (n = 6) comparing the effects of amphetamine in the presence and absence of SCH23390.
To further characterize the receptor subtype mediating the actions of DA on inhibitory synaptic transmission, we examined the effects of a number of different agonists. The broad-spectrum DA agonist 6,7-ADTN (50 μm) mimicked the effects of DA and amphetamine by causing a significant (21 ± 3%) reduction in the IPSPs (n = 4, p < 0.02) (Fig.4A). In contrast, the specific D2 receptor agonist quinpirole (10 μm) had no effect (3 ± 2% depression, n = 14, p > 0.1) (Fig. 4B). Surprisingly, the D1 agonist SKF38393 (20 μm) was also without significant effect on IPSPs (0 ± 1% depression, n = 9, p > 0.5) (Fig. 4C). A different D1 agonist, dihydrexidine (10–40 μm), had no significant effects on either IPSPs or EPSPs, despite the fact that dopamine (75 μm) reduced the magnitude of the response in the same cells (n = 3 for IPSPs and n = 5 for EPSPs; maximum depression in dihydrexidine was 10 ± 5% compared with 21 ± 4% and 38 ± 9% reductions in dopamine for IPSPs and EPSPs, respectively; p > 0.05 for dihydrexidine andp < 0.05 for dopamine; data not shown). However, SKF38393 (20 μm) was able to significantly attenuate the actions of DA (75 μm) on IPSPs (22 ± 3% depression induced by DA alone, 11 ± 3% depression induced by DA in the presence of SKF38393, n = 9, p < 0.001; data not shown). These results suggest that D1 agonists bind to the DA receptor responsible for depressing inhibitory synaptic transmission but do not function as full agonists.

Effects of dopamine agonists on IPSPs.A, The traces are taken from a single experiment during baseline, during the application of 6,7-ADTN (50 μm) and during washout. The bottom graph is a summary of four experiments in which 6,7-ADTN was applied. B, The traces are taken from the experiment shown in the middle graph. Dopamine (75 μm) depressed the IPSPs, whereas in the same cell, application of the D2 agonist quinpirole (10 μm) was without effect. The bottom graph is a summary of 14 experiments in which quinpirole was applied. C, The traces are taken from the experiment shown in the middle graph. Dopamine (75 μm) depressed the IPSPs, whereas in the same cell, SKF38393 (20 μm) had minimal effect. The bottom graph is a summary of nine experiments in which SKF38393 was applied.
In the VTA, DA enhances the release of GABA by acting at a D1 receptor (Cameron and Williams, 1993). If DA has a similar effect in the NAc, the depression of IPSPs could be attributable to GABA acting on presynaptic GABAB receptors, which depress IPSPs in the NAc (Uchimura and North, 1991). To test this possibility, we examined the effects of the GABAB antagonist CGP35348 on the DA-induced depression of IPSPs. In the absence of CGP35348, DA (75 μm) caused a depression of 23 ± 4%, whereas reapplication of DA in the presence of CGP35348 (500 μm) still depressed IPSPs by 21 ± 4% (n = 3) (Fig.5A,E). Previously, when examining excitatory synaptic transmission, a similar lack of effect of CGP35348 on DA’s actions was found (Nicola et al., 1996). Another transmitter that may mediate the effects of DA on synaptic transmission is adenosine. In VTA slices prepared from animals administered cocaine or morphine chronically, DA depresses inhibitory synaptic transmission via a mechanism involving adenosine acting at presynaptic adenosine A1 receptors (Bonci and Williams, 1996). Indeed, a similar mechanism involving the NMDA receptor-dependent release of adenosine has been suggested recently to underlie the synaptic effects of DA in the NAc (Harvey and Lacey, 1996a). However, we found that the adenosine receptor antagonist 8-CPT (20 μm) did not reduce the depressant action of DA on either EPSPs or IPSPs. DA depressed IPSPs by 23 ± 7% in control conditions and by 26 ± 4% in the same cells in the presence of 8-CPT (p > 0.9, n = 3) (Fig.5B,E). Similarly, DA depressed the EPSP by 26 ± 1% in control conditions and by 35 ± 4% in the presence of CPT (p > 0.3, n= 2) (Fig. 5C,E). Furthermore, under our conditions, depression of neither EPSPs nor IPSPs requires the activation of NMDA receptors, because DA was capable of depressing the EPSP by 32.3 ± 1% in the presence ofd,l-APV (75 μm) (Fig.5D,E), and all of our experiments involving IPSPs are performed with this concentration ofd,l-APV in the bathing medium. Thus, the effects of DA on inhibitory or excitatory synaptic transmission in the NAc do not appear to require activation of GABAB, adenosine, or NMDA receptors.
