

Abstract
NMDA receptor-induced excitotoxicity has been hypothesized to mediate abnormal choline (Cho) metabolism that is involved in alterations in membrane permeability and cell death in certain neurodegenerative disorders. To determine whether NMDA receptor overactivation modulates choline metabolism in vivo, we investigated the effects of NMDA on interstitial choline concentrations using microdialysis. Perfusion of NMDA by retrodialysis increased dialysate choline (∼400%) and reduced dialysate acetylcholine (Ach) (∼40%). Choline levels remained increased for at least 2.5 hr, but acetylcholine returned to pretreatment values 75 min after NMDA perfusion. The NMDA-evoked increase in dialysate choline was calcium and concentration dependent and was prevented with 1 mmAP-5, a competitive NMDA antagonist, but was not altered by mepacrine, a phospholipase A2 inhibitor. NMDA increased extracellular choline levels four- to fivefold in prefrontal cortex and hippocampus, produced a slight increase in neostriatum, and did not modify dialysate choline in cerebellum. Perfusion with NMDA for 2 hr produced a delayed, but not acute, reduction in choline acetyltransferase activity in the area surrounding the dialysis probe. Consistent with a lack of acute cholinergic neurotoxicity evoked by this treatment, basal acetylcholine levels were unaltered by 2 hr of continuous NMDA perfusion. Prolonged NMDA perfusion produced a 34% decrease in phosphatidylcholine content in the lipid fraction of the tissue surrounding the dialysis probe. These results show that NMDA modulates choline metabolism, eliciting a receptor-mediated, calcium-dependent, and region-specific increase in extracellular choline from membrane phospholipids that is not mediated by phospholipase A2 and precedes delayed excitotoxic neuronal cell death.
Excitotoxicity produced by overactivation of glutamatergic neurotransmission has been proposed as a mechanism that might mediate the cell death observed in several chronic neurodegenerative pathologies (Rothman and Olney, 1987; Olney, 1989; Farooqui and Horrocks, 1994a). Evidence from in vitrostudies using primary cell cultures indicates that among the different glutamate receptors, the NMDA subtype plays a key role in activating the mechanisms that lead to the neuronal cell death induced by excitotoxicity (Choi, 1994; Rothman and Olney, 1995). Intracellular calcium overload, produced mainly by calcium entry through the NMDA receptor, activates several calcium-dependent enzymatic pathways, including phospholipases, proteases, and nucleases (Farooqui and Horrocks, 1991, 1994b). Sustained activation of some of these lytic enzymes would result ultimately in an irreversible cell breakdown process. Studies performed in primary cell cultures have shown that both β-amyloid peptide and altered metabolism of tau protein, hallmarks of chronic degenerative processes such as Alzheimer’s disease, may contribute to an increase in the susceptibility of neurons to excitotoxic damage (Koh et al., 1990; Pizzi et al., 1995). However, although NMDA receptor overactivation is one of the early events in the biochemical cascade leading to excitotoxic cell death, the mechanisms responsible for the slow progression of the neurodegenerative process and for the special vulnerability of specific neuron populations have not been established.
Excitotoxic NMDA receptor overactivation has been proposed to be involved in the abnormal phospholipid metabolism observed in both acute neural trauma and chronic degenerative disorders such as Alzheimer’s disease (Farooqui and Horrocks, 1994b). Significant reductions in phosphatidylcholine (PtdCho) levels have been reported in certain neurodegenerative diseases (Nitsch et al., 1992), but the ability of excitotoxic mechanisms to modulate PtdCho metabolism has not been demonstrated.
In addition to alterations in phospholipid metabolism, some neurodegenerative disorders such as Alzheimer’s disease are characterized by an early loss of cholinergic function. Cholinergic neurons are unique in that under certain conditions they use choline (Cho) from membrane phospholipids to synthesize acetylcholine (Ach) (Wurtman, 1992). This metabolic capability, which permits cholinergic neurons to sustain neurotransmission at the expense of membrane building (Ulus et al., 1989), may lead to an actual decrease in the quantity of membrane per cell. This metabolic process has been proposed to underlie the particular vulnerability of cholinergic neurons (Wurtman, 1992). On the basis of this hypothesis, the present studies were undertaken to determine whether NMDA receptor overactivationin vivo is associated with alterations in Cho metabolism. Thus, the effects of local administration by retrodialysis of NMDA on extracellular Ach and Cho levels were investigated using an in vivo microdialysis technique. Cholinergic neurodegeneration was assessed by measuring choline acetyltransferase (ChAT) activity in the area surrounding the dialysis probe. We now report that NMDA increases extracellular Cho concentrations in vivo. This effect is calcium dependent and region specific and precedes a delayed reduction in ChAT activity around the dialysis probe.
