

Abstract
Inflammatory pain involves the sensitization of both primary afferent and spinal cord neurons. To explore the neurochemical changes that contribute to inflammatory pain, we have examined the expression and ligand-induced internalization of the substance P receptor (SPR) in the spinal cord in acute, short-term, and long-term inflammatory pain states. These inflammatory models included unilateral injection of formalin (8–60 min), carrageenan (3 hr), and complete Freund’s adjuvant (CFA; 3 d) into the rat hindpaw as well as adjuvant-induced polyarthritis (21 d). In acute inflammatory pain there is ongoing release of substance P (SP) as measured by SPR internalization in lamina I neurons at both 8 and 60 min after formalin injection. Although there is no tonic release of SP in short-term inflammatory pain, at 3 hr after carrageenan injection, SP is released in response to both noxious and non-noxious somatosensory stimulation with SPR internalization being observed in neurons located in both laminae I and III-IV. In long-term inflammatory pain models (CFA and polyarthritis) the same pattern of SP release and SPR activation occurs as is observed in short-term inflammation with the addition that there is a significant upregulation of the SPR in lamina I neurons. These results suggest that SPR internalization might serve as a marker of the contribution of ongoing primary afferent input in acute and persistent pain states. These stereotypical neurochemical changes suggest that there are unique neurochemical signatures for acute, short-term, and long-term inflammatory pain.
- carrageenan
- complete Freund adjuvant
- formalin
- inflammation
- internalization
- pain
- spinal cord
- substance P receptor
Chronic inflammation is responsible for a variety of persistent pain states including arthritis, back pain, and temporomandibular joint disorder. Although significant progress has been made in understanding the peripheral inflammatory response, the neurochemical changes within the spinal cord that are involved in the generation and maintenance of chronic inflammatory pain are poorly understood. In response to persistent inflammatory pain, normally innocuous sensory stimuli are perceived as painful (allodynia), and mildly noxious sensory stimuli are perceived as highly painful (hyperalgesia). Both hyperalgesia and allodynia are thought to arise from a sensitization of peripheral nociceptors (peripheral sensitization) and spinal dorsal horn neurons (central sensitization) (Treede et al., 1992).
Peripheral sensitization is believed to result from the release of proinflammatory substances at the site of injury, including bradykinin, prostaglandins, serotonin, ATP, and protons (Beck and Handwerker, 1974;Schaible and Schmidt, 1988; Thayer et al., 1988; Steen et al., 1992;Bevan and Geppetti, 1994; for review, see Dray, 1994). These substances can directly activate and/or sensitize the peripheral nociceptors and are at least partly responsible for the sensitization of primary afferent neurons and the resulting hyperalgesia and allodynia (for review, see Dray, 1995).
The sustained activity of primary afferent fibers that occurs after peripheral sensitization also induces an increase in the efficacy of synaptic transmission between primary afferent fibers and dorsal horn neurons, a process referred to as central sensitization (Mendell, 1966;Woolf, 1983, 1994; see references in Dickenson, 1990, 1995). The detailed mechanisms that underlie central sensitization are not fully understood; however several in vitro and in vivopharmacological studies have implicated a cooperation between substance P (SP) and NMDA-mediated events in the development and maintenance of inflammation-induced central sensitization (Chizh et al., 1995; Cumberbatch et al., 1995; see references in Urban et al., 1994).
In an effort to understand the mechanisms involved in the peripheral and central sensitization associated with inflammatory pain, we explored the expression and internalization of the substance P receptor (SPR) in the spinal dorsal horn in four well-characterized and widely used experimental models of inflammatory pain in the rat. These were produced by unilateral subcutaneous injection of formalin, carrageenan, or complete Freund’s adjuvant (CFA) into the hindpaw. In addition, we examined the neurochemical changes in an animal model of polyarthritis induced by CFA injection into the base of the tail. Each of these inflammatory models is characterized by a different onset and time course of nociceptive inputs and responses. Using this approach, we compared the previous electrophysiological and behavioral data with the alterations in the amount and/or site of release of SP from primary afferent neurons, the number and location of SPR-expressing spinal neurons that are activated by this released SP, and the populations of neurons showing upregulation of the SPR. Together, these data suggest that each type of inflammatory pain (acute, short-term, and long-term) is characterized by a unique neurochemical signature within the spinal cord.
Neurochemical signature of inflammatory pain
MATERIALS AND METHODS
Experimental animals. Experiments were performed on 124 adult male Sprague Dawley rats (Harlan, Madison, WI), weighing 225–250 gm. Rats were kept in a vivarium, maintained at 22°C, with a 12 hr alternating light/dark cycle and were given food and waterad libitum. All procedures were approved by the Animal Care Committees at the Veterans Affairs Medical Center and the University of Minnesota.
All subcutaneous injections in the rat hindpaw were performed under anesthesia (50 mg/kg sodium pentobarbital, i.p.). In addition to a group of naive rats used as normal controls, each experimental group (formalin, carrageenan, CFA, and polyarthritis) had an associated control group receiving an injection of saline (formalin, carrageenan, and CFA controls) or mineral oil (polyarthritis controls) and surviving the same amount of time.