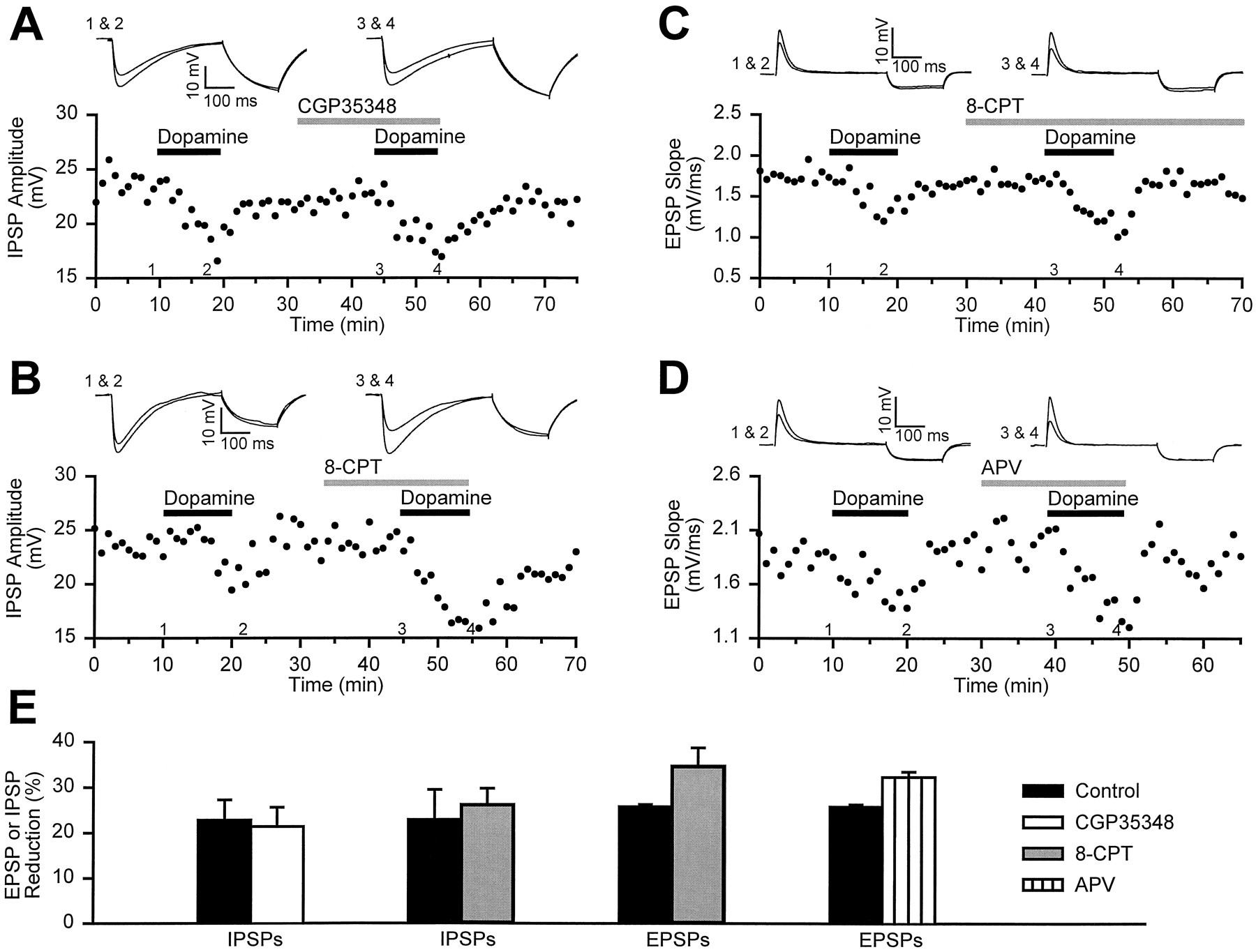
GABAB, adenosine, and NMDA receptor antagonists do not reduce the effects of dopamine on EPSPs and IPSPs. A, The traces (top) are IPSPs taken from an experiment (bottom) in which dopamine (75 μm) was applied both in the absence and then in the presence of the GABAB antagonist CGP35348 (500 μm). B, As illustrated by the traces (top) taken from an example experiment (bottom), the adenosine antagonist 8-CPT does not reduce the effects of dopamine on IPSPs. C, An experiment identical to that in B, except that EPSPs were examined instead of IPSPs. D, The NMDA receptor antagonistd,l-APV (75 μm) does not reduce the magnitude of the depression of EPSPs caused by dopamine (75 μm). E, A summary graphillustrates that none of these antagonists reduced the ability of dopamine to depress EPSPs and IPSPs (n ≥ 2 for each antagonist). Dopamine was applied twice in each cell, once before application of antagonist and once in the presence of the antagonist.
Mechanism of DA actions on IPSPs
To determine whether DA reduces inhibitory synaptic transmission by a postsynaptic reduction in the sensitivity to synaptically released GABA or a presynaptic depression of GABA release, we initially examined the effects of DA on the ratio of the amplitudes of IPSCs elicited by paired-pulse stimulation. When two stimuli are given in rapid succession, the probability of transmitter release in response to the second stimulation is altered (Zucker, 1989). The ratio of the second response to the first inversely correlates with the probability of release and is therefore usually affected by manipulations that alter release probability (Zucker, 1989; Manabe et al., 1993). To examine the effects of DA on the paired-pulse ratio, cells were voltage-clamped at 0 mV and paired pulses (50 msec interstimulus interval) were delivered alternately with single pulses. As shown in Figure 6, DA (100–150 μm) significantly increased the paired-pulse ratio to 180 ± 22% (p < 0.05) of the baseline ratio (0.74 ± 0.11, n = 6). This result is consistent with a DA-induced decrease in the probability of GABA release, although it does not rule out contributions of additional postsynaptic mechanisms.