MATERIALS AND METHODS
Animals and surgery. Male Wistar rats (Specific pathogen free Wistar/Han-Ico, IFFA-CREDO; L’Arbresle, Lyon, France), weighing 275–300 gm, were housed four per cage with food and water freely available. Animals were kept under standardized temperature, humidity, and illumination conditions (12 hr light/dark cycle, light beginning at 7 A.M.) for at least 5 d before use. Rats were anesthetized with chloral hydrate (Panreac, 400 mg/kg, 2 ml/kg, i.p.) and placed in a small-animal stereotaxic frame (Stoelting, Chicago, IL) with the incisor bar set at −3.3 mm. Custom-made, 5 mm membrane length, concentric dialysis probes were placed according to the atlas of Paxinos and Watson (1986), at the following coordinates (in centimeters relative to Bregma), in separate groups of rats: prefrontal cortex [anteroposterior (AP) +0.32, lateral (L) −0.08, ventral (V) −0.64.)]; ventral hippocampus (AP −0.56, L −0.50, V −0.86); neostriatum (AP +0.04, L −0.30, V −0.83); cerebellum (AP −1.05, L −0.08, V −0.72). At the end of each experiment, rats were perfused with methylene blue through the probe to verify location. All procedures involving animals and their care were conducted in conformity with institutional guidelines that are in compliance with national and international (European Economic Community Council Directive 86/609,OJ L 358,1; December 12, 1987) (National Institutes of Health Guide for the Care and Use of Laboratory Animals, National Institutes of Health Publication no. 85–23, 1985) laws and policies.
Microdialysis. Microdialysis was performed in freely moving animals 24 hr after surgery. A Krebs’–Ringer’s bicarbonate solution (KRB) [(in mm): KCl 1.4, NaCl 120, KH2PO4 1.3, NaHCO3 20, CaCl2 1.2, MgCl2 0.83, d-glucose 10 in 5 mm phosphate buffer, pH 7.4] was filtered through a 0.2 μm nylon membrane filter (Gelman Sciences, Ann Arbor, MI) and perfused through the inflow line at a flow rate of 0.7 μl/min, using a slow-speed peristaltic pump (Syringe Infusion Pump 22, Harvard Apparatus, South Natick, MA). The KRB solution contained 0.5 μm neostigmine, an acetylcholinesterase inhibitor, to recover detectable dialysate concentrations of Ach. Preliminary studies performed in our laboratory to characterize the effects of different concentrations of neostigmine in the perfusion fluid showed that the presence of 0.5 μm neostigmine in the perfusate does not significantly modify basal dialysate Cho levels. Under the conditions and flow rate used in our microdialysis technique, the lowest concentration of neostigmine in the perfusate that produced a significant reduction of dialysate Cho was 10 μm.
After a 75 min equilibration period, consecutive 15 min (10.5 μl) dialysate samples were collected, stored at −20°C, and assayed for Ach and Cho content. Drug treatment was administered by dissolving the corresponding compound at the specified concentration in the perfusion fluid. Although diffusion characteristics of probes may be different in brain matter than when tested in vitro (Benveniste et al., 1989), recovery for Ach and Cho in a static solution maintained at 37°C with a 5 mm membrane was assessed to provide an estimate of extracellular concentrations under the conditions used in our experiments. The average percentage in vitro recovery (±SEM) obtained with our probes against 0.6 μm Ach and 8 μm Cho when perfused at 0.7 μl/min was Ach = 41 ± 5%, Cho = 43 ± 8%.
Determination of Ach and Cho. Dialysate Ach and Cho levels were determined using a Bioanalytical Systems (BAS, West Lafayette, IN) HPLC with an electrochemical detector in conjunction with an enzyme reactor. Ach and Cho were separated in a BAS/Sepstik microbore column with a mobile phase consisting of 50 mmNaH2PO4, 0.005% Kathon, and 0.5 mm EDTA, pH 8.5 (adjusted with NaOH), at a flow rate of 140 μl/min. Ach and Cho were enzymatically converted to hydrogen peroxide by a post-column immobilized enzyme reactor (containing immobilized acetylcholinesterase and Cho oxidase). The resulting peroxide was measured electrochemically on a platinum electrode at +500 mV (vs an Ag/AgCl reference electrode, BAS-LC4B). In these conditions, the variation coefficient (CV) for repeated injections of low (1 pmol) or high (50 pmol) amounts of Ach and Cho was ∼8 and 4%, respectively. The detection limit at 2.5 nA was 100 fmol at a signal-to-noise ratio of 3:1.
ChAT assay. Immediately after dialysis, or 7 d later when required, rats were killed, and their brains were removed and frozen on dry ice. A micropunch steel tube (1.2 mm diameter; Stoelting) was introduced in the prefrontal cortex after the probe path, and the tissue sample was weighed and stored at −80°C for determination of ChAT. Additional samples were taken from the contralateral prefrontal cortex in each animal and processed in parallel. The method was adapted from Fonnum (1975) after Lehmann et al. (1993) with minor modifications. Tissue was homogenized in 50 vol of ice-cold 10 mm phosphate buffer (PB), pH 7.4. Homogenate was treated with Triton X-100 (0.02%, 30 min on ice). Ten microliters of Triton X-100-treated homogenate were incubated with 10 μl of substrate solution for 30 min at 37°C in a 1.2 ml microtube. Final concentrations in the incubation mixture were as follows (in mm): choline chloride 10, NaCl 300, PB 50, EDTA 10, physostigmine hemisulfate 0.1, acetyl-CoA 0.5; and [3H]acetyl-CoA (3 μCi/ml, specific activity 5 Ci/mmol). Reaction was terminated by placing tubes on ice, and 150 μl of ice-cold 10 mm PB followed by 1 ml of extraction/scintillation mixture (1.5% w/v tetraphenylborate in econofluor/acetonitrile 85:15 v/v) were added. Tubes were shaken gently, the two phases were allowed to separate, and radioactivity was counted by liquid scintillation. The protein concentration in the homogenate was estimated by the bicinchoninic acid method (Pierce, Rockford, IL). Data (nanomoles of [3H]Ach per milligrams of protein per hour) were expressed as percentage of the contralateral (untreated) prefrontal cortex values for each animal.