The formalin model. Formalin injection (Dubuisson and Dennis, 1977; for review, see Tjolsen et al., 1992) induces a biphasic stereotypical nocifensive behavior and corresponding electrophysiological (Dickenson and Sullivan, 1987a,b) responses. These responses are divided into an early and short-lasting first phase (0–10 min) caused by a primary afferent discharge produced by the stimulus, followed by a quiescent period, and then a second, prolonged phase (15–60 min) of tonic pain. In the present study, rats received formalin (5%, pH 6.9, in saline, 50 μl, s.c.; Sigma, St. Louis, MO) in the plantar surface of the hindpaw and were allowed to survive 8 min (n = 8) or 1 hr (n = 8; controls received saline, 50 μl, n = 6 and n = 6, respectively). The extent of peripheral edema was expressed as percent of the control values, i.e., the diameters of both paw (site of injection) and ankle (edema diffusion) were measured immediately before perfusion using a caliper square. Because of the high number of spinal internalized SPR-positive neurons after formalin injection, the effects of additional stimulation in formalin-injected animals were not examined.
The carrageenan model. Injection of λ-carrageenan (Winter et al., 1962) in the rat hindpaw produces an acute, restricted inflammation associated with thermal and mechanical hyperalgesia and allodynia that peak 3 hr after carrageenan injection (Kayser and Guilbaud, 1987; Hargreaves et al., 1988; Kayser et al., 1991). In the present study, rats received carrageenan (2%, pH 6.8, in saline, 100 μl, s.c.; Sigma) in the plantar surface of the hindpaw and survived 3 hr (n = 11; controls received saline, 100 μl,n = 7). The acute effects of carrageenan injection were evaluated by perfusing rats 10 min after carrageenan injection (n = 3). As in the formalin model, extent of peripheral edema was measured. In addition, the changes in spinal SPR expression and internalization during inflammation-induced allodynia and hyperalgesia were studied by evaluating the effects of non-noxious and noxious mechanical stimulation in carrageenan- and saline-injected rats (see below).
The CFA model. Injection of CFA (Millan et al., 1988) in the rat hindpaw produces a long-lasting pain syndrome, peaking at 3 d and associated with thermal and mechanical hyperalgesia and allodynia (Ma and Woolf, 1996). In the present study, rats received CFA (50%, pH 7.0, in saline, 100 μl, s.c.; Sigma) in the plantar surface of the hindpaw and were allowed to survive 3 d (n = 11; controls received saline, 100 μl, n = 7). The acute effects of CFA injection were evaluated by perfusing rats 10 min after CFA injection (n = 3). As in the formalin model, extent of peripheral edema was measured. In addition, the changes in spinal SPR expression and activation during inflammation-induced allodynia and hyperalgesia were studied by evaluating the effects of non-noxious and noxious mechanical stimulation in CFA- and saline-injected rats (see below).
The polyarthritis model. Injection of Mycobacterium butirycum into the base of the rat tail (Pearson and Wood, 1959) results in an initial inflammatory response within hours; in addition, significant physiological, behavioral, and electrophysiological changes appear 10 d after the injection and last several weeks, peaking at 3–4 weeks (Colpaert et al., 1980; De Castro Costa et al., 1981;Colpaert and Van den Hoogen, 1982; Menetrey and Besson, 1982; see references in Besson and Guilbaud, 1988). In the present study, polyarthritis was induced by subcutaneous injection of heat-killedMycobacterium butirycum into the base of the tail (performed at the breeding center, 10 mg/ml, 100 μl, Charles River, Saint Aubin Les Elbeuf, France). Rats were kept for 3 weeks after inoculation (n = 10; controls received vehicle alone,n = 10). As in the formalin model, extent of peripheral edema was measured. To reduce discomfort, animals were housed three per large cage with food directly available on the cage floor. Because of their general impairment and the bilateral nature of the inflammation, the effects of additional mechanical stimulation were not evaluated.
Mechanical stimulation. Under pentobarbital anesthesia, carrageenan (3 hr; n = 4)-, CFA (3 d; n= 4)-, and saline-injected (3 hr and 3 d; n = 4/group) rats were stimulated with a non-noxious mechanical stimulus (light stroking of the dorsal hindpaw every second for 2 min using the wooden handle of a brush applying a pressure approximately equivalent to 3.6 × g or with a noxious mechanical stimulation (a noxious pressure was applied for 30 sec with a hemostat placed on the distal part of the hindpaw between the footpads). Rats were perfused 5 min after the end of the mechanical stimulation.