Dopamine increases the paired-pulse ratio.A, IPSCs in response to single stimuli (top) and paired stimuli (bottom) (50 msec interstimulus interval) before and during application of dopamine (150 μm). The single and paired stimuli were alternately applied to the slice. In the paired-pulse traces, the IPSCs to the second pulse were obtained after subtracting an averaged IPSC in response to the single stimuli. In the paired-pulse trace in dopamine (bottom right), the first IPSC was scaled to the first IPSC in the absence of dopamine. B, Summary (n = 6) of the time course of the effects of dopamine (100–150 μm) on the paired-pulse ratio (top graph) and the amplitude of the IPSC in response to the first pulse (bottom graph).
To determine whether DA may also depress GABA receptor function or number, we recorded spontaneous mIPSCs in the presence of TTX (1.5 μm). The electrode solution for these experiments contained either cesium gluconate (n = 6, holding potential of 0 mV) or CsCl (n = 4, holding potential of −80 mV). Picrotoxin (100–200 μm) reduced mIPSC frequency to zero under both recording conditions (data not shown), indicating that the mIPSCs were not contaminated by excitatory or nonsynaptic events. As shown in Figure 7, DA (75–100 μm) did not change the mIPSC amplitude distribution (Fig.7B) or mean mIPSC amplitude (Fig. 7C) (6 ± 3% depression, p > 0.05, n = 10). These findings indicate that the effects of DA on synaptic transmission are unlikely to involve a postsynaptic reduction in the sensitivity of the cell to synaptically released GABA. However, in contrast to the robust depression in mEPSC frequency we observed in previous work (Nicola et al., 1996), mIPSC frequency was not reduced by DA (113 ± 8%, p > 0.05) (Fig.7A,C).
