

Abstract
Agrin plays a key role in directing the differentiation of the vertebrate neuromuscular junction. Understanding agrin function at the neuromuscular junction has come via molecular genetic analyses of agrin as well as identification of its receptor and associated signal transduction pathways. Agrin is also expressed by many populations of neurons in brain, but its role remains unknown. Here we show, in cultured cortical neurons, that agrin induces expression of the immediate early gene c-fos in a concentration-dependent and saturable manner, as expected for a signal transduction pathway activated by a cell surface receptor. Agrin is active in cortical neurons at picomolar concentrations, is Ca2+ dependent, and is inhibited by heparin and staurosporine. Despite marked differences in acetylcholine receptor (AChR)-clustering activity, all alternatively spliced forms of agrin are equally potent inducers of c-fos in cortical neurons. A similar, isoform-independent response to agrin was also observed in cultures prepared from the hippocampus and cerebellum. Only agrin with high AChR-clustering activity was effective in cultured muscle, whereas non-neuronal cells were agrin insensitive. Although consistent with a receptor tyrosine kinase model similar to the muscle-specific kinase–myotube-associated specificity component complex in muscle, our data suggest that CNS neurons express a unique agrin receptor. Evidence that neuronal signal transduction is mediated via an increase in intracellular Ca2+ means that agrin is well situated to influence important Ca2+-dependent functions in brain, including neuronal growth, differentiation, and adaptive changes in gene expression associated with synaptic remodeling.
The postsynaptic apparatus of the vertebrate neuromuscular junction is characterized by high concentrations of acetylcholine receptor (AChR) and other molecules that support its function in synaptic transmission. During development, differentiation of the postsynaptic apparatus is triggered by signals supplied by motor nerves. Convincing evidence now exists that one such signal is the extracellular matrix protein agrin (for review, see Hall and Sanes, 1993; Sanes, 1997). Neurotransmitter receptors are also concentrated in the postsynaptic membranes of neuron–neuron synapses (Craig et al., 1994). This and other similarities with the neuromuscular junction imply that differentiation of interneuronal synapses might be controlled by similar mechanisms.
A possible role for agrin in neuron–neuron synapse formation was first suggested by the observation that brain extracts contain agrin (Godfrey et al., 1988; Magill-Solc and McMahan, 1988; Godfrey, 1991). Further support for this hypothesis came with the demonstration that agrin is expressed by virtually all populations of neurons in the CNS (O’Connor et al., 1994; Stone and Nikolics, 1995) with the highest levels in developing brain coinciding with periods of synapse formation (Hoch et al., 1993; Li et al., 1997). Moreover, like the neuromuscular junction, agrin is present at neuron–neuron synaptic contacts (Kröger et al., 1996; Mann and Kröger, 1996). However, recent studies, showing that synapses form between neurons cultured from mice carrying a mutation in the agrin gene that disrupts neuromuscular junction formation (Li et al., 1999; Serpinskaya et al., 1999), have raised questions regarding agrin’s role in neuron–neuron synaptogenesis and the function it might serve in the developing brain.
To gain insight into agrin’s function in the CNS, we sought to identify and characterize a signal transduction pathway that could mediate its effects there. Without a specific function for agrin in brain, we searched for an alternate cellular response that might be harnessed as a reporter of agrin-dependent signaling. Immediate early genes (IEGs) are characterized by their rapid responses to a wide range of cellular stimuli and have been used to monitor neural activation in a variety of experimental paradigms including behavioral testing, during seizures, and after injury or treatment with growth factors (Herrera and Robertson, 1996). Many IEGs are themselves transcription factors, and recent studies showing that both the ε-subunit of the AChR and the utrophin gene (Jones et al., 1996; Gramolini et al., 1998) are under agrin-dependent transcriptional control are consistent with the possibility that changes in IEG expression are part of the normal cellular response to agrin. Accordingly, using the prototypical IEGc-fos as a reporter, we have examined the response of cells cultured from mouse cortex to treatment with agrin. The results of these studies demonstrate the existence of an agrin-dependent signaling pathway in cortical neurons whose biochemical properties are similar to, but distinct from, that mediating agrin-induced AChR clustering in skeletal muscle. These data provide evidence of a neuronal receptor for agrin whose characterization represents an important first step in a systematic approach to establishing the function of agrin in the mammalian CNS.
MATERIALS AND METHODS
Tissue culture. Mouse cortical cultures were prepared from somatosensory cortices of newborn or 1-d-old pups by a minor modification of the procedure described by Li et al. (1997). Neurons were plated into 24-well plastic tissue culture plates (Corning, Corning, NY) or glass coverslips coated with poly-d-lysine (Collaborative Biomedical Products) and maintained in Neurobasal Medium (NBM) + B27 supplements (Life Technologies, Gaithersburg, MD) at 37°C in a humidified 5% CO2 atmosphere for 1 d. On the following day, the culture medium was replaced with NBM + B27 supplements that had been conditioned for 24 hr (cNBM) by 1- to 2-week-old non-neuronal feeder cell cultures prepared from cortices of postnatal day 1 (P1)–P5 mice (Li et al., 1997) and subsequently fed cNBM every 3–4 d thereafter. Hippocampal and cerebellar cultures were prepared by the same procedure. All experiments were performed on 10- to 15-d-old cultures.
Chick myotube cultures were prepared from pectoral muscles of 10–11 d White Leghorn chick embryos by the method of Fischbach (1972). Myoblasts were plated onto laminin-coated (Collaborative Biomedical Products) 35 mm plastic culture dishes or 24-well tissue culture plates in MEM (Life Technologies) supplemented with 10% horse serum, 5% chick embryo extract, 50 U/ml penicillin (Life Technologies), and 50 μg/ml streptomycin (Life Technologies) and maintained at 37°C in a humidified 5% CO2 atmosphere. Two days after plating, cultures were treated with 0.1% cytosine arabinoside (Sigma, St. Louis, MO) for 2 d before returning to normal growth medium. Chick skin fibroblasts were prepared and maintained in a similar manner but were not treated with cytosine arabinoside.
Expression of recombinant agrin. Agrin was produced by transient transfection of COS-7 cells with p-cytomegalovirus expression vector constructs encoding the soluble C-terminal half of rat agrin [C-Ag12,4,8, C-Ag12,4,0, and C-Ag12,0,0; referred to hereafter as C-Ag4,8, C-Ag4,0 and C-Ag0,0, respectively (Ferns et al., 1993)]. COS-7 cells were grown to 50–80% confluence in 60 mm tissue culture dishes in DMEM (Life Technologies) supplemented with 10% calf serum, 25 U/ml penicillin, 25 μg/ml streptomycin, and 2 mml-glutamine and transfected with 1 μg of plasmid DNA using the Effectene Transfection Reagent (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Control, sham-transfected cells were treated in an identical manner in the absence of DNA. Cultures were incubated overnight, then washed with fresh medium and incubated for an additional 6–8 hr before being passaged into 100 mm tissue culture dishes containing 10 ml of growth medium. Agrin-containing and control conditioned media were harvested 2–4 d later.