Determination of Cho in lipid fractions. NMDA (600 μm) was administered by retrodialysis for 12 hr in prefrontal cortex. Immediately after dialysis, rats were killed, and their brains were removed and frozen on dry ice. Micropunch tissue samples from the area surrounding the dialysis probe and from an equivalent area in the contralateral side were obtained as described above. Lipid extraction was performed according to Gonzalez-Sastre and Folch-Pi (1968). Hydrolysis of Cho containing phospholipids was adapted from Lee et al. (1993). Tissue was homogenized in 50 vol of ice-cold distilled water with a Polytron homogenizer. Lipids were extracted by adding 4 ml of chloroform/methanol/H2O (2:1:1 v/v) to 200 μl of homogenate. Phases were separated by centrifugation for 10 min at 1500 × g. Aliquots (1 ml) of the lower organic phases were dried by centrifugation under vacuum, resuspended in 6m HCl (200 μl), and hydrolyzed by incubation at 95°C for 1 hr. Samples were neutralized with 5 m NaOH (200 μl) and diluted 1:100 with mobile phase. Cho concentration in these samples was determined with HPLC as described above. Preliminary experiments using this procedure showed that Cho originates from the hydrolysis of PtdCho in the lipid fraction because sphingomyelin is not hydrolyzed under these conditions. Data (nanomoles of PtdCho per milligrams of wet weight) were expressed as percentage of the contralateral (untreated) prefrontal cortex values for each animal.
Data analysis. Results were expressed in dialysate concentrations of Ach or Cho (picomoles per microliters). The mean of at least three basal samples was calculated, and values were expressed as percentage of basal mean. Data were analyzed by repeated measures multivariate ANOVA (MANOVA), including the mean of the two pretreatment samples and the corresponding NMDA-treated samples as the within-subject factor. Post hoc comparisons were performed by a paired t test or independent t test for between groups comparisons when appropriate.
RESULTS
Mean basal Ach and Cho dialysate levels were 0.035 ± 0.005 and 1.65 ± 0.25 pmol/μl, respectively. Basal levels remained unchanged during 4 hr of perfusion in control untreated animals (Fig.1A,B). Perfusion with NMDA (300 μm) for 30 min reduced extracellular Ach levels to 57% of basal values and produced a threefold increase in dialysate Cho levels (p < 0.01, MANOVA) (Fig.1A,B). When NMDA was removed from the perfusion fluid, Ach rapidly returned to basal levels but Cho levels remained increased for at least 2 hr (p < 0.01, MANOVA) (Fig. 1B).

Effects of 30 min perfusion with 300 μm NMDA through the microdialysis probe on extracellular Ach (A) and Cho (B) levels in prefrontal cortex. Symbols: □, control; ▪, NMDA. Basal levels (dialysate picomoles per microliter) and number of animals per group (mean ± SEM) were as follows: Ach, □, 0.0354 ± 0.0046 (n = 5); ▪, 0.0349 ± 0.0037 (n = 6); Cho, □, 1.6503 ± 0.2497 (n = 5); ▪, 2.1078 ± 0.4149 (n = 6). The horizontal barrepresents the period of NMDA perfusion.
Continuous perfusion with NMDA (300 μm) reduced Ach levels by 34%, an effect similar to that observed during transient perfusion of NMDA (p < 0.05, MANOVA) (Fig.2A). During continuous NMDA perfusion, Ach returned to basal values 75 min after the beginning of NMDA treatment, with NMDA still present in the perfusion fluid (Fig.2A). In contrast, continuous perfusion with NMDA produced a fourfold increase in dialysate Cho levels (Fig.2B). This increase was sustained for at least 2 hr of continuous NMDA treatment (p < 0.05, MANOVA). The effects of NMDA on Ach and Cho levels were completely prevented by the inclusion of AP-5 (1 mm) in the perfusion fluid (significant AP-5 × NMDA interaction; p < 0.01). AP-5 by itself induced an increase in Ach levels (p < 0.05, MANOVA), with no changes in Cho levels (Fig. 2A,B). All experiments were performed in the presence of 0.5 μm neostigmine in the perfusion fluid to achieve detectable dialysate Ach levels. Previous studies have shown that neostigmine, in the concentration range used in our experiments, does not significantly influence basal extracellular Cho levels (Marshall and Wurtman, 1993). Initial studies performed in our laboratory indicated that the presence or absence of 0.5 μm neostigmine did not have a significant influence on the Cho levels evoked by NMDA (data not shown).

Effects of continuous perfusion with 300 μm NMDA through the microdialysis probe on extracellular Ach (A) and Cho (B) levels in prefrontal cortex: prevention by AP-5. Symbols: ▪, NMDA; ▾, AP-5; ▴, AP-5 + NMDA. Basal levels (dialysate picomoles per microliter) and number of animals per group (mean ± SEM) were as follows: Ach, ▪, 0.0376 ± 0.0039 (n = 6); ▾, 0.0341 ± 0.0059 (n = 4); ▴, 0.0275 ± 0.0025 (n = 4). Cho, ▪, 1.8928 ± 0.2959 (n = 6); ▾, 0.7028 ± 0.1246 (n = 4); ▴, 1.4444 ± 0.4496 (n = 4). Horizontal bars represent the period of continuous perfusion with NMDA or AP-5.