Given the surprising dichotomy in the effects of DA on mEPSC and mIPSC frequency, we wanted to ensure that slices in which no change in mIPSC frequency was observed were competent to express DA-induced changes in mEPSC frequency. To accomplish this, we simultaneously recorded mEPSCs and mIPSCs in the same cell (Fig. 8) by maintaining the holding potential at −15 to −25 mV, using a cesium gluconate electrode solution and bathing the slice in TTX (1.5 μm) and d-APV (50 μm) (but no AMPA or GABA receptor antagonists). Under these conditions, both inward and outward miniature synaptic currents could be resolved (Fig.8A). Application of DNQX (10–20 μm) selectively abolished the inward currents, and picrotoxin (200 μm) abolished the outward currents, indicating that inward currents were mEPSCs and outward currents were mIPSCs. Application of DA (100 μm, n = 3) caused a clear decrease in the frequency of mEPSCs (49 ± 4% reduction,p < 0.01), whereas the frequency of mIPSCs remained unchanged (8 ± 14% reduction, p > 0.5) (Fig.8A,C). Also consistent with the previous results, no change in either mEPSC or mIPSC amplitude was elicited by DA (Fig. 8). This clear differential effect of DA on the frequency of miniature events suggests that DA depresses excitatory and inhibitory synaptic transmission by different presynaptic mechanisms.

Dopamine does not reduce mIPSC frequency in cells in which an effect of dopamine on mEPSC frequency can be observed.A, Consecutive 10 sec traces before (left) and during application of dopamine (100 μm) taken from a cell recorded with a cesium gluconate-based electrode solution and held at −20 mV. The external bathing solution contained d-APV (50 μm) but no DNQX or picrotoxin. Inward currents are mEPSCs, and their frequency is reduced in dopamine. Dopamine had no effect on the outward currents, which are mIPSCs. B, Cumulative probabilityhistograms of mEPSC amplitudes (left) and mIPSC amplitudes (right) show that neither amplitude distribution was affected by dopamine. C, Summary (n = 3) of the effects of dopamine on simultaneously monitored mEPSC and mIPSC frequency and amplitude. Theasterisk indicates a statistical difference from baseline (p < 0.01). Mean baseline values for mEPSC frequency and amplitude were, respectively, 1.1 ± 0.1 Hz and 4.8 ± 0.2 pA; for mIPSCs they were 0.6 ± 0.2 Hz and 6.6 ± 0.3 pA.
Presynaptic modulation of transmitter release may occur through at least two general mechanisms: (1) the influx of Ca2+through voltage-dependent Ca2+ channels in the synaptic terminal may be reduced by modulation of Ca2+ or K+ channels, or (2) the release machinery may be altered at some point after calcium entry into the terminal (Thompson et al., 1993). Blockade of presynaptic K+ channels with Ba2+ will broaden the presynaptic action potential waveform and may therefore interact with the former mechanism, but is unlikely to interact with the latter (Thompson and Gähwiler, 1992). Therefore, we examined the effects of DA on evoked IPSPs and EPSPs in the presence and absence of Ba2+ to determine whether this manipulation differentially changes the DA-mediated inhibition of inhibitory and excitatory synaptic transmission (Fig. 9). For monosynaptic IPSPs (recorded in DNQX and APV), DA was first applied in normal external solution and then in solution containing 1 mm Ba2+ (Fig. 9A). In the absence of Ba2+, DA (75 μm) depressed the IPSP by 42 ± 9%, whereas in the presence of Ba2+, DA reduced the IPSP by 16 ± 6%, a significant reduction in the effectiveness of DA (p < 0.01, n = 5). For EPSPs (recorded in picrotoxin), however, DA’s effects were identical whether or not Ba2+ was present (Fig. 9B). In control conditions, DA (75 μm) reduced the EPSP by 27 ± 6%, and in 1 mm Ba2+, DA reduced the EPSP by 26 ± 6% (p > 0.1,n = 5). These results provide evidence that DA modulates inhibitory synaptic transmission by modulating a presynaptic ionic conductance, whereas DA modulates excitatory transmission by interference with a process that occurs independent of the entry of Ca2+.