The AChR-clustering activity of COS-7 cell–conditioned media containing C-Ag4,8 was assayed on 5- to 7-d-old cultured chick myotubes. Cultures were treated with C-Ag4,8 overnight followed by incubation with 2 × 10−8 M rhodamine-conjugated α-bungarotoxin (Molecular Probes, Eugene, OR) for 1 hr at 37°C in culture medium. Cultures were subsequently rinsed in PBS, fixed for 20 min at room temperature in 4% paraformaldehyde in PBS, washed three times for 10 min each in PBS, and then mounted and viewed at 250× magnification under epifluorescent illumination. AChR clusters were counted blind with respect to treatment in five random fields in each culture dish. AChR-clustering activity was expressed as the mean number of AChR clusters per field ± SEM, determined from triplicate cultures for each treatment.
Because C-Ag4,0 and C-Ag0,0lack AChR-clustering activity, their concentration in COS-7 cell–conditioned media was determined using a two-site sandwich ELISA (Gesemann et al., 1995; O’Toole et al., 1996) using an anti-agrin monoclonal antibody Agr 530 [StressGen (Ferns et al., 1993; Hoch et al., 1994)] and a rabbit antiserum raised against a synthetic peptide corresponding to amino acids 1862–1893 of the mouse agrin sequence (Rupp et al., 1992). These antibodies recognize distinct epitopes common to all agrin isoforms. The specificity of the rabbit anti-agrin serum was confirmed by Western blot analysis (data not shown). Ninety-six–well ELISA plates (Nunc, Naperville, IL) were coated overnight at 4°C with 100 μl/well of 2 mg/ml Agr 530 in PBS followed by washing in PBS and blocking with 4% bovine serum albumin (BSA) in PBS for 1 hr at room temperature. Control wells were treated similarly but without Agr 530. Triplicate wells were incubated in serial dilutions of C-Ag–containing conditioned medium for 2 hr at room temperature with continuous agitation. Wells were rinsed followed by incubation in rabbit anti-agrin serum (diluted 1:200 in PBS) for 1.5 hr and then washed and incubated in alkaline phosphatase–labeled goat anti-rabbit antibody (diluted 1:500 in PBS) for a further 1.5 hr at room temperature. Wells were washed in PBS, and bound alkaline phosphatase goat anti-rabbit antibody was detected by incubation with 100 μl of 1 mg/ml p-nitrophenyl phosphate (pNPP) in 0.8 mdiethanolamine buffer, pH 9.8, containing 0.24 mmMgCl2 according to the manufacturer’s protocol (Southern Biotechnology, Birmingham, AL). Substrate conversion was stopped by the addition of 25 μl of 0.5 m NaOH, and the concentration of the soluble yellow reaction product was determined by measuring the absorbance at 405 nm. Concentrations of C-Ag4,0 and C-Ag0,0 were determined on the basis of a C-Ag4,8 standard of known AChR-clustering activity run in parallel. Consistent with previous reports (Ferns et al., 1993), expression levels of all three constructs were similar and differed by <25%. To facilitate comparison between the different isoforms, concentration dependence curves were plotted in agrin units (AU) where 1 AU is the amount of C-Ag4,8 (or molar equivalent of a C-Agz0 isoform) required to produce a half-maximal increase in AChR clusters on cultured chick muscle.
Immunodepletion of agrin from COS-7 cell–conditioned media.Agrin-containing COS-7 cell–conditioned media were incubated with anti-agrin monoclonal antibody Agr 530 at a concentration of 50 μg/ml for 2 hr at room temperature. Rabbit anti-mouse IgG (Vector Laboratories, Burlingame, CA) was added to a final concentration of 75 μg/ml, and the mixture was incubated for 2 hr followed by incubation with 40 μl of a 1:1 suspension of formalin-treatedStaphylococcus aureus cells (Sigma) in PBS for an additional 2 hr at room temperature. Immune complexes were subsequently separated by centrifugation, and the cleared supernatant was used for AChR-clustering and Fos induction assays as described. Control precipitations were treated in an identical manner but in the absence of Agr 530.
Immunohistochemistry. Cultures were prepared for immunohistochemistry by briefly rinsing with PBS at room temperature followed by fixation in 4% paraformaldehyde in PBS on ice for 1 hr. Cells were subsequently washed three times in PBS and then permeabilized and blocked in PBS containing 0.1% Triton X-100 and 4% BSA. In experiments in which the DAB reaction was used to visualize Fos, cultures were incubated overnight at 4°C in the anti-Fos rabbit antiserum Ab-2 [Oncogene Research Products; diluted 1:1500 in PBS containing 2% BSA, 0.1% Triton X-100, and 0.02% NaN3 (PBS-BTA)] and then washed three times in PBS followed by incubation in biotinylated anti-rabbit IgG (Vector Laboratories; diluted 1:200 in PBS-BTA) for 2 hr at room temperature. Cultures were washed three times in PBS before incubation for 30 min in the ABC reagent for DAB with nickel intensification (Vector Laboratories) according to the manufacturers instructions, mounted, and examined using transmitted illumination on a Nikon Optiphot-2 microscope.
To identify cells expressing Fos, double-immunofluorescence staining was performed using Ab-2 (diluted 1:400 in PBS-BTA) in combination with monoclonal antibody SMI-52 (Sternberger Monoclonals; diluted 1:400 in PBS-BTA) to detect the neuron-specific marker MAP-2 or with monoclonal antibody G-A-5 (Sigma; diluted 1:200 in PBS-BTA) for the glial cell marker GFAP. Cultures were labeled with the primary antibodies overnight at 4°C and then washed in PBS before being incubated in FITC-labeled goat anti-rabbit and Texas Red–labeled horse anti-mouse IgG (Vector Laboratories; diluted 1:200 in PBS-BTA) for 2 hr at room temperature. After washing in PBS, cultures were mounted in Fluoromount (Southern Biotechnology) and examined using epifluorescent illumination on a Nikon Optiphot-2 microscope.
Quantitative analysis of c-fos expression.Cortical cultures were treated at room temperature with recombinant agrin, control medium from sham-transfected COS-7 cells, or other agents, dissolved in PBS or NBM, and then briefly rinsed in NBM before being returned to their original medium for 2 hr at 37°C. Cultures were rinsed with PBS, fixed, permeabilized, and blocked as described for immunohistochemistry, followed by overnight incubation in Ab-2 (diluted 1:1000 in PBS-BTA) at 4°C. Cultures were washed three times for 10 min each in PBS, incubated with alkaline phosphatase–conjugated goat anti-rabbit antibody (Southern Biotechnology; diluted 1:1000 in PBS-BTA) for 2 hr at room temperature, and then washed three times for 10 min each in PBS as described above. After the last PBS wash, cultures were incubated in 200 μl of pNPP in 0.8 mdiethanolamine buffer, pH 9.8, containing 0.24 mmMgCl2. The reaction was stopped by addition of 50 μl of 0.5 m NaOH, and the optical density of the soluble yellow reaction product was measured at 405 nm. Preincubation of Ab-2 with a 10-fold molar excess of peptide antigen blocked >95% of Ab-2 binding measured by conversion of pNPP substrate. Specific Fos immunoreactivity was defined as total OD405 minus nonspecific OD405 observed in the presence of an equivalent concentration of control medium and was expressed in arbitrary OD405 units or normalized to the maximal level of Fos expression within an experiment to facilitate comparison between agrin isoforms.