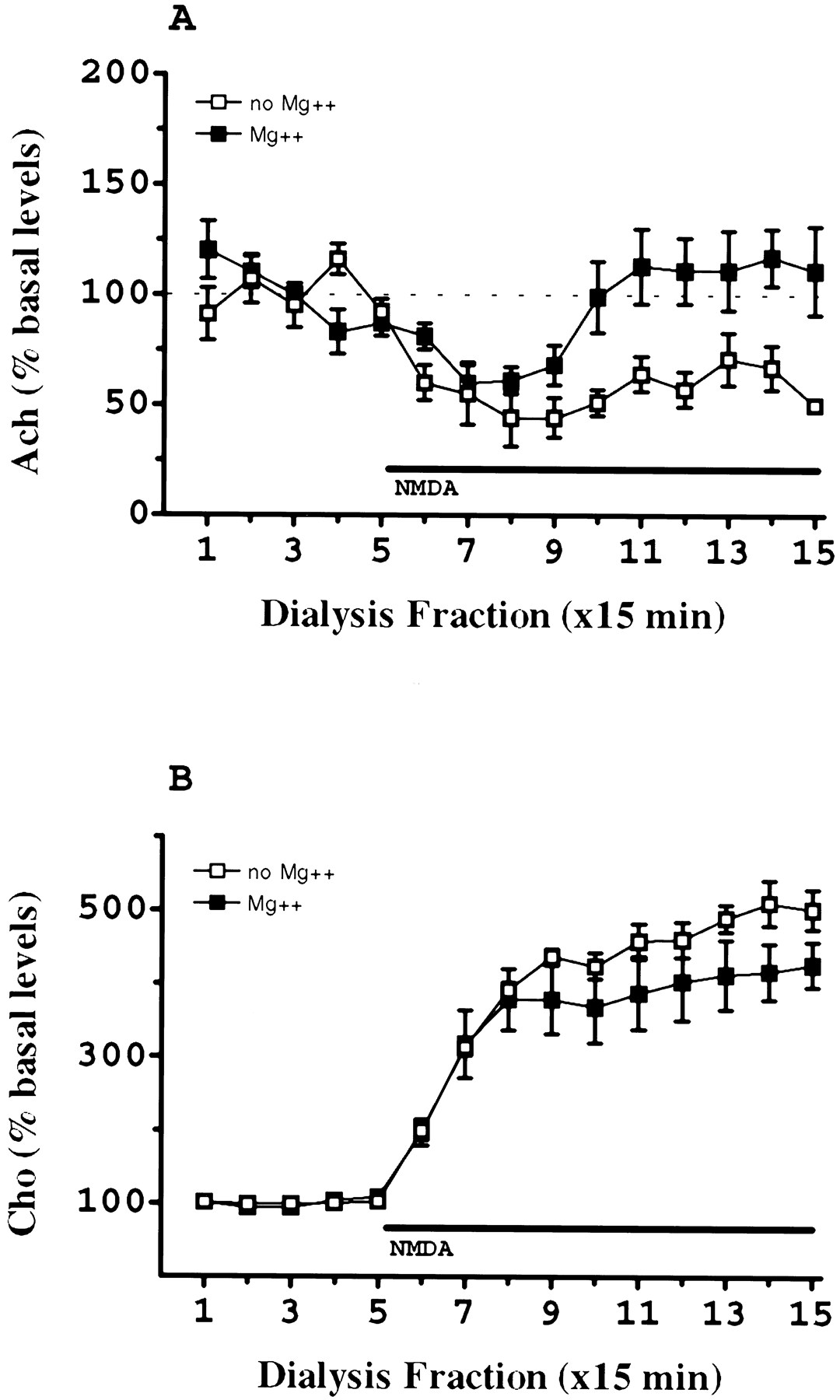
Removal of magnesium from the perfusion fluid did not significantly modify the effects of NMDA on dialysate Cho levels (p > 0.05, MANOVA) (Fig.3B). Similarly, the percentage reduction in Ach levels evoked by NMDA was not significantly altered by the removal of magnesium from the perfusion fluid. However, the absence of magnesium in the dialysate blocked the restoration of Ach levels to basal values observed during perfusion of NMDA in the presence of magnesium (Fig. 3A).

Influence of the omission of Mg2+ in the perfusion fluid on the effects of continuous perfusion with 300 μm NMDA on dialysate Ach (A) and Cho (B) levels in prefrontal cortex. Symbols: ▪, Mg2+; □, no Mg2+. Basal levels (dialysate picomoles per microliter) and number of animals per group (mean ± SEM) were as follows: Cho, ▪, 1.67 ± 0.12 (n = 5); □, 1.50 ± 0.10 (n = 5); ACh, ▪, 0.0389 ± 0.005 (n = 5); □, 0.0500 ± 0.008 (n = 5). The horizontal barrepresents the period of NMDA perfusion.
To determine whether the effects of NMDA were dependent on the extracellular concentration of Ca2+, NMDA (300 μm) was perfused in a Krebs’-Ringer’s solution buffer in the absence of Ca2+. The removal of Ca2+ from the perfusion fluid significantly inhibited the increase in dialysate Cho levels induced by NMDA (significant NMDA × calcium condition interaction;p < 0.01, MANOVA) (Fig.4), but did not alter basal Cho levels (1.57 ± 0.11 and 1.89 ± 0.35 pmol/μl in control and Ca2+-free group, respectively). Because removal of Ca2+ from the perfusion fluid may not be sufficient to produce a substantial decrease in extracellular Ca2+, a Ca2+ chelator was included in the dialysis buffer. Inclusion of 5 mm EGTA in the perfusion fluid completely inhibited the NMDA-induced increase in dialysate Cho. However, EGTA produced an effect by itself and significantly increased basal Cho levels (3.41 ± 0.21 vs 1.57 ± 0.11 in EGTA and control group; t = −8.54; p < 0.01 t test) (Fig. 4). Basal Ach levels decreased below detectable values in Ca2+-free and 5 mm EGTA groups, indicating that dialysate Ach levels reflect a Ca2+-dependent neurotransmitter release (data not shown).