Ba2+ reduces the effects of dopamine on inhibitory, but not excitatory, synaptic transmission.A, Traces (top) were taken from the example experiment (middle) at the times indicated. Dopamine (75 μm) was applied to pharmacologically isolated IPSPs in cells held at 0 mV, and then reapplied in the presence of BaCl2 (1 mm). The summary graph (bottom, n = 5) compares the dopamine-induced depression of IPSPs in the absence and presence of Ba2+. B, Traces of reversed EPSPs (top) were taken from the example experiment (middle) at the times indicated. The effects of dopamine (75 μm) on EPSPs were examined first in the absence and then in the presence of Ba2+. The cell was held at +25 mV in the presence of d-APV (50 μm) and picrotoxin (50 mm). The summary graph (bottom, n = 5) compares the dopamine-induced depression of EPSPs in the absence and presence of Ba2+.
To test this hypothesis further, we elevated the external K+ concentration from 1.6 to 22–25 mmand examined mIPSCs recorded in TTX. In high K+, mIPSC frequency was increased by four- to eightfold over the frequency in normal K+ (Fig.10A,B). Brief application of the Ca2+ channel blocker Cd2+ (100 μm) reversibly reduced the increased mIPSC frequency to its value in normal K+, whereas Cd2+ application in normal K+ had no effect on the frequency of mIPSCs recorded in the same cells (n = 3) (Fig. 10B). This indicates that the increased mIPSC frequency in elevated K+ was attributable to Ca2+influx through voltage-dependent Ca2+ channels, whereas mIPSCs recorded in normal K+ are independent of Ca2+ channel activity (Doze et al., 1995;Scanziani et al., 1995). DA (100 μm) reduced the frequency of Ca2+-dependent mIPSCs recorded in high K+ by 18 ± 4% (p < 0.05, n = 4) while leaving their amplitude unchanged (3 ± 4% depression, p > 0.5) (Fig. 10). In the same cells, the frequency of Ca2+-independent mIPSCs recorded in normal K+ was not reduced by DA (Fig.10B). Thus, DA is capable of modulating only those inhibitory synaptic events that are dependent on Ca2+ entry, suggesting that DA depresses the degree of Ca2+ influx into GABAergic terminals.
In contrast, DA appears to modulate glutamate release at excitatory synapses whether or not the release is dependent on presynaptic Ca2+ influx (Fig.8A,C) (Nicola et al., 1996). To test this conclusion further, we recorded mEPSCs in the presence of the Ca2+ channel blockers Cd2+ (100 μm, n = 4) or Co2+ (5 mm, n = 4). In these experiments, TTX was not used, and complete abolition of the evoked EPSC by the Ca2+ channel blocker was observed before beginning the experiment. When amphetamine (10 μm) was applied, mEPSC frequency was reduced by 41 ± 7% (p< 0.005), whereas mEPSC amplitude remained unchanged (2 ± 3% depression, p > 0.2, n = 8) (Fig.11). Previous work has demonstrated that amphetamine reduces mEPSC frequency by increasing extracellular DA levels (Nicola et al., 1996). Thus, unlike the modulation of inhibitory synaptic transmission, DA can presynaptically decrease excitatory synaptic transmission in the NAc by a mechanism that does not involve decreasing Ca2+ entry into the presynaptic terminal through voltage-dependent Ca2+ channels.