RESULTS
“Active” agrin induces c-fos expression in cultured cortical neurons
The observation that functional synapses form between cultured cortical neurons isolated from agrin-deficient mice (Li et al., 1999) led us to ask which, if any, cells in brain might be responsive to agrin. Accordingly, we used induction of the IEG c-fos to report activation of an agrin-dependent signal transduction pathway in cultured cells isolated from postnatal mouse cortex. Initial studies used a soluble form of recombinant agrin, C-Ag4,8, that has high AChR-aggregating activity in cultured muscle cells and corresponds structurally to the C-terminal half of a naturally occurring agrin isoform in brain (Ferns et al., 1993).
Staining with the anti-Fos antibody Ab-2 revealed a marked increase in the level of Fos expression in cultures treated with C-Ag4,8 compared with that in those treated with control medium from sham-transfected COS-7 cells (Fig.1). The morphology of the Fos-positive cells suggested that neurons but not non-neuronal cells in the cultures were responsive to agrin, an impression that was confirmed by experiments in which cultures were double labeled for Fos together with the neuron-specific marker MAP-2 or the glial cell marker GFAP (Fig. 1). Neuronal nuclei in agrin-treated cultures were intensely labeled for Fos, whereas staining of non-neuronal cell nuclei was indistinguishable from the basal level of expression seen in neurons and non-neuronal cells treated with control medium. The lack of Fos expression in non-neuronal cells is not caused by the absence of a functional pathway for c-fos induction because other treatments, such as exposure to 20 μm forskolin for 4 hr or 60 mm K+for 10 min, increased Fos levels in non-neuronal cells (data not shown).

Neuron-specific induction of c-fosexpression by C-Ag4,8. a, Right, Cortical cultures were incubated for 10 min with C-Ag4,8 at a concentration determined previously to provide maximal induction of AChR clusters on cultured chick myotubes.Left, Control cultures were treated with an equivalent concentration of medium from sham-transfected COS-7 cells. Treated cultures were returned to cNBM, incubated for 2 hr at 37°C, and then fixed and labeled using the anti-Fos antibody Ab-2. Whereas basal levels of c-fos expression in control cultures are low, staining for Fos is markedly increased after treatment with C-Ag4,8. The majority of cells are darkly stained with prominent round nuclei and neuronal morphology (arrows). Cells that appear unaffected by C-Ag4,8 treatment have more irregularly shaped nuclei and non-neuronal morphology (arrowheads). b, Cultures were treated with either control (left) or C-Ag4,8–containing (middle,right) medium followed by double labeling for either Fos (fluorescein channel) and MAP-2 (Texas Red channel) or Fos and GFAP (Texas Red channel). In C-Ag4,8–treated cultures, levels of Fos expression evident in MAP-2–labeled neurons (arrows) are high, whereas Fos levels of MAP-2–negative non-neuronal cells (arrowheads) are comparable with basal levels seen in control cultures. Conversely, after treatment with C-Ag4,8, levels of c-fos expression are low in GFAP-labeled non-neuronal cells but high in the GFAP-negative neurons. Scale bars, 50 μm.
Immunohistochemical data showing that C-Ag4,8induces a neuron-specific increase in Fos expression imply the existence of a signal transduction pathway activated by occupancy of a specific receptor present in neuron cell membranes. To characterize the biochemical properties of this ligand–receptor interaction and associated signal transduction pathway in more detail, we measured levels of neuronal c-fos induction in the cultures using a quantitative high-throughput enzyme-linked assay. The results of these experiments show that C-Ag4,8 induction ofc-fos is concentration dependent and saturable (Fig.2a) as expected for a signaling pathway activated via a cell surface receptor. Fos expression curves were well fit by a single-site nonlinear regression model (R2 = 0.99) predicting an EC50of 2.3 AU for the preparation of C-Ag4,8 used, similar to its EC50 for AChR clustering when tested on cultured chick myotubes. Nonspecific binding of Ab-2, determined by preabsorption with its peptide antigen, accounted for <5% of the total binding. Levels of Fos expression in control cultures treated with medium from sham-transfected COS-7 cells (Fig. 2a) were low and similar to naive cultures, suggesting that C-Ag4,8 and not some other component of the conditioned medium was responsible for the increase in Fos levels. This interpretation was confirmed by the observation that immunoprecipitation of C-Ag4,8 from the conditioned medium removed >90% of both the AChR-clustering and the Fos-inducing activities (Fig. 2b) and is consistent with a similar specific activity for agrin in both assays.