Effects of continuous perfusion with 300 μm NMDA through the microdialysis probe on extracellular Cho levels in prefrontal cortex: calcium dependence. Symbols: ▪, NMDA; ▴, no Ca2+ + NMDA; ▾, no Ca2+ + EGTA 5 mm + NMDA. Basal levels (dialysate picomoles per microliter) and number of animals per group (mean ± SEM were as follows: Cho, ▪, 1.5505 ± 0.1038 (n = 6); ▴, 1.8098 ± 0.3171 (n = 4); ▾, 3.3017 ± 0.3750 (n = 4). The horizontal barrepresents the period of NMDA perfusion.
The effects of NMDA on extracellular Cho levels were concentration dependent (significant NMDA and dose group effects and NMDA × dose group interaction; p < 0.001, MANOVA) (Fig.5B). Concentration dependency studies for the effects of NMDA on modulation of Ach release were not performed. NMDA decreased extracellular Ach levels (significant NMDA effect; p < 0.001, MANOVA) (Fig. 5A), but the effect was already maximal at the lowest concentration of NMDA used in these experiments (no significant effect of dose group or NMDA × dose interaction; p > 0.05, MANOVA) (Fig.5A).

Effects of continuous perfusion with NMDA through the microdialysis probe on extracellular Ach (A) and Cho (B) levels in prefrontal cortex: dose–response. Symbols: ▪, control; ▵, NMDA 50 μm; ▿, NMDA 100 μm; ⋄, NMDA 300 μm; ○, NMDA 600 μm. Basal levels (dialysate picomoles per microliter) and number of animals per group (mean ± SEM) were as follows: Ach, ▪, 0.0354 ± 0.0046 (n = 5); ▵, 0.0416 ± 0.0186 (n = 3); ▿, 0.0410 ± 0.0054 (n = 5); ⋄, 0.0509 ± 0.0078 (n = 5); ○, 0.0420 ± 0.0052 (n = 3); Cho, ▪, 1.6503 ± 0.2497 (n = 5); ▵, 2.0743 ± 0.8061 (n = 3); ▿, 1.9570 ± 0.4533 (n = 5); ⋄, 1.4892 ± 0.1027 (n = 5); ○, 0.8294 ± 0.1501 (n = 3). The horizontal barrepresents the period of NMDA perfusion.
The effects of continuous NMDA (300 μm) perfusion on extracellular Cho levels were remarkably different depending on the brain region investigated (Fig. 6). The areas studied were prefrontal cortex, neostriatum, ventral hippocampus, and cerebellum. Significant NMDA and region effects as well as NMDA × region interaction (all p < 0.001, MANOVA) were observed, showing pronounced regional differences in NMDA-evoked Cho release (Fig. 6B). One-way ANOVA between groups showed that 1 hr after beginning NMDA treatment (sample 9), the Cho increase evoked by NMDA was significantly different in all brain areas (Student–Newman–Keuls, p < 0.05). Two hours after the beginning of NMDA perfusion (sample 13), no significant differences were observed in dialysate Cho levels between prefrontal cortex and ventral hippocampus. However, the effects of NMDA on Cho release in these two brain regions were significantly higher than those observed in neostriatum and cerebellum (Fig. 6B). No significant differences were observed between these last two regions in NMDA-evoked dialysate Cho levels. The maximum increase in Cho levels evoked by NMDA for each one of the regions investigated was as follows (mean percentage basal levels ± SEM): prefrontal cortex 512 ± 29, ventral hippocampus 481 ± 40, neostriatum 221 ± 27, cerebellum 136 ± 13.

Effects of continuous perfusion with 300 μm NMDA through the microdialysis probe on extracellular Ach (A) and Cho (B) levels in different brain areas. Symbols: ▪, prefrontal cortex; ▵, neostriatum; ▿, ventral hippocampus; ⋄, cerebellum. Basal levels (dialysate picomoles per microliter) and number of animals per group (mean ± SEM) were as follows: Ach, ▪, 0.0509 ± 0.0078 (n = 5); ▵, 0.0766 ± 0.0196 (n = 4); ▿, 0.0257 ± 0.0030 (n = 4); ⋄, below detectable range; Cho, ▪, 1.4892 ± 0.1027 (n = 5); ⋄, 1.0367 ± 0.1301 (n = 4); ▿, 0.7708 ± 0.0675 (n = 4); ⋄, 3.2175 ± 0.6227 (n = 5). The horizontal barrepresents the period of NMDA perfusion.
Consistent with the absence of cholinergic terminals in cerebellum, basal Ach levels were below the detectable range in this brain area. NMDA reduced Ach levels in the other three brain regions studied (Fig.6A) (significant NMDA effect, p < 0.01, and significant effects of area, p < 0.03; no NMDA × area interaction; MANOVA). When independent analyses were performed for each sample, the effect of NMDA on Ach levels was found to be more pronounced in prefrontal cortex (samples 9, 10, and 11) (Fig. 6A).