Integrative consequences of dual modulation of EPSPs and IPSPs
Depression of both excitatory and inhibitory synaptic transmission by DA may influence information processing by NAc cells. In vivo iontophoresis studies using single unit recording of striatal cells have shown that local adminstration of DA reduces baseline “noise” firing more than firing evoked by cortical stimulation or behavioral events (Johnson et al., 1983; Rolls et al., 1984). Reduction by DA of both EPSPs and IPSPs may contribute to this enhancement of the signal-to-noise ratio. NAc (and striatal) cells form dense inhibitory synaptic connections with neighboring cells. Strong excitatory activation of many NAc cells is therefore likely to incur a greater degree of inhibition from neighboring NAc cells than weak excitation of only a few NAc cells; that is, strong inputs will be reduced proportionally by GABAergic inhibition to a greater extent than will weak inputs. DA might therefore reduce the magnitude of strong excitatory responses less than that of weak responses, because IPSPs that serve to reduce the strong response will be depressed along with the EPSPs. To test whether DA has differential effects on synaptic potentials resulting from strong and weak inputs, we used a Cs gluconate-based electrode solution with the Na+channel blocker QX314 to record compound synaptic potentials in the absence of antagonists of excitatory or inhibitory synaptic transmission. The synaptic potential was recorded at −70 mV, and the slice was stimulated alternately (5 sec between stimuli) at two different stimulus strengths. One stimulus strength was ∼5–10 times higher than the other, but the magnitude of the response to the higher stimulus was usually less than twice as large as the response to the lower stimulus strength (Fig. 12A), and the time course of decay tended to be shorter for the larger response.
As shown in Figure 12C, on average (n = 7), the response to the higher stimulus was attenuated by DA (100 μm) less than the response to the lower stimulus (21 ± 3% depression for the larger, 45 ± 8% for the smaller). After recovery of the DA effect, picrotoxin was applied (Fig.12B). Picrotoxin (200 μm) caused a much greater increase in the response for the higher stimulus than for the lower stimulus (150 ± 12% of baseline for the larger, 102 ± 8% for the smaller), indicating that the larger response was more attenuated by GABAergic inhibition than the smaller response. When DA was applied to the same cells in the presence of picrotoxin (with the same stimulus strengths used to elicit the responses in the absence of picrotoxin), the inhibition of the larger and smaller responses was approximately equal (31 ± 6% depression for the larger, 38 ± 9% for the smaller) (Fig. 12C). For the higher stimulus strength, blockade of inhibition resulted in an increase in the magnitude of the depression elicited by DA (p < 0.003), whereas for the lower stimulus strength, there was no difference in DA’s effectiveness in the presence of picrotoxin (p > 0.7). Thus, as a result of reciprocal inhibition among NAc cells, DA depresses strong inputs less than weak ones.
DISCUSSION
We have found that DA and the psychostimulant amphetamine, acting via release of DA, depress inhibitory synaptic transmission in the NAc via a presynaptic mechanism that is distinct from the presynaptic modulation of excitatory synaptic transmission by DA in the same structure. The site of action of this modulation is likely to be the GABAergic terminals of the axons of NAc medium spiny cells. There are few, if any, GABAergic inputs to the NAc from other brain nuclei (Christie et al., 1987), whereas, as in the dorsal striatum, NAc medium spiny neurons are GABAergic and exhibit dense local axonal arborizations (Chang and Kitai, 1985; Pennartz et al., 1991; O’Donnell and Grace, 1993; Kawaguchi et al., 1995) that likely give rise to the large numbers of GABA-containing axon terminals found in ultrastructural studies of the NAc (Pickel et al., 1988).