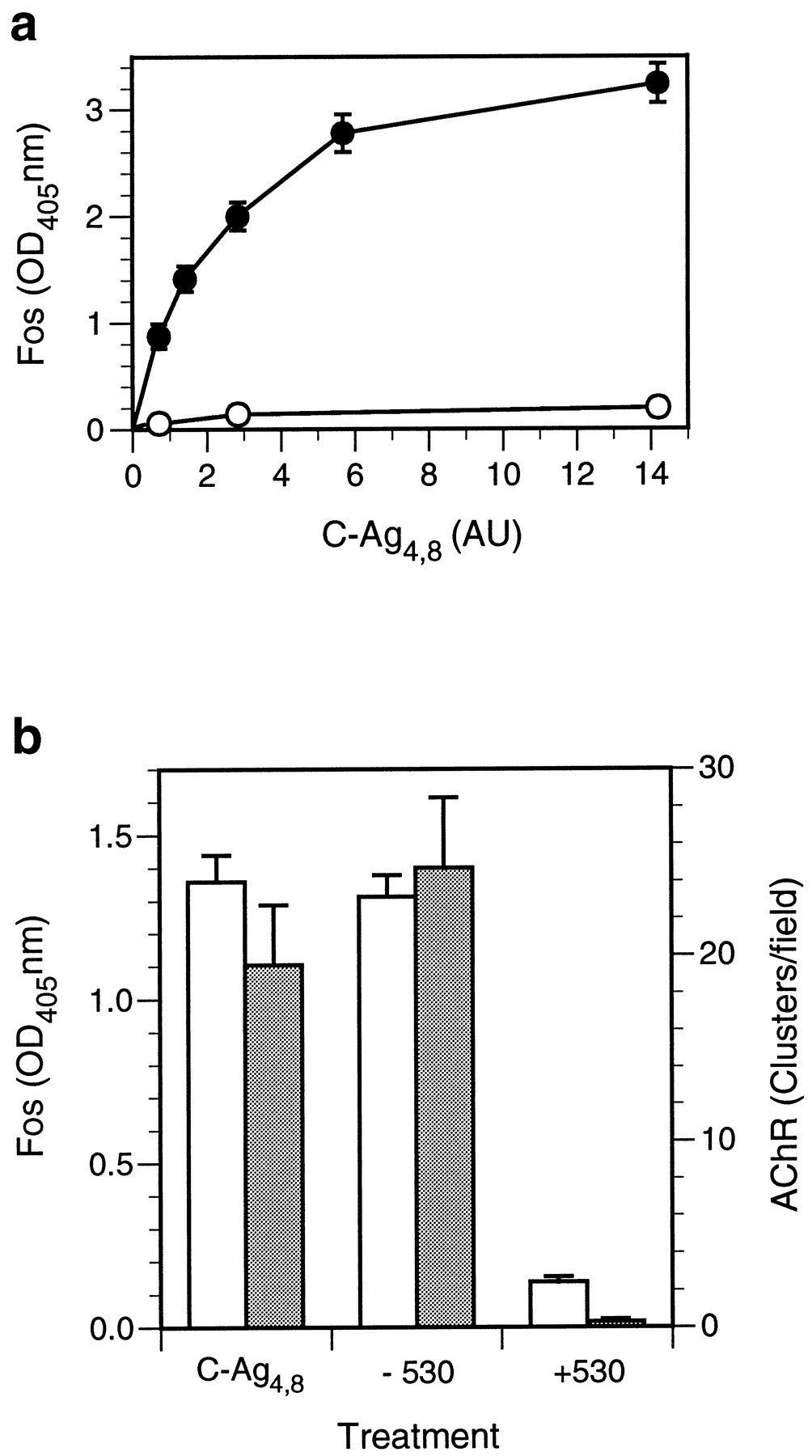
c-fos induction in cultured cortical neurons by C-Ag4,8 is concentration dependent and saturable.a, Cortical cultures were treated with C-Ag4,8 or control medium in NBM for 10 min at room temperature, returned to cNBM, and incubated for 2 hr at 37°C after which Fos levels were determined using the quantitative assay described in Materials and Methods. Levels of Fos expression, shown in arbitrary OD405 units, increased in a C-Ag4,8concentration–dependent manner (filled circles). Basal levels of Fos expression (open circles) were low and did not change significantly with an increasing concentration of control medium added to the culture. The graph shows Ab-2–specific binding; nonspecific binding, determined by labeling C-Ag4,8– or control medium–treated cultures with Ab-2 preadsorbed to an excess of peptide antigen, was <5% of total binding and has been subtracted. The C-Ag4,8 concentration is given in AU where one unit is the amount of C-Ag4,8 required to induce a half-maximal increase in AChR clusters assayed on cultured chick myotubes. Each point represents the mean ± SEM for triplicate determinations. Similar results were seen in two other experiments. b, To test the specificity of C-Ag4,8 induction of Fos expression, we immunoprecipitated an aliquot of C-Ag4,8–containing medium with the anti-agrin monoclonal antibody Agr 530. The chart showsc-fos–inducing (open bars; left scale) or AChR-clustering (shaded bars;right scale) activity measured in cortical neuron cultures and cultured chick myotubes, respectively, after treatment with a saturating concentration of C-Ag4,8medium (C-Ag4,8) or an equivalent amount of C-Ag4,8 medium immunoprecipitated in the absence (−530) or presence (+530) of Agr 530. Greater than 90% of thec-fos–inducing and AChR-clustering activities are removed from the C-Ag4,8 medium by immunoprecipitation with Agr 530, confirming that C-Ag4,8 is responsible forc-fos induction in cortical neurons. Basal levels of Fos observed in cortical cultures treated with sham-conditioned medium and spontaneous AChR clusters have been subtracted. Error bars represent the mean ± SEM for triplicate determinations. Similar results were also seen in a second experiment.
Agrin-induced tyrosine phosphorylation of MuSK occurs within 1–2 min (Glass et al., 1996), and incipient signs of AChR clustering appear on skeletal muscle fibers between 1 and 2 hr of treatment with agrin (Wallace, 1988). Preliminary experiments revealed that with continuous exposure to C-Ag4,8, maximal levels of Fos expression in cortical neurons were achieved 2–4 hr after the onset of treatment (data not shown). To investigate the time course of agrin induction of c-fos in more detail, we exposed cortical cultures to a saturating concentration of C-Ag4,8for different lengths of time and then returned the cultures to the incubator in normal growth medium for an additional 2 hr before assaying Fos levels. As can be seen in Figure3, brief exposure to agrin is sufficient to trigger a detectable increase in Fos expression, with half-maximal induction occurring at ∼5 min. The activation kinetics of the agrin signal transduction pathway in neurons is, therefore, similar to that reported for MuSK (Glass et al., 1996) and reminiscent of other well characterized receptor tyrosine kinase/ligand systems (for review, seeFantl et al., 1993).

Time course of C-Ag4,8 induction ofc-fos. Cortical cultures were incubated in a saturating concentration of C-Ag4,8 for the indicated length of time and then washed and returned to the incubator in cNBM for 2 hr before levels of Fos expression were assayed. Induction ofc-fos is relatively rapid and half-maximal after only 5 min of exposure to C-Ag4,8. The graph shows C-Ag4,8–specific c-fos induction; levels of Fos in sister wells treated with sham-conditioned medium have been subtracted. Error bars represent the mean ± SEM for triplicate determinations. Similar results were also seen in a second experiment.
Biochemical properties of C-Ag4,8 signaling in neurons
Agrin’s interaction with components of the muscle cell surface is influenced by constituents of the extracellular milieu. In particular, agrin-induced AChR clustering is dependent on extracellular Ca2+ and inhibited by heparin (Wallace, 1988, 1990). To define more precisely the nature of agrin’s interaction with its putative neuronal receptor, we studied the Ca2+ requirements and heparin sensitivity of C-Ag4,8–induced changes in c-fosexpression in cultured cortical neurons.
To examine the Ca2+ dependence of agrin-induced c-fos expression, we incubated cultures for 10 min at room temperature in a saturating concentration of C-Ag4,8 either in PBS containing 10 mm EDTA (Ca2+-free) or in PBS containing Ca2+ at known concentrations. Cultures were subsequently washed with fresh growth medium and returned to the incubator for 2 hr before fixation and assay of Fos levels. As can be seen in Figure4a, c-fos induction was completely blocked in the absence of Ca2+ but increased rapidly with increasing Ca2+ concentration in the extracellular medium. Removal of Ca2+ had no effect on the number or the morphology of neurons or on the basal levels of Fos in cultures treated with control medium. Moreover, C-Ag4,8 induction of c-fos was unaffected in cultures that were allowed to recover overnight after exposure to PBS containing 10 mm EDTA (data not shown). Nonlinear regression using a single-site model (R2 = 0.97) predicts that half-maximal C-Ag4,8 induction ofc-fos occurs at ∼36.5 ± 0.95 μm Ca2+ (mean ± SEM; n = 2), ∼10-fold lower than that reported for agrin-induced AChR clustering in muscle (Wallace, 1988). These data suggest that although similar, agrin receptors in neurons and muscle may be distinct.