To determine whether dialysate choline levels could be modulated by voltage-dependent Ca2+ entry induced by nonspecific depolarization, the effects of continuous perfusion with 100 mm KCl on dialysate Cho were investigated in prefrontal CTX (Fig. 7). This treatment significantly increased Ach levels (maximum increase 598 ± 55; percentage basal levels ± SEM; p < 0.01, MANOVA), showing a depolarization-dependent neurotransmitter release. However, despite this remarkable increase in Ach, Cho levels were not affected by this treatment (p > 0.05, not significant; MANOVA).

Effects of continuous perfusion with 100 mm KCl through the microdialysis probe on extracellular Ach and Cho levels in prefrontal cortex. Symbols: ▪, Ach; ▴, Cho. Basal levels (dialysate picomoles per microliter) and number of animals per group (mean ± SEM) were as follows: Ach, 0.0479 ± 0.0054 (n = 6); Cho, 2.5981 ± 0.2860 (n = 6). The horizontal barrepresents the period of KCl perfusion.
To determine whether the effect of NMDA on Cho release was the result of acute necrosis induced by cell swelling, NMDA was perfused in a hypertonic Krebs’-Ringer’s solution buffer containing 150 mm sucrose. Under this condition, the effect of NMDA on Cho release was not affected, indicating that the increase in dialysate Cho was not dependent on cell swelling (results not shown). Alternatively, NMDA-evoked Cho release could be simultaneous with or precede neuronal cell death. To investigate this possibility, we measured ChAT activity as an index of neuronal cell death in the area surrounding the dialysis probe. Continuous perfusion of 300 μm NMDA for 2.5 hr in prefrontal cortex produced a delayed but not acute cholinergic neurotoxicity. As shown in Figure8, treatment with NMDA did not significantly modify ChAt activity in micropunch samples of the tissue surrounding the dialysis probe, obtained immediately after the dialysis experiment, compared with control KRB-perfused animals. However, when micropunch tissue samples were obtained 1 week after the dialysis experiment, ChAT activity was significantly reduced compared with control, KRB-perfused animals (p < 0.05, ANOVA).

Effects of continuous perfusion with 300 μm NMDA for 2.5 hr on ChAT activity measured in a micropunch tissue sample taken around the microdialysis probe location in the prefrontal cortex. Tissue was dissected immediately after dialysis (0 d) or 7 d after. Data were expressed as percentage activity relative to the contralateral prefrontal cortex for each animal. No significant differences were found in both control (KRB-perfused) groups, and they were pooled for analysis. Number of animals: KRB (8), NMDA 0 d (4), NMDA 7 d (4). *, significantly different from the other two groups;p < 0.05, Student–Newman–Keuls.
To investigate the source of Cho released by NMDA, we measured the Cho content in the lipid fraction from tissue samples surrounding the dialysis probe. The Cho released by NMDA during a 2 hr perfusion protocol represents a small fraction of the membrane Cho reservoir. To be able to detect significant changes in membrane PtdCho levels, the concentration of NMDA and the duration of perfusion were increased. Prolonged perfusion with NMDA (600 μm) for 12 hr induced an increase in dialysate Cho levels (450%) that was sustained and remained significantly elevated for the whole period of perfusion (Fig.9). This treatment produced a significant reduction (34%) (p < 0.05, two-tailedt test) of the Cho content in the lipid fraction of the tissue surrounding the membrane probe compared with the equivalent area of the contralateral side (Fig. 9). In control animals, perfusion with Krebs’-Ringer’s solution buffer did not significantly modify dialysate Cho levels, and no significant reduction in the content of Cho in the lipid fraction was observed (Fig. 9).

Effects of a prolonged perfusion with 600 μm NMDA for 12 hr on dialysate Cho levels. Symbols: □, control perfusion with KRB (n = 6); ▪, NMDA (600 μm) perfusion (n = 10).Inset: Content of Cho in the lipid fraction of a micropunch tissue sample taken around the microdialysis probe location in the prefrontal cortex after perfusion with NMDA (600 μm) for 12 hr. Tissue was dissected immediately after dialysis. Data were expressed as percentage relative to the contralateral prefrontal cortex for each animal. Cho content in the lipid fraction expressed in nanomoles per milligram of wet weight was as follows: controls (n = 6), contralateral side = 14 ± 1, probe side 13 ± 1; NMDA treatment (n = 10), contralateral side = 16 ± 1, probe side 10 ± 0.5. *, significantly different from control group; p < 0.05, Student–Newman–Keuls.
To investigate the mechanism involved in the NMDA-evoked increase in dialysate Cho, mepacrine (300 μm and 10 mm), a nonspecific inhibitor of phospholipase A2 and C at low and high concentrations, respectively (Lapetina et al., 1981), was perfused 30 min before and during perfusion with NMDA. Mepacrine (300 μm) did not inhibit the increase in dialysate Cho evoked by NMDA. Instead, mepacrine produced a slight nonsignificant potentiation of the effects of NMDA on dialysate Cho (Fig.10). This potentiation became significant when a higher (10 mm) concentration of mepacrine was used in the dialysate. NMDA (300 μm) increased dialysate Cho to 281 ± 18% of basal values. In comparison, in the presence of 10 mm mepacrine, NMDA (300 μm) increased dialysate Cho to 608 ± 103% of basal levels.