Previous studies of the NAc in vivo (Yim and Mogenson, 1988) and in slices (Pennartz et al., 1992a) reported that DA depressed putative IPSPs. These studies did not, however, determine the receptor subtype mediating this effect or the locus of its action. Our pharmacological experiments using subtype-specific antagonists indicate that a D1-like receptor mediates the effects of DA on inhibitory synaptic transmission. We ruled out the possibility that DA worked indirectly to depress IPSPs by causing a D1-mediated enhanced release of GABA (Cameron and Williams, 1993) or adenosine (Bonci and Williams, 1996; Harvey and Lacey, 1996a). Thus, the critical D1-like receptors appear to be localized directly on the GABAergic terminals. A similar presynaptic D1-mediated effect has been found on GABAergic inhibitory responses recorded in magnocellular basal forebrain cells (Momiyama and Sim, 1996) as well as on excitatory synaptic transmission in the NAc (Pennartz et al., 1992a; Harvey and Lacey, 1996b; Nicola et al., 1996) and basal forebrain (Momiyama et al., 1996). D1-mediated reduction in synaptic transmission that may be either pre- or postsynaptic has also been reported in the prefrontal (Law-Tho et al., 1994) and entorhinal (Pralong and Jones, 1993) cortex. Thus, whereas D2-like receptors function as autoreceptors on dopaminergic cell bodies and terminals (Wolf and Roth, 1987), D1-like receptors appear to be targeted to nerve terminals where they can regulate either GABA or glutamate release.
A curious result was that neither D1- nor D2-specific agonists mimicked the effect of DA on inhibitory synaptic transmission, although the D1 agonist SKF38393 did appear to act as a weak antagonist of the DA-induced depression. One possible explanation for the apparent discrepancy in the effects of D1 agonists and antagonists is that in the NAc, D1 receptors are modified such that SKF38393 is able to bind the receptor but unable to activate it. An alternate possibility is that the DA receptor responsible for the effects observed here is a new member of the D1-like family. Several lines of evidence point to the existence of such receptors. Neurochemical data suggests a dissociation between the localization of dopamine-stimulated adenylate cyclase activity and binding sites for [3H]-SCH23390 (Mailman et al., 1986; Andersen et al., 1990). Furthermore, behavioral studies have found that the ability of D1 agonists to promote D1 antagonist-sensitive behaviors and their ability to stimulate adenylate cyclase activity in vitro are not well correlated (Murray and Waddington, 1989; Arnt et al., 1992; Downes and Waddington, 1993;Starr and Starr, 1993; Deveney and Waddington, 1995; Gnanalingham et al., 1995; Waddington and Deveney, 1996). Lastly, there is some evidence for a D1-like receptor in the striatum that is coupled to phosphoinositide turnover (Mahan et al., 1990; Undie and Friedman, 1990, 1992; Undie et al., 1994) and two novel adenylate cyclase-coupled D1-like receptors have been cloned recently from Drosophila(Sugamori et al., 1995; Feng et al., 1996). Interestingly, these receptors exhibit a pharmacological profile similar to that reported here.
The major finding of this study is that the presynaptic mechanisms by which DA receptors depress inhibitory and excitatory synaptic transmission are different. DA clearly depressed the frequency of mEPSCs under conditions in which the mEPSCs were not dependent on the influx of Ca2+ via voltage-dependent Ca2+ channels. This finding suggests that DA depresses glutamate release via a mechanism that is independent of the entry of Ca2+ into excitatory terminals. In contrast, DA had no effect on the frequency of mIPSCs unless the frequency was increased by tonic activation of voltage-dependent Ca2+ channels. Thus, DA appears to act on GABAergic nerve terminals in the NAc via modulation of an ionic conductance. Consistent with this conclusion, the K+ channel blocker Ba2+ significantly reduced the ability of DA to depress IPSPs, but not EPSPs, presumably because Ba2+ prolonged the presynaptic action potential and enhanced the Ca2+ influx into the terminal.