C-Ag4,8 induction ofc-fos is Ca2+-dependent and blocked by heparin. Cortical cultures were incubated for 10 min in a saturating level of C-Ag4,8 in the presence of Ca2+(a) or heparin (b) at the indicated concentrations and then returned to the incubator in cNBM for 2 hr before determination of Fos levels by enzyme-linked assay as described. Graphs show C-Ag4,8–specificc-fos induction (circles); levels of Fos in sister wells treated with sham-conditioned medium have been subtracted. Induction of c-fos is Ca2+-dependent and blocked by heparin, similar to agrin-induced AChR clustering in muscle. Treatment with heparin alone (b; square) at 500 μg/ml had no significant effect. Each data pointrepresents the mean ± SEM for triplicate determinations.
A more complete parallel with agrin action in muscle was observed when we tested the ability of heparin to block neuronal c-fosinduction by C-Ag4,8 treatment. Incubation of cortical cultures with a saturating concentration of C-Ag4,8 in the presence of various concentrations of heparin revealed that heparin is an effective antagonist of C-Ag4,8 activity in neurons (Fig. 4b). Half-maximal inhibition of c-fos induction was observed at a heparin concentration of ∼20 μg/ml, similar to that reported for heparin blockade of agrin-induced AChR clustering in skeletal muscle cells (Wallace, 1990; Hopf and Hoch, 1997). In contrast, levels of Fos expression in cultures treated with 500 μg/ml heparin and sham-conditioned medium were similar to basal levels observed in control cultures treated with sham-conditioned medium alone. Inhibition of C-Ag4,8 induction of c-fosexpression was almost completely reversible; C-Ag4,8–induced Fos levels were reduced by only 21.2 ± 1.1% (mean ± SEM; n = 3) when cultures treated with 500 μg/ml heparin and then allowed to recover overnight were compared with untreated cultures.
Changes in intracellular Ca2+ levels regulate agrin-induced AChR clustering in muscle (Megeath and Fallon, 1998). To investigate a possible role for Ca2+ as an intracellular messenger of agrin receptor activation in neurons, we incubated cortical cultures with the membrane-permeant calcium chelator BAPTA- AM (Tsien, 1981) before treatment with C-Ag4,8. Clamping intracellular Ca2+ at basal levels inhibited C-Ag4,8–induced c-fosexpression in a BAPTA-AM concentration–dependent manner (Fig.5a). Half-maximal inhibition was observed at a BAPTA-AM concentration of ∼20 μm. The use of BAPTA-AM for studies of intracellular signaling by Ca2+ has been documented in a number of different systems (Stern, 1992; Roberts, 1993; Gu and Spitzer, 1995), and in agreement with these findings, we found no evidence of acute toxicity of the drug that might prejudice our findings. BAPTA-AM did not affect basal levels of c-fosexpression in the cultures, and inhibition produced by exposure to the maximal concentration of the drug was reversed (74.7 ± 11.1%, mean ± SEM; n = 3), compared with untreated cultures, after overnight recovery. The results indicate that intracellular Ca2+ fluxes are necessary for C-Ag4,8 induction of c-fos.

Intracellular Ca2+ and protein kinase activation are required for C-Ag4,8signaling. Cortical cultures were equilibrated for 1 hr in BAPTA- AM (a) or 10 min in staurosporine (b) followed by coincubation for 10 min with a saturating concentration of C-Ag4,8. a, BAPTA-AM–treated cultures were subsequently washed with NBM and then returned to cNBM media for 2 hr at 37°C before determination of Fos levels. b, Cultures treated with staurosporine were incubated with staurosporine alone for an additional 10 min after removal of C-Ag4,8 before being returned to cNBM.Graphs show C-Ag4,8–specific Fos expression (circles); Fos induction in sister cultures treated with sham-conditioned medium containing BAPTA-AM or sham-conditioned medium alone has been subtracted. Treatment with staurosporine alone at the highest concentration used (b; square) had no significant effect. Each data point represents the mean ± SEM for triplicate determinations. Similar results were seen in at least one other experiment for each treatment.
Protein kinase activation is an intrinsic component of the agrin signal transduction in muscle. Agrin triggers phosphorylation of MuSK (Glass et al., 1996; Hopf and Hoch, 1998b), and the protein kinase inhibitor staurosporine blocks agrin-induced AChR clustering (Wallace, 1994). To examine a potential role for protein phosphorylation in agrin signaling in neurons, we tested the ability of staurosporine to inhibit C-Ag4,8 induction of c-fos. Cultured neurons were preincubated for 10 min in staurosporine alone followed by coincubation for 10 min with a saturating concentration of C-Ag4,8 and then washed and incubated for an additional 10 min in staurosporine alone before being returned to cNBM before assaying for Fos. The results of these experiments (Fig.5b) show a clear concentration-dependent inhibition of C-Ag4,8 induction of c-fos by staurosporine. Half-maximal inhibition was achieved at a staurosporine concentration of 1 nm, similar to that reported for inhibition of agrin-induced AChR clustering in muscle (Wallace, 1994). Based on morphology, no obvious adverse effects were evident after the relatively brief treatment with staurosporine, and Fos levels in neurons exposed to the highest concentration of staurosporine used were similar to those seen in cultures receiving control medium. Inhibition by staurosporine was also reversible in that levels of C-Ag4,8–induced Fos determined for cultures treated with the maximal concentration of staurosporine for 30 min followed by overnight recovery were reduced by only 30.2 ± 2.3% (mean ± SEM; n = 3) compared with that in untreated cells. These data suggest that activation of one or more protein kinases is required for C-Ag4,8 induction of c-fos.
“Inactive” agrin isoforms induce c-fos expression in cortical neurons
Alternative splicing represents an important mechanism for regulating agrin’s AChR-clustering activity and binding to cell surface components (Ruegg et al., 1992; Ferns et al., 1993; Hoch et al., 1994; Gesemann et al., 1995, 1996; O’Toole et al., 1996). To examine the role of alternative splicing in agrin signal transduction in cortical neurons, we tested the ability of two alternatively spliced soluble forms of agrin, C-Ag4,0 and C-Ag0,0, which lack AChR-clustering activity, to induce c-fos.