Effect of mepacrine on NMDA-induced dialysate Ach (A) and Cho (B). Mepacrine was perfused for 30 min before NMDA. Pilot experiments showed that mepacrine passed through the dialysis membrane. NMDA (300 μm) and mepacrine (300 μm) were co-perfused in prefrontal cortex for 1.25 hr. Symbols: ▪, perfusion with NMDA (300 μm); ▵, perfusion with mepacrine (300 μm); ○, perfusion with NMDA (300 μm) + mepacrine (300 μm). Basal levels of Ach (dialysate picomoles per microliter) and number of animals per group (mean ± SEM) were as follows: ▪, 0.0558 ± 0.0069 (n= 4); ▵, 0.0455 ± 0.0083 (n = 3); ○, 0.0578 ± 0.0087 (n = 4). Basal levels of Cho (dialysate picomoles per microliter) and number of animals per group (mean ± SEM) were as follows: ▪, 0.9593 ± 0.1478 (n = 4); ▵, 1.3475 ± 0.0904 (n = 3); ○, 1.3200 ± 0.2788 (n = 4). Horizontal bars represent period of drug perfusion.
DISCUSSION
These results show that local perfusion with NMDA by retrodialysis produces a sustained increase in dialysate Cho. This effect of NMDA exhibits marked brain regional differences (Fig. 6B). The NMDA-evoked increase in dialysate Cho precedes delayed excitotoxic cholinergic cell death and is blocked with AP-5, a competitive NMDA receptor antagonist. Interestingly, when NMDA was perfused for a short period of time (30 min), Cho levels remained significantly increased for at least 2 hr after discontinuation of NMDA perfusion (Fig.1B).
Perfusion with a Ca2+-free medium or with a Ca2+-free medium in the presence of 5 mmEGTA completely blocked the NMDA-evoked increase in dialysate Cho, indicating that this effect is dependent on the extracellular concentration of calcium (Fig. 4). The fact that a strong depolarizing stimulus (i.e., 100 mm KCl), which is able to induce a sixfold increase in extracellular Ach (reflecting activation of voltage-dependent calcium inflow) and does not modify extracellular Cho levels (Fig. 7), provides strong evidence that the NMDA-evoked Cho release is specifically dependent on extracellular calcium influx through NMDA receptors. Moreover, perfusion with 100 mmKCl, despite inducing a pronounced Ach release, did not significantly modify basal Cho levels (Fig. 7), showing that the amount of Cho originated from the hydrolysis of Ach does not contribute significantly to dialysate Cho levels in the presence of 0.5 μmneostigmine.
The perfusion fluid used in our experiments is a Krebs’-Ringer’s solution containing Mg2+. The omission of Mg2+ from this solution did not modify either basal or NMDA-evoked Cho release (Fig. 3B). This result was initially unexpected because it is well established that NMDA receptors are subject to a voltage-dependent channel blockade by Mg2+ (Mcbain and Mayer, 1994). However, omission of Mg2+ from the perfusion fluid may not be enough to reduce the extracellular Mg2+ to a level that is sufficiently low to diminish Mg2+ blockade at NMDA receptors (Bogdanov and Wurtman, 1997). Nevertheless, removal of Mg2+ from the perfusion fluid blocked the restoration of Ach to basal values observed during perfusion with NMDA in the presence of Mg2+ (Fig. 3A).
The NMDA-evoked increase in dialysate Cho could be interpreted as a result of acute osmotic lysis produced by neuronal cell swelling secondary to a depolarization-mediated influx of Na+, Cl−, and water (Choi, 1994). However, results from several experiments argue against this interpretation. Continuous perfusion of NMDA in a hypertonic dialysis fluid containing 150 mm sucrose, which inhibits acute osmotic lysis, did not modify the effects of NMDA on dialysate Ach and Cho (results not shown). Moreover, continuous perfusion with 100 mm KCl, which evokes a strong depolarization and a pronounced release of Ach, did not significantly alter Cho dialysate levels. Furthermore, there was no significant cholinergic cell death, measured by ChAT activity, in the area surrounding the dialysis probe, immediately after perfusion with NMDA compared with an equivalent area of the contralateral side or compared with an area surrounding probes perfused without NMDA (Fig. 8), suggesting that the Cho released by NMDA could not be attributed to rapidly triggered excitotoxicity or necrosis. In contrast, local perfusion of NMDA in the prefrontal cortex at the concentration used in the present experiments produced delayed cholinergic cell death, shown by a significant decrease in ChAT activity in micropunch samples dissected 7 d after NMDA perfusion, when compared with control, KRB-perfused animals. These results strongly indicate that Cho release evoked by NMDA precedes and is not a consequence of NMDA-evoked neurotoxicity.
Continuous perfusion of NMDA produced a transient decrease in Ach that is in marked contrast with the sustained increase in dialysate Cho (Fig. 2A). This finding indicates that these two effects of NMDA are mediated by independent mechanisms and strongly suggests that the sustained increase in Cho levels is not caused by inhibition of high-affinity Cho uptake. Ach release is highly dependent on Cho uptake. Inhibition of Cho uptake would produce a more pronounced and permanent reduction of Ach release because of a sustained precursor deficit (Vickroy and Malphurs, 1994). Accordingly, recent experiments have shown that perfusion with hemicholinium-3, a Cho uptake inhibitor, increases dialysate Cho levels but completely inhibits basal Ach release (Ikarashi et al., 1997). Moreover, the finding that the NMDA-evoked changes in Ach levels (which were qualitative and quantitatively similar in cortex, striatum, and hippocampus) (Fig.6A) did not parallel changes in dialysate Cho (significantly lower in the striatum) (Fig. 6B) provides additional evidence against an involvement of the Cho uptake system in these effects of NMDA.