Results and conclusions very similar to those reported here have come from the study of the GABAB receptor agonist baclofen on synaptic transmission in the hippocampus (Scanziani et al., 1992;Thompson and Gähwiler, 1992; Doze et al., 1995). In hippocampal pyramidal cells, baclofen presynaptically reduced the magnitude of evoked EPSCs and IPSCs, but reduced the frequency only of mEPSCs, leaving mIPSC frequency unchanged. As was the case for DA in the NAc, baclofen reduced mIPSC frequency in high K+ (Doze et al., 1995), and Ba2+ reduced the ability of baclofen to depress IPSPs, but not EPSPs, in pyramidal cells (Thompson and Gähwiler, 1992). Measurement of presynaptic Ca2+ influx at the granule cell to Purkinje cell synapses in the cerebellum provides additional direct evidence that modulation of Ca2+ influx and modulation of a Ca2+-independent step in the release process are two distinct mechanisms by which presynaptic receptors can influence transmitter release (Dittman and Regehr, 1996).
Other investigators using intracellular recording from NAc neurons have reported that DA may cause small D1-mediated hyperpolarizing and D2-mediated depolarizing responses by modulation of a K+ conductance (Uchimura et al., 1986; Higashi et al., 1989; Uchimura and North, 1990). We did not observe significant postsynaptic effects of DA; however, this may be a consequence of our recording conditions, which often included replacement of K+ in the electrode solution with Cs+. Indeed, it is possible that if a similar D1-mediated effect occurred in the terminals of NAc axons, it could contribute to the reduction in Ca2+ influx, which we propose is responsible for the depression of GABA release by DA. In neostriatal cells, DA acting at D1 receptors has been reported to reduce both a voltage-dependent sodium current (Surmeier et al., 1992;Schiffmann et al., 1995) and voltage-dependent Ca2+currents (Surmeier et al., 1995). If similar effects occur in NAc cells, these too may contribute to the effects of DA on inhibitory synaptic transmission.
Understanding the neural mechanisms responsible for the behaviors that involve DA and the NAc will require an understanding of the functional consequences of DA’s actions on the output of NAc neurons. To begin to examine the effects of DA on the integrative activity of NAc cells, we recorded compound synaptic potentials elicited by different stimulus strengths in an attempt to approximate the mixture of excitation and inhibition to which NAc cells are exposed in vivo. DA was more effective in reducing synaptic potentials evoked with small stimulus strengths than with large stimuli. When GABAergic inhibition was blocked with picrotoxin, however, this difference was no longer apparent. An explanation for these results is that the responses to larger stimulus strengths were already reduced by inhibition resulting from activation of neighboring NAc cells. Reduction by DA of both EPSPs and IPSPs would therefore result in a greater reduction of compound potentials resulting from weak inputs than from stronger ones. Such a differential effect of DA may contribute to the apparent decrease in spontaneous random firing of NAc cells in vivo that develops during the self-administration of cocaine, as well as the concomitant changes in single unit activity that are specifically associated with the act of lever-pressing and the receipt of cocaine (Carelli et al., 1993; Carelli and Deadwyler, 1996).
Although the synaptic interactions among GABAergic, glutamatergic, and dopaminergic systems in the NAc are complex, behavioral studies suggest that modulation of excitatory synaptic transmission in the NAc may have behavioral consequences different from those of modulating inhibitory synaptic transmission (Goeders et al., 1989; Goeders, 1992; Pulvirenti et al., 1992; Jackson and Westlind-Danielsson, 1994). This raises the possibility that the distinct mechanisms by which DA affects inhibitory and excitatory synaptic transmission may provide a basis for the development of additional pharmacological strategies for the study and manipulation of behaviors that involve the NAc. It will also be of interest to determine how these presynaptic mechanisms are modified by chronic administration of drugs of abuse and thereby contribute to the development of addiction.
Footnotes
Correspondence should be addressed to Dr. Robert C. Malenka, Department of Psychiatry, LPPI, Box 0984, University of California, San Francisco, CA 94143.
This work was supported by grants from the National Institute on Drug Abuse and the National Institute of Mental Health. We thank Roger Nicoll and members of the Malenka lab for helpful advice and encouragement.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}