Initial immunohistochemical studies showed that similar to that with C-Ag4,8, treatment with either C-Ag4,0 or C-Ag0,0 also triggered a marked increase in the neuronal levels of Fos expression but had no effect on Fos levels in non-neuronal cells (data not shown). Quantitative analysis demonstrated that Fos induction by both recombinant proteins was concentration dependent and saturable (Fig.6a). This response was agrin specific in that levels of Fos expression in the cultures treated with conditioned medium from sham-transfected COS-7 cells were not increased. Moreover, immunoprecipitation of C-Ag4,0 or C-Ag0,0 with Agr 530 depleted its Fos-inducing activity (Fig. 6b), confirming the agrin dependence of c-fos induction in these experiments. Together these data show that the EC50 values for C-Ag4,0 and C-Ag0,0 are similar to each other and comparable with that determined for C-Ag4,8, in marked contrast to the >1000-fold difference in AChR-clustering activity reported for these constructs in cultured muscle cells (Ferns et al., 1993). Consistent with these observations, no difference was detected among Fos levels in neurons from sister cultures treated with saturating concentrations of C-Ag4,8 or with either of the C-Agz0 isoforms (data not shown).

Agrin isoforms that lack AChR-clustering activity induce c-fos expression in cortical neurons.a, Cortical cultures were exposed to C-Ag4,0(open circles)– or C-Ag0,0(filled circles)–containing media for 10 min, and c-fos expression was assayed. Nonspecific induction of c-fos determined in control cultures treated with sham-conditioned medium has been subtracted. Induction of c-fos by both C-Ag4,0 and C-Ag0,0 is concentration-dependent and saturable. Regardless of their AChR-clustering activities, the different agrin isoforms exhibit similar specific activities in terms ofc-fos induction in cortical neurons. b,To confirm the specificity of action of C-Ag4,0(open bars)– and C-Ag0,0(shaded bars)–containing media, the ability of Agr 530 to immunoprecipitate thec-fos–inducing activity was tested in a manner similar to that described in Figure 2. Greater than 80% of thec-fos–inducing activity was precipitated by the antibody. To facilitate comparison, we expressed results as the percent of the maximal level of Fos induction for a given isoform within each experiment. Data show the mean ± SEM for triplicate determinations. Similar results were obtained in at least one other experiment for each group.
The ability of agrin to increase c-fos expression in cortical neurons, regardless of exon usage at the z-site, led us to ask whether a similar agrin signal transduction pathway might be present in other cell types. To examine this question, we measured Fos levels in cultured hippocampal and cerebellar neurons, as well as in chick myotubes, fibroblasts, and COS-7 cells, after treatment with a saturating concentration of C-Ag4,8 or C-Ag0,0 (Fig. 7). Consistent with our inability to detect agrin-induced increases in Fos staining among non-neuronal cells in cortical cultures, Fos levels in fibroblasts and COS-7 cells were not affected by treatment with agrin. In contrast, both C-Ag4,8 and C-Ag0,0 increased Fos expression in hippocampal and cerebellar cultures, with no difference evident in the potencies of the two isoforms. Interestingly, agrin induction of c-fos in muscle paralleled its AChR-clustering activity in that treatment with C-Ag4,8 but not C-Ag0,0upregulated Fos levels in cultured myotubes. These data provide evidence of a neuron-specific agrin receptor that is distinct from the agrin receptor in skeletal muscle.
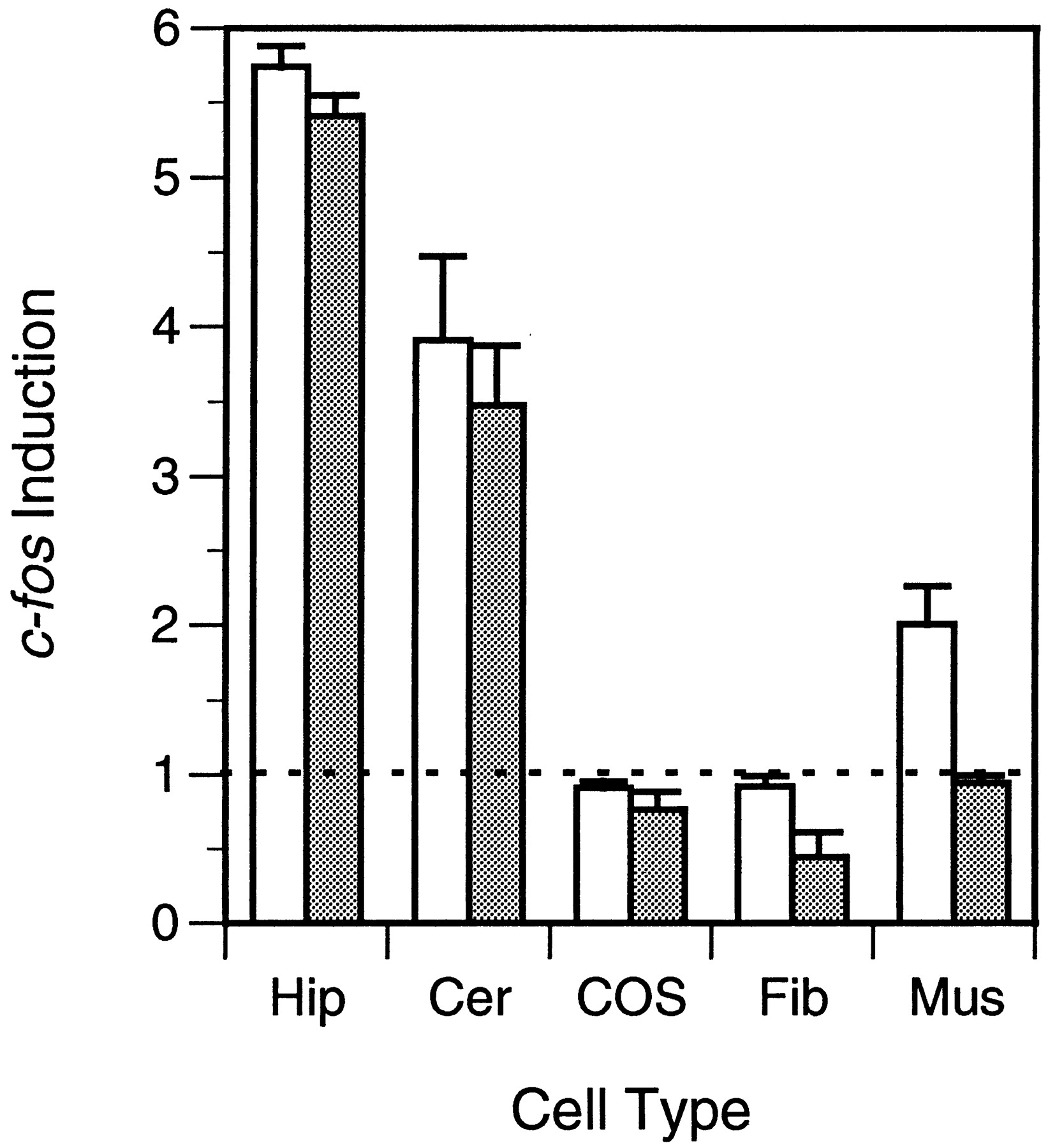
Induction of c-fos by different agrin isoforms is cell-specific. Fos levels were measured in cultured hippocampal (Hip), cerebellar (Cer), COS-7 (COS), chick fibroblast (Fib), and muscle (Mus) cells at 2 hr after a 10 min treatment with a saturating concentration of C-Ag4,8 (open bars)– or C-Ag0,0 (shaded bars)–containing media. Induction ofc-fos is expressed as the ratio of Fos expressed in agrin-treated cultures over that observed in control cultures treated with sham-conditioned medium. A value of 1 represents no induction. Fos expression in hippocampal and cerebellar cultures was increased by a similar amount after treatment with either agrin isoform. In contrast, only C-Ag4,8 induced c-fos in muscle, and neither isoform was effective in fibroblasts or COS-7 cells. Error bars represent the mean ± SEM for triplicate determinations. Similar results were obtained in one other experiment for each group.