Immunohistochemistry. At the appropriate time point (8 and 60 min after formalin, 10 min and 3 hr after carrageenan, 10 min and 3 d after CFA, 21 d after the inoculation of polyarthritis and 5 min after acute mechanical stimulation) the animals were deeply anesthetized with pentobarbital (50 mg/kg, i.p.) and perfused intracardially with 200 ml of 0.1 m PBS followed by 500 ml of a solution of 4% formaldehyde and 12.5% picric acid in 0.1m PBS. The spinal cord was then removed and post-fixed for 16 hr in the same fixative and cryoprotected for 24 hr in 30% sucrose in 0.1 m phosphate buffer. Serial frozen sections, 60-μm-thick, were cut with a microtome and collected in PBS to be processed immunohistochemically as free-floating sections.
The tissue sections were incubated for 30 min at room temperature in a blocking solution of 1% normal goat serum in PBS with 0.3% Triton X-100 and were then incubated overnight (SPR antibody) or for 4 hr (SP antibody) at room temperature in the primary antiserum. SPR was detected with a polyclonal rabbit anti-SPR antibody (1:5000, raised in our laboratory). SP was detected with a polyclonal guinea pig anti-SP antibody (1:1000, a kind gift from J. Maggio). The incubated sections were washed three times for 10 min in PBS and incubated in the secondary antibody solution for 3 hr at room temperature. Secondary antibodies conjugated to fluorescent markers Cy3 (used with SPR) and FITC (used with SP) were used at 1:600 and 1:150, respectively. Finally, the sections were washed three times for 10 min in PBS, mounted on gelatin-coated slides, air-dried, dehydrated via an alcohol gradient (70, 90, and 100%), cleared in Xylene, and coverslipped. To confirm the specificity of the primary antibody, controls were performed; preabsorption with the corresponding synthetic peptide (see Fig. 2A) or omission of any stage in the protocol abolished the staining. Because staining intensity might vary between experiments, control sections were included in each run of staining.
Quantification of immunofluorescence levels and SPR internalization. Sections from the lumbar spinal cord were analyzed by fluorescent and confocal microscopy to characterize SPR and SP immunofluorescence and SPR internalization, using an MRC-1024 confocal imaging system (Bio-Rad, Hercules, CA) and an Olympus BH-2 microscope equipped for epifluorescence (Mantyh et al., 1995; Allen et al., 1997).
In all cases, immunofluorescence levels for SP and SPR and the percentage of SPR immunoreactive (SPR-IR) neurons demonstrating internalization were determined. Because an increase in SPR immunofluorescence could be explained by an increase in the number of SPR-IR neurons, the number of SPR-IR neurons was also counted. Analyses were performed at the lumbar level (L4), the main projection site for the primary afferent fibers innervating the hindpaws (Molander et al., 1984; Molander and Grant, 1985; LaMotte et al., 1991), in laminae I-II and III-IV of the dorsal horn, ipsilateral and contralateral to the stimulation. Sagittal sections were viewed through a 1 cm2 eyepiece grid divided into 100 1 × 1 mm units. In cell bodies that do not contain internalized SPR, the SPR immunoreactivity is uniformly distributed on the cell surface; in contrast, in the neurons that have internalized the SPR, the cytoplasm contained bright, immunofluorescent endosomes. An endosome was defined as an intense SPR-immunoreactive intracellular organelle between 0.1 and 0.7 μm in diameter that was clearly not part of the external plasma membrane. Unstimulated cells contained less than five endosomes per cell. In the present study, neurons containing 20 or more endosomes were considered to be internalized. Importantly, because neurons with <20 endosomes were not counted, it is possible that subtle changes in the magnitude of internalization were missed.
Immunofluorescence intensities were obtained with a confocal fluorescent imaging system and analyzed using NIH Image 1.7. These results were confirmed using a 12 bit SPOT2 digital camera (Diagnostic Instruments, Sterling Heights, MI) on an Olympus BX-60 fluorescence microscope with Image Pro Plus version 3.0 software (mediaCybernetics, Silver Spring, MD). The response of the digital camera was measured using 540/560 nm Inspeck fluorescent bead standards (Molecular Probes, Eugene, OR). A ratio was established between the output of the camera and a given relative fluorescence of the beads. The camera response was determined to be linear, thus establishing that a doubling of the camera grayscale output represents a doubling of label present in the tissue.
Statistical tests. Student’s t test was used to compare immunohistochemical measures in each inflammatory model with its respective control. For the effects of mechanical stimulation in carrageenan- or CFA-injected animals on SPR internalization and the development of peripheral edema, a one-way ANOVA was used. To evaluate the correlation between the extent of peripheral edema and SPR internalization at the spinal level, a Pearson’s correlation coefficient was performed. For multiple comparisons, the Fisher’s Protected Least Significant Difference (PLSD) post hoc test was used; significance at p < 0.05. In all cases, the investigator responsible for plotting and counting the SPR-IR neurons was blind to the experimental situation of each animal.