The NMDA-evoked reduction in Ach release observed in our experiments is consistent with recent results showing that local perfusion with both NMDA and AMPA in the hippocampus causes a significant decrease in the output of Ach (Moor et al., 1996). In addition, our results show that the reduction of Ach output by local administration of NMDA is not restricted to the hippocampus and occurs also in cortex and striatum (Fig. 6A). The possibility that this effect is produced at NMDA receptors located in cholinergic nerve terminals has been considered unlikely (Moor et al., 1996). The decrease in Ach output caused by local administration of NMDA has been proposed to be mediated by local GABAergic interneurons (Moor et al., 1996). The mechanism of this effect of NMDA is currently unknown, but recent studies have shown that glutamatergic inputs to cholinergic neurons are modulated by GABAergic interneurons (Giovannini et al., 1997).
Significant differences were observed in basal dialysate concentrations of Cho among the brain areas investigated (see legend to Fig. 6). These differences are directly related to the different capacities of Cho uptake systems among the brain areas investigated (Manaker et al., 1986). Thus, the cerebellum, which exhibits the highest concentration of basal dialysate Cho, contains little high-affinity uptake capacity compared with the other brain regions (Swann et al., 1986). However, the differences in basal dialysate Cho concentrations were not related to differences in NMDA-evoked Cho release.
The effect of NMDA on dialysate Cho exhibited marked differences depending on the brain region investigated. Thus, NMDA evoked a pronounced increase in extracellular Cho in prefrontal cortex and hippocampus, but the effect was much less pronounced in neostriatum (Fig. 6). This region specificity cannot be explained simply by the distribution of NMDA receptors, because NMDA did not alter dialysate Cho in the cerebellum, a brain area with a high density of NMDA receptors (Monaghan et al., 1989). In addition, this regional heterogeneity argues against a peripheral origin of the Cho released in response to perfusion with NMDA.
The mechanism responsible for the NMDA-evoked increase in interstitial Cho is likely to involve calcium-dependent activation of phospholipases that cleave Cho-containing phospholipids (Farooqui and Horrocks, 1994b,1995; Bazan et al., 1995). In support of this interpretation, prolonged perfusion with NMDA reduced the Cho content in the lipid fraction from tissue samples surrounding the dialysis probe (Fig. 9), showing that NMDA reduces membrane choline containing phospholipids. However, perfusion of NMDA in the presence of a concentration of mepacrine (300 μm), reported to inhibit phospholipase A2(Lazarewicz et al., 1990, 1992), did not modify the increase in Cho evoked by NMDA (Fig. 10). Instead, mepacrine produced a significant increase in Ach release and a slight nonsignificant potentiation of the effects of NMDA on dialysate Cho (Fig. 10). This potentiation was subsequently confirmed using a high (10 mm) concentration of mepacrine, previously reported to inhibit both phospholipase A2 and C (Lapetina et al., 1981). These results indicate that the increase in dialysate Cho evoked by NMDA is not mediated by phospholipases A2 or C. Whether the effect of NMDA on dialysate Cho is mediated by phospholipase D is currently under investigation.
Although it is well established that NMDA receptor overactivation leads to neurodegeneration by an increase in intracellular calcium, the sequence of events triggered by the calcium-dependent mechanisms involved in this process have not yet been established. The present results indicate that NMDA receptor activation initiates a calcium-dependent and region-specific release of Cho from membrane phospholipids that, if sustained, may lead to alterations in membrane permeability and subsequent cell death. Accordingly, the brain areas in which NMDA receptor activation induces little or no release of Cho, such as the cerebellum, contain cell populations that are more resistant to NMDA-evoked excitotoxic cell death (Meldrum and Garthwaite, 1990). Moreover, this relationship between NMDA-evoked Cho release and delayed excitotoxic cell death might explain the special vulnerability of cholinergic neurons that die in certain neurodegenerative disorders (Wurtman, 1992; Bierer et al., 1995). Cholinergic neurons obtain Cho from PtdCho to sustain Ach synthesis and release (Lee et al., 1993). Because cholinergic neurons are unique in using Cho from membrane phospholipids for Ach synthesis (Wurtman, 1992;Lee et al., 1993), an additional reduction of Cho produced by NMDA receptor activation may render them more vulnerable to cell death compared with noncholinergic neurons.
In summary, the results reported here indicate that the NMDA-evoked increase in dialysate Cho is not mediated by hydrolysis of the Ach pool or by changes in high affinity Cho uptake. The fact that prolonged perfusion with NMDA produces a significant reduction of the Cho content in the lipid fraction of the tissue area surrounding the dialysis probe (Fig. 9) indicates that NMDA evokes a direct release of Cho from the phospholipid pool that is followed by cell death. Consistent with these results is the finding that extracellular Cho levels are increased by treatments that produce glutamate release and subsequent excitotoxic cell death, such as seizures induced by different convulsant treatments (Jope and Gu, 1991), energy deprivation (Djuricic et al., 1991), or hypoxia (Klein et al., 1993).
Footnotes
This work was supported by Grants DGICYT, PB94–0017, and CICYT, SAF98–0063, from Ministerio de Educación y Cultura of Spain to R.T. A.Z. is a Severo Ochoa Fellow from the Ferrer Internacional Foundation.
Correspondence should be addressed to Dr. Ramon Trullas, Neurobiology Unit, Institut d’Investigacions Biomèdiques de Barcelona, Consejo Superior de Investigaciones Cientı́ficas, Jordi Girona 18, 08034 Barcelona, Spain.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}