“Inactive” and “active” agrin isoforms activate a common signaling pathway
The observation that different agrin isoforms have the same specific activity as inducers of neuronal c-fos suggests that they activate a common signal transduction pathway. To test this hypothesis we examined the biochemical profiles of c-fosinduction by C-Ag4,0 and C-Ag0,0 and compared them with that obtained for C-Ag4,8.
Our first experiments looked at the Ca2+dependence of c-fos induction by C-Ag4,0 and C-Ag0,0proteins. The results of these studies clearly show that, like C-Ag4,8, induction of c-fos by isoforms lacking inserts at the z-site is also Ca2+ dependent (Fig.8a). We note, however, that the EC50 values determined for C-Ag4,0 and C-Ag0,0 of 0.17 ± 0.08 mm (n = 3) and 0.15 ± 0.07 mm (n = 3), respectively, were higher than that for C-Ag4,8, raising the possibility that splicing at the z-site may play a role in regulating Ca2+-dependent binding of agrin to neurons. Similarly, induction of c-fos by C-Ag4,0 or C-Ag0,0 was also inhibited by heparin in a concentration-dependent manner with half-maximal inhibition occurring at ∼25 μg/ml (Fig.8b). Previous studies have suggested that heparin inhibition of agrin-induced AChR clustering involves binding of heparin directly to a region on agrin that requires exon 28 at the y-site as well as to a component on the muscle cell surface (Gesemann et al., 1996; O’Toole et al., 1996; Hopf and Hoch, 1997). Our observation that 32.1 ± 12.8% (mean ± SEM; n = 3 experiments) of C-Ag0,0 induction of c-fos expression appears to be heparin resistant suggests that a two-site model (Hopf and Hoch, 1997) may also apply to heparin inhibition of agrin action in neurons. The fact that heparin blocks c-fos induction of C-Ag0,0 that lacks the heparin-binding domain suggests that all agrin isoforms interact with a common component on the neuronal cell surface.

Biochemical properties of c-fosinduction by C-Agz0 isoforms. Extracellular Ca2+ dependence (a), blockade by heparin (b), the requirement for intracellular Ca2+ (c), and inhibition by staurosporine (d) of C-Ag4,0(open circles)– and C-Ag0,0(filled circles)–dependent induction of c-fos were examined. With the exception of a minor heparin-resistant component of the C-Ag0,0–dependent increase in Fos expression, marked similarity is evident in the biochemical profiles of the two isoforms. For comparison, results are expressed as the percent of the maximal level of Fos induction for a given isoform within each experiment. Data represent the mean ± SEM for triplicate determinations. Similar results were obtained in at least one other experiment in each group.
If it is true that different agrin isoforms activate a single receptor or receptor complex, then all agrin proteins should activate a common intracellular signaling pathway. To test this prediction we examined the ability of BAPTA-AM and staurosporine to block c-fosinduction by C-Ag4,0 and C-Ag0,0. As can be seen in Figure 8, cand d, both drugs were equally effective in antagonizing Fos expression by either agrin isoform. These results support the conclusion that increased intracellular Ca2+ and protein kinase activation are also required components of the signal transduction pathway activated by C-Ag4,0 and C-Ag0,0 in cortical neurons.
DISCUSSION
Agrin is expressed by many populations of CNS neurons, but its function in brain remains unclear. As part of a systematic approach to this question, we used induction of c-fos as an assay of cellular activation to identify cells in cortical tissue that might be targeted by agrin. Our results demonstrate that neurons, but not non-neuronal cells, respond to agrin and provide evidence of a functional agrin receptor in neurons. Activation of this putative neuronal agrin receptor occurs at agrin concentrations similar to those that induce AChR clustering in muscle. The existence of an agrin-signaling pathway supports a role for agrin in brain, and its similarity with the transduction pathway in muscle suggests that some components of the pathway and its effectors may be common to both tissues. Agrin-dependent changes in the expression of IEGs such asc-fos represent a mechanism by which agrin released by neurons could influence expression of other genes important in neuron–neuron interactions.
Our data demonstrate that agrin induction of c-fos in cortical neurons is mediated by an increase in intracellular Ca2+. Studies in other systems have identified two DNA regulatory elements, the cAMP-response element (CRE) and the serum-response element (SRE), that control expression ofc-fos by Ca2+ signals (Ghosh and Greenberg, 1995). A recent report, showing that agrin induces phosphorylation of CRE-binding protein (CREB) in hippocampal neurons, provides direct evidence of a role for the CRE in agrin signaling (Ji et al., 1998). Interestingly, phosphorylation of CREB appears to be isoform specific in that only agrin containing an insert at the z-site is effective (Ji et al., 1998). Our observation that all agrin isoforms induce expression of c-fos, even in hippocampal neurons, suggests that regulation of gene expression via the SRE is also an important pathway for agrin signaling and implies the existence of either a second agrin receptor or, as has been suggested for the p75NTR (Bothwell, 1996), a single receptor whose signaling is ligand dependent. Analysis of agrin induction ofc-fos identified both cell- and isoform-specific agrin receptors, and we would, therefore, have expected to detect similar differences among neurons if they existed. However, with the exception of a fourfold difference in Ca2+dependence of c-fos induction by C-Ag4,8 and C-Ag4,0 or C-Ag0,0, we found little evidence of more than one class of agrin receptors in cortical neurons. Such differences, however, are consistent with a single agrin receptor in which C-Ag4,8 activates the SRE and/or CRE and C-Ag4,0 or C-Ag0,0activates the SRE only.
On the basis of its pattern of expression during development and in mature brain, agrin has been proposed to play a role in neuron–neuron synapse formation. Recent studies, however, have shown that functional glutamatergic and GABAergic synapses can form between cultured cortical neurons that lack agrin-containing inserts at the z-site but express agriny4z0 (Li et al., 1999;Serpinskaya et al., 1999). On the basis of these observations, it was concluded that agrin proteins with high AChR-clustering activity are not required for the initial stages of neuron–neuron synapse formation, but a possible role for inactive agrin could not be eliminated. In fact, agrin isoforms lacking inserts at the z-site can influence the distribution of AChR at developing neuromuscular junctions (Pun and Tsim, 1997; Godfrey et al., 1999), and our results suggest that agrinz0 isoforms could also play a role in neuron–neuron synaptogenesis. Because of these findings, it will be interesting to learn whether clustering of neurotransmitter receptors or other aspects of neuron–neuron synapse formation are perturbed in animals in which agrin expression is blocked completely.