RESULTS
Distribution of SPR immunoreactivity in the spinal segment L4
Under normal or unstimulated experimental conditions, a distinct pattern of spinal SPR immunoreactivity is observed. The densest SPR staining is found in lamina I (Fig.1A), and SPR immunoreactivity covers almost the entire dendritic and somatic surface of each neuron that expresses the receptor (Fig.2B). In contrast, lamina II contains almost no SPR-IR cell bodies (Fig.1A), but is traversed by SPR-IR dendrites originating from SPR-IR neurons located, in part, in laminae III-IV (Fig.1B). SPR immunoreactivity is also observed in spinal neurons localized in laminae V and VI and around the central canal (lamina X). In all these regions, the SPR immunoreactivity is predominantly associated with the plasma membrane, with few SPR-IR endosomes present in the cytoplasm. In the present study, SPR staining was evaluated in all the spinal regions, but the results are reported for laminae I–IV because changes were only observed in these laminae.

Laminar distribution of SPR immunoreactivity in the spinal cord and upregulation of SPR expression in lamina I in long-term inflammatory pain state. Confocal image illustrating the laminar distribution of SPR immunoreactivity in a coronal section of the L4 spinal segment, 3 d after a unilateral CFA injection (A). This image is from 60-μm-thick tissue section acquired with a 10× lens. Scale bar, 200 μm. Confocal images of lamina III SPR-IR neurons contralateral (B) and ipsilateral (C) to CFA injection. The densest SPR staining is found in lamina I. In contrast, lamina II contains almost no SPR-IR neuronal cell bodies, but is traversed by SPR-IR dendrites originating from SPR-IR neurons located in laminae III-V. SPR immunoreactivity is also observed in laminae V-VI and around the central canal. In long-term inflammation, upregulation of the SPR appears confined to lamina I neurons (see box inA and arrow pointing at a lamina I SPR-IR neurons in C). These images, obtained from 60-μm-thick tissue sections, are projected from 25 optical sections acquired at 0.8 μm intervals with a 60× lens. Scale bar, 20 μm.

Normally innocuous mechanical stimulation induces SPR internalization in lamina I SPR-expressing neurons in carrageenan- and CFA-induced inflammatory pain but not in saline-injected animals. Confocal images of lamina I SPR-IR neurons ipsilateral to the injection of (B) saline, (D) carrageenan (3 hr), and (F) CFA (3 d), observed after innocuous mechanical stimulation. A is a preabsorbed control for SPR primary antibody, showing the specificity of the primary antibody against SPR. C andE present the basal SPR immunoreactivity observed after carrageenan (3 hr) and CFA (3 d) injections, respectively. Note that innocuous mechanical stimulation does not induce any SPR internalization in saline-injected animals, whereas this same stimulus induces SPR internalization in CFA- (F) and carrageenan-injected (D) rats. Note also the increase in immunofluorescence level in lamina I neurons after CFA injection (E), whereas no changes are observed after carrageenan injection. These images, obtained from 60-μm-thick tissue sections, are projected from 18 optical sections acquired at 0.8 μm intervals with a 60× lens. Scale bar, 5 μm.
Acute inflammation: ongoing release of SP
Formalin injection induced a unilateral peripheral edema observable at 1 hr after the injection (115 ± 1% and 109 ± 2% of saline control values for the paw and ankle diameters, respectively; p < 0.0001), edema was not detectable at an earlier time point (8 min; 99 ± 1% and 100 ± 1% of saline control values, respectively). Neither spinal SPR nor SP immunofluorescence levels changed when comparing: (1) formalin-injected rats to their respective saline controls or (2) the ipsilateral and contralateral sides in formalin-injected animals (see Fig. 4). SPR internalization was not observed in saline control animals. In contrast, SPR internalization was observed in laminae I-II but not in laminae III-IV (Figs. 3A,4), ipsilateral to the formalin injection. Eight minutes after formalin, 70–75% of laminae I-II SPR-IR neurons showed internalization. Furthermore, at 1 hr after formalin, 55–60% of laminae I-II SPR-IR neurons were still containing internalized SPR. For laminae III-IV neurons, SPR-IR endosomes were observed in their dorsally directed dendrites located in laminae I and II (Fig. 3A), but not in their cell bodies.
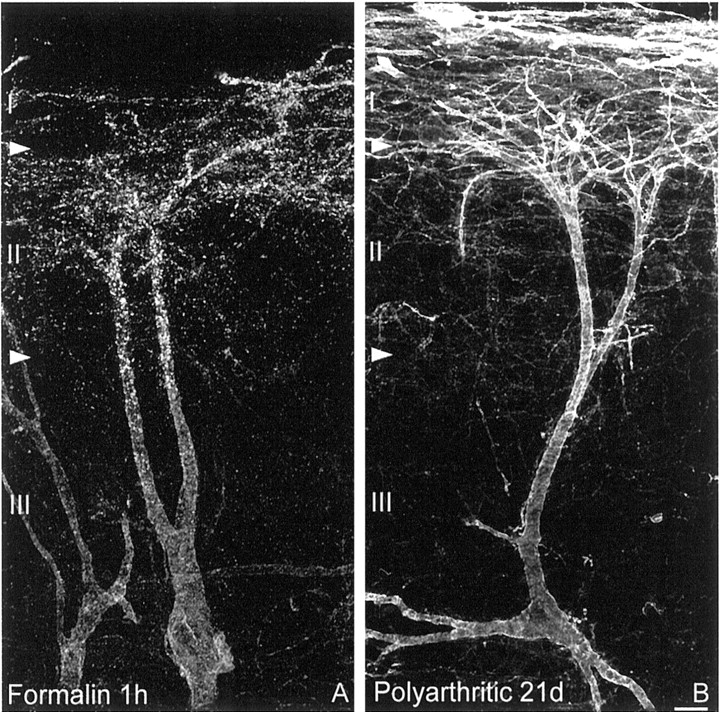
SPR internalization in acute inflammation and SPR upregulation in long-term inflammation are confined to lamina I spinal neurons. Confocal images of lamina III SPR-IR neurons observed 1 hr after formalin injection (A) and 21 d after induction of polyarthritis (B). SPR internalization in laminae I and II and in dorsally directed dendrites of lamina III neurons characterizes the neurochemical signature of acute inflammatory pain, whereas SPR upregulation in lamina I neurons is observed in long-term inflammatory pain. As in CFA, the SPR upregulation observed in polyarthritis is confined to neurons located in lamina I of the dorsal horn and is not observed in lamina III SPR-IR neurons. These images, obtained from 60-μm-thick tissue sections, are projected from 25 optical sections acquired at 0.8 μm intervals with a 60× lens. Scale bar, 20 μm.

Quantification of SPR internalization and levels of SPR immunoreactivity in short- and long-term inflammatory pain states. Results are expressed as percent increase compared to respective saline control values for SPR immunofluorescence levels in spinal lamina I (mean ± SEM) and percent of internalized SPR-IR lamina I neurons in saline-injected animals (saline), 8 min (Form 8 min), and 1 hr (Form 1 hr) after formalin injection, 10 min (Carra 10 min) and 3 hr (Carra 3 hr) after carrageenan injection, 10 min (CFA 10 min) and 3 d (CFA 3 d) after CFA injection and in CFA-induced polyarthritic rats (Poly 21d). Student’s t test, ***p < 0.001 compared to respective controls.
In addition, no change in the number of SPR-IR neurons was observed in laminae I-II (8.2 ± 1.7 and 7.8 ± 0.8 neurons per section compared to 7.4 ± 0.8 and 7.5 ± 0.4 in respective saline controls) and III-IV (3.7 ± 0.4 and 3.8 ± 0.4 neurons per section compared to 3.8 ± 0.1 and 4.2 ± 0.3 in respective saline controls).
Short-term inflammation: alteration of SPR internalization in response to non-noxious and noxious mechanical stimulations
Ten minutes after carrageenan injection, we observed a small degree of SPR internalization in the spinal dorsal horn, mainly localized in dendrites and in few cell bodies of lamina I neurons (Fig.4).
Three hours after carrageenan injection, we detected an extended unilateral peripheral edema in both paw and ankle of the injected hindpaw (increase of 50 ± 1% and 39 ± 2% compared to saline control values, respectively; p < 0.0001). Spinal SPR or SP immunofluorescence levels remained unchanged when comparing: (1) carrageenan-injected rats to their respective saline controls (Figs. 2C, 4) and (2) the ipsilateral and contralateral sides in carrageenan-injected rats. In addition, no alteration in the number of SPR-IR neurons in laminae I-II (8.8 ± 1.2 neurons per section compared to 8.5 ± 0.8 in saline controls) and III-IV (5.5 ± 0.9 neurons per section compared to 5.4 ± 0.4 in saline controls), and no SPR internalization in laminae I-II (Fig. 2C) or laminae III-IV was observed in saline- or carrageenan-injected animals.
Non-noxious mechanical stimulation did not induce spinal SPR internalization in saline-injected rats (Fig. 2B). In contrast, this normally non-noxious stimulus induced SPR internalization in SPR-IR lamina I neurons in carrageenan-injected rats (Fig. 2D). SPR internalization was observed in both the dendrites and cell body of SPR-IR neurons localized throughout the lumbar enlargement (L1–L6), ipsilateral to the stimulation. After non-noxious stimulation, 57 ± 3% of the SPR-IR lamina I neurons showed SPR internalization (Fig. 5;p < 0.0001 compared to saline control values). No SPR internalization was detected in laminae III-IV.

Quantification of SPR internalization in cell bodies of laminae I and III-IV SPR-IR neurons after innocuous and noxious stimulation in carrageenan- and CFA-induced inflammatory pain states. Results are expressed as percent of internalized SPR-IR neurons in laminae I and III after innocuous mechanical stimulation and in laminae I, III, and IV after noxious mechanical stimulation, in saline-, carrageenan-, and CFA-treated rats. Note that there is greater SPR internalization in lamina I in carrageenan-injected rats after innocuous stimulation compared to CFA-injected rats (p < 0.01) and that this difference is also observed in lamina III neurons after noxious stimulation (p < 0.0001). In addition, SPR internalization in lamina IV is only observed in carrageenan-injected animals. In contrast, noxious mechanical stimulation induces a maximal SPR internalization in lamina I neurons in both carrageenan- and CFA-injected rats. No SPR internalization is observed in lamina III-IV after non-noxious stimulation in any of the inflammatory pain states examined. One-way ANOVA and Fisher PLSD, **p < 0.01, ***p < 0.001 compared to the saline-injected group.
In saline control rats, noxious mechanical stimulation induced SPR internalization in 76 ± 6% of the SPR-IR neurons in lamina I (Fig. 5). In addition, in carrageenan-injected animals, noxious mechanical stimulation induced a maximum SPR internalization in lamina I (100% of SPR+ neurons show internalization, p< 0.01 compared to control values). In laminae III-IV spinal neurons, noxious mechanical stimulation did not induce any SPR internalization in control rats (Figs. 5,6A). In contrast, this noxious stimulation induced SPR internalization in SPR-IR lamina III neurons in carrageenan-injected animals (Fig. 5). After noxious stimulation, 63 ± 2% of the SPR-IR lamina III neurons (p < 0.0001 compared to saline control values, 2 ± 1%) and 45 ± 5% of the SPR-IR lamina IV neurons (p < 0.0001 compared to saline control values, 0 ± 0%) showed SPR internalization in carrageenan-injected animals (Fig. 5), ipsilaterally to the stimulation.
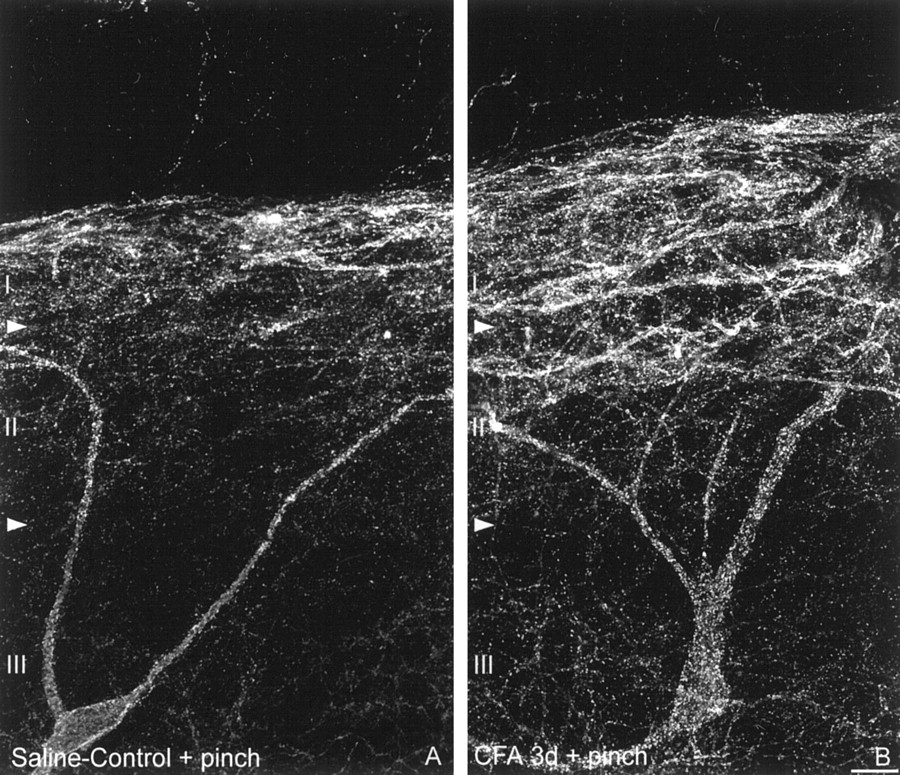
Noxious mechanical stimulation induces SPR internalization in lamina III neurons in long-term inflammatory pain states. Confocal images of lamina III SPR-IR neurons ipsilateral to the injection of (A) saline and (B) CFA after noxious mechanical stimulation. Note that after noxious mechanical stimulation in saline-treated rats, SPR internalization is observed in cell bodies and dendrites located in lamina I and II. In contrast, in CFA-inflamed rats, the same stimulation induces a significant SPR internalization in laminae I and II as well as in the cell bodies and dendrites of lamina III neurons. These images, obtained from 60-μm-thick tissue sections, are projected from 25 optical sections acquired at 0.8 μm intervals with a 60× lens. Scale bar, 20 μm.
Long-term inflammation: increase in spinal SPR immunofluorescence and alteration of SPR internalization in response to non-noxious and noxious mechanical stimulations
Ten minutes after CFA injection, we observed a small degree of SPR internalization in the spinal dorsal horn, mainly localized in dendrites and in few cell bodies of lamina I neurons (Fig. 4).
Three days after CFA injection, we detected a unilateral peripheral edema, smaller than observed after carrageenan injection (increase of 30 ± 1% and 18 ± 1% compared to saline control values for the paw and ankle diameters, respectively; p < 0.0001). Consistent with our previous results using this model at this time point, spinal SPR immunofluorescence levels were significantly increased in laminae I-II (Figs. 1A,C,2E), ipsilateral to the CFA injection (Fig. 4;Abbadie et al., 1996). No significant change in SP immunofluorescence levels in laminae I-II was detected. In laminae I-II, SPR immunofluorescence levels were increased by 94% (p < 0.0001 compared to saline controls) and by 76% (p < 0.0001 compared to the contralateral side of CFA-injected rats). In contrast, no change in SPR immunofluorescence was observed in laminae III-IV (Fig.1A,C). In addition, in the absence of superimposed stimulation, no SPR internalization was detected in the inflamed animals (Figs. 1C, 2E, 4).
It is important to note that the number of lamina I SPR-IR neurons remained the same after CFA injection, ruling this out as a possible explanation for the increase in SPR immunofluorescence observed in lamina I. The total number of SPR-IR neurons per section in laminae I-II was 7.8 ± 0.5 (compared to 8.1 ± 0.1 in saline controls) and in laminae III-IV 3.5 ± 0.6 (compared to 4.3 ± 0.5 in saline controls).
Non-noxious mechanical stimulation induced SPR internalization in SPR-IR lamina I neurons in CFA-injected animals (Fig.2F). SPR internalization was mainly observed in dendrites and in a few cell bodies of SPR-IR neurons in lumbar segments (L3–L5), ipsilateral to the stimulation. After non-noxious stimulation, 32 ± 7% of the SPR-IR lamina I neurons showed SPR internalization in CFA-injected animals, (Fig. 5; p < 0.003 compared to saline control values). Interestingly, the number of SPR-IR neurons showing SPR internalization in lamina I was significantly lower than in carrageenan-injected animals (p < 0.007). In addition, there was a positive correlation between the number of SPR-internalized lamina I neurons and the size of the peripheral edema as measured at both the paw and ankle (r = 0.95; p < 0.0001). The correlation between edema magnitude and degree of SPR internalization observed after non-noxious stimulation in inflammatory conditions suggest that SPR internalization is at least partly caused by peripheral sensitization present in inflammatory states. Finally, SPR internalization was not detected in laminae III-IV SPR-IR.
In CFA-injected animals, noxious mechanical stimulation induced a maximum SPR internalization in lamina I (97 ± 2% of SPR-IR neurons are internalized; p < 0.01 compared to saline control values 76 ± 6%, no difference between CFA- and carrageenan-injected groups). In laminae III-IV spinal neurons, noxious mechanical stimulation did not induce any SPR internalization in saline control rats (Figs. 5, 6A). In contrast, noxious stimulation induced SPR internalization in SPR-IR lamina III neurons in CFA-injected animals (Figs. 5, 6B). After noxious stimulation, 32 ± 5% of the SPR-IR lamina III neurons showed SPR internalization in CFA-injected animals, (Fig. 5; p < 0.001 compared to saline control values, 2 ± 1%), and the number of SPR-IR neurons showing SPR internalization in lamina III was significantly lower than in carrageenan-injected animals (p < 0.001). There was a positive correlation between the number of SPR internalized lamina III neurons and the magnitude of the peripheral edema as measured at both the paw and ankle (r = 0.96; p < 0.0001). The correlation between edema magnitude and degree of SPR internalization observed after noxious stimulation in inflammatory conditions suggests once again that SPR internalization is at least partly caused by peripheral sensitization present in inflammatory states inducing an increased release of SP and subsequently a larger diffusion of SP. In contrast to carrageenan inflammation, SPR internalization was not observed in lamina IV neurons after noxious mechanical stimulation in CFA-injected rats.
Long-term inflammation: bilateral increase in SPR immunofluorescence in polyarthritic rats
Three weeks after the inoculation with CFA, a generalized inflammatory reaction developed (polyarthritis), inducing a wide spread peripheral edema that was evident in both hindpaws (increase of 53 ± 1% and 51 ± 1% compared to control values for the paw and ankle diameters, respectively; p < 0.0001). No significant change in spinal SP immunofluorescence levels was observed. However, in the same spinal cord sections, SPR immunofluorescence levels increased bilaterally, by 29% (p < 0.0001) and 31% (p < 0.0001) in laminae I-II, for each side of the dorsal horn, compared with control rats. In contrast, no change in SPR immunofluorescence levels was observed in laminae III-IV (Fig. 3B). In addition, no SPR internalization was observed in resting, nonstimulated animals (Fig.4).
As in CFA rats, no modification in the number of SPR-IR neurons was observed. The total number of SPR-IR neurons per section was 7.2 ± 0.9 in laminae I-II (compared to 8.2 ± 0.4 in controls) and 4.6 ± 0.3 in laminae III-IV (compared to 4.7 ± 0.6 in controls).
To further determine which cells were the origin of the upregulation of the SPR in lamina I, SPR immunofluorescence levels in cell bodies of laminae I and III neurons and in the dendrites of lamina III neurons, extending to lamina I or localized in lamina III were measured. In control rats as well as in inflamed rats, there were no differences in SPR immunofluorescence levels originating from lamina III neurons, but we observed an increase in SPR immunofluorescence levels in lamina I neurons in inflamed animals, suggesting that the increase in SPR immunofluorescence in lamina I in long-term inflammation is primarily caused by an increase in SPR immunofluorescence in cell bodies and dendrites of lamina I SPR-IR neurons.
DISCUSSION
Acute inflammatory pain
Formalin induces a stereotypical biphasic response, consisting of an early short-lasting painful response followed by a prolonged period of tonic/persistent pain (Dubuisson and Dennis, 1977; Dickenson and Sullivan, 1987a,b). Although it is generally agreed that the first phase results from a direct action of formalin on nociceptive primary afferent fibers, the factors contributing to the second phase have not been fully defined. In the present report, we observed a significant SPR internalization in lamina I of the spinal dorsal horn at the end of the first phase (8 min), a time where there is almost no detectable peripheral edema. At 60 min after the initial injection of formalin, SPR internalization was still observed in a majority of lamina I SPR-IR neurons, at which time significant peripheral edema has developed.
It is of interest to compare this pattern of internalization with that produced by a single injection of capsaicin (Mantyh et al., 1995). Capsaicin induces a rapid, marked SPR internalization that is confined to lamina I neurons, and this SPR internalization is largely resolved 60 min after capsaicin injection. In contrast, 60 min after formalin injection there remains a significant SPR internalization in lamina I neurons, suggesting that there is an ongoing release of SP from primary afferent terminals. These results suggest that during both the first and second phases of the formalin response there is ongoing primary afferent input from C fibers and release of SP in the spinal cord. These data are in accord with previous reports demonstrating that SPR antagonists could block both the first and second phases of the formalin response in spinal neurons (Chapman and Dickenson, 1993) and that peripheral injection of local anesthetics after the end of the first phase reduces electrophysiological, behavioral, and anatomical correlates of the second phase (Dickenson and Sullivan, 1987a; Taylor et al., 1995; Puig and Sorkin, 1996; Abbadie et al., 1997a). These results provide further evidence that a major component of the second phase of the formalin response is caused by ongoing activity of primary afferent neurons. Together these results suggest that the neurochemical signature of acute inflammation is very similar, although with a longer time course, to what is produced by a brief chemical noxious stimulus, i.e., a stimulus that produces acute pain under nonpathological conditions.
Short-term inflammatory pain
Within 3 hr after carrageenan injection, peripheral edema, allodynia, and hyperalgesia had fully developed. In this condition, SPR internalization was observed early, i.e., 10 min after carrageenan injection, in dendrites and in a few lamina I SPR-IR neurons. This indicates that SP release occurs in the early stage of carrageenan inflammation. However, there is a major difference between carrageenan-induced inflammation (short-term) and formalin-induced inflammation (acute). Specifically, we found that formalin injection induced SPR internalization in lamina I neurons even at 1 hr after injection, whereas there was no evidence of ongoing SPR internalization at 3 hr after carrageenan injection. This lack of ongoing SPR internalization suggests either that there is no significant release of SP (using SP-induced SPR internalization as an assay of SP release) from primary afferents, even though a significant edema has developed at this time point, or that the system had desensitized, i.e., the receptor no longer responds. This absence of ongoing SPR internalization agrees with the observation that there is no spontaneous activity of dorsal horn neurons 3 hr after carrageenan injection (Stanfa et al., 1992; see however, Kocher et al., 1987), although a large number of spinal neurons express the Fos protein in both laminae I-II and III-IV 3 hr after carrageenan injection (Honore et al., 1995). If spinal Fos induction is a reflection of neuronal activation (Hunt et al., 1987; Munglani and Hunt, 1995; Doyle and Hunt, 1999), it seems reasonable to conclude that there must be maintained nociceptive input from the periphery to the spinal cord during the development of carrageenan inflammation. Whether this is caused by SP, released immediately after carrageenan injection (the Fos protein half-life is ∼2 hr), or other neurotransmitters such as excitatory amino acids is unclear.
What is apparent at 3 hr after carrageenan injection is that normally non-noxious mechanical stimulation of the carrageenan-inflamed hindpaw, which does not induce SPR internalization in normal animals, now induces massive SPR internalization in lamina I neurons. Furthermore, noxious mechanical stimulation, which only induces SPR internalization in lamina I in normal animals, now induces SPR internalization in neurons located in laminae I-II and III-IV.
A key question raised by these observations is whether the activation of these laminae III-IV SPR-IR neurons is caused by increased release and/or diffusion of SP from terminals that reside in laminae I-II or from de novo synthesis and release of SP from primary afferent neurons that terminate in laminae III-