A number of muscle cell surface components have been identified that interact with agrin, including N-CAM (Tsen et al., 1995), laminin (Denzer et al., 1997), heparin-binding growth-associated molecule [HBGAM/pleiotrophin (Daggett et al., 1996)], dystroglycan (Bowe et al., 1994; Campanelli et al., 1994; Gee et al., 1994), and MuSK (Glass et al., 1996), each of which might serve as a neuronal receptor for agrin. Because the recombinant proteins used in the present study consist only of agrin’s 95 kDa C-terminal portion, proteins such as laminin, N-CAM, and HBGAM that bind domains within its N terminal (Sanes et al., 1998) are unlikely to be essential components of the putative receptor described here. However, dystroglycan- and MuSK-binding sites are present in the recombinant agrin used, and the possible role that these molecules might serve in agrin signaling in cortical neurons is considered below.
Dystroglycan is the major agrin-binding protein in muscle and was initially believed to represent the receptor responsible for agrin-induced AChR clustering (Bowe et al., 1994; Campanelli et al., 1994; Gee et al., 1994). Although this now seems doubtful (Sugiyama et al., 1994; Bowen et al., 1996), evidence suggests that dystroglycan is involved at some level in the AChR-clustering pathway (Jacobson et al., 1998). Dystroglycan is also expressed in brain (Górecki et al., 1994; Schofield et al., 1995), and some of the biochemical characteristics of agrin induction of c-fos are consistent with the possibility that it might be the putative neuronal receptor for agrin. Several lines of evidence, however, make this unlikely. First, dystroglycan mRNA levels in cortex are below detection byin situ hybridization (Górecki et al., 1994) and, therefore, unlikely to be expressed at a significant level by cortical neurons in culture. Second, despite marked differences in their affinity for dystroglycan (Sugiyama et al., 1994; Gesemann et al., 1996), no difference was evident in the EC50values for c-fos induction by the different agrin isoforms. Third, C-Ag0,0 did not increase Fos levels in cultured muscle, suggesting that agrin binding to dystroglycan does not influence c-fos expression. Finally, although heparin does not block binding of agriny0 isoforms to dystroglycan (Gesemann et al., 1996; O’Toole et al., 1996), it is an effective inhibitor of C-Ag0,0 induction ofc-fos. Thus, although it will be important to determine whether C-terminal fragments of agrin that lack domains required for dystroglycan binding are able to induce c-fos in cortical neurons, our data do not support a model in which dystroglycan is a necessary component of the putative neuronal receptor for agrin.
In muscle, an initial step in agrin-induced AChR clustering is the rapid phosphorylation of the transmembrane tyrosine kinase MuSK (Glass et al., 1996). Consistent with this model of agrin action in muscle, activation of the putative neuronal receptor occurs at the same agrin concentration that triggers MuSK phosphorylation and AChR clustering (Hopf and Hoch, 1998a,b). Agrin induction of c-fos in cortical neurons is also rapid and blocked by the protein kinase inhibitor staurosporine. Despite these similarities, however, MuSK is unlikely to be a component of the putative neuronal receptor for agrin. MuSK is not expressed in either developing or mature brain (Valenzuela et al., 1995), and although heparin is an effective inhibitor of Fos induction, it has no effect on agrin-induced MuSK phosphorylation (Hopf and Hoch, 1998a). Finally, only agrin isoforms active in AChR clustering (i.e., those that include an insert at the z-site) induce MuSK phosphorylation (Glass et al., 1996; Hopf and Hoch, 1998b), in sharp contrast to the indiscriminant behavior of the putative neuronal receptor whose activation can be effected by any agrin isoform with equal potency.
The fact that the EC50 and other biochemical parameters of c-fos induction are comparable with those reported for AChR clustering suggests that components of the muscle and neuronal receptors might be shared. Agrin activation of MuSK in skeletal muscle requires an accessory component referred to as the myotube-associated specificity component (MASC) (Glass et al., 1996). Interestingly, although MuSK is activated only by agrin-containing inserts at the z-site, in the presence of MASC, MuSK forms complexes with all agrin proteins (Glass et al., 1996). This observation has been used to suggest a model in which MuSK activation is regulated by its interaction with different MASC–agrin complexes (Glass et al., 1996). A similar model for the putative neuronal receptor for agrin would suggest MASC to be a common feature of both systems but predict coupling to a receptor tyrosine kinase other than MuSK in neurons. Possible candidates would include the receptor-like tyrosine kinases Ror1 and Ror2 (Masiakowski and Carroll, 1992) that are highly expressed in brain and display significant homology in their ectodomains to MuSK, suggesting they might recognize a similar ligand.
Agrin induction of c-fos was blocked by the membrane-permeant calcium chelator BAPTA AM, implying that an increase in intracellular Ca2+ is a necessary step in agrin signaling in neurons. Activation of the putative neuronal receptor also requires external Ca2+, but further experiments will be needed to determine whether the increase in intracellular Ca2+ derives from extracellular or intracellular stores or both. It is interesting to note that a transient increase in intracellular Ca2+ also regulates agrin-induced AChR clustering in muscle (Megeath and Fallon, 1998), further evidence that although not identical, agrin signaling in muscle and neurons is mediated by closely related pathways. Calcium plays a key role in a number of intracellular signaling pathways in neurons that could be modulated by agrin-induced increases in intracellular Ca2+. For example, intracellular Ca2+ transients are important regulators of neural differentiation, axonal growth, and guidance (Doherty and Walsh, 1994; Spitzer et al., 1994), and several studies have provided evidence that agrin may serve as a stop and differentiation signal for some populations of neurons (Campagna et al., 1995; Gautam et al., 1996; Chang et al., 1997). Interestingly, this activity it not isoform dependent and appears to be a property of the C-terminal half of the protein, consistent with our findings for the activation of the putative agrin receptor in cortical neurons. Calcium is also an important mediator of activity-dependent changes that underlie long-term alterations in synaptic physiology associated with learning and memory (Bito et al., 1997). If, as is the case for MuSK in muscle, agrin receptors are concentrated at synaptic sites on neurons, then their activation might be expected to influence local events associated with synaptic remodeling. Clearly it will be important to identify these putative receptors for agrin and determine their distribution on neurons.
Footnotes
This work was supported by National Institutes of Health Grant NS33213 to M.A.S. We thank Dr. Michael Ferns for his generous gift of the C-Ag constructs, Dr. Diane K. O’Dowd for critical reading of this manuscript, and Bernadette Nicolas for expert technical assistance.
Correspondence should be addressed to Dr. Martin A. Smith, Department of Anatomy and Neurobiology, Irvine Hall, Room 110, University of California at Irvine, Irvine, CA 92697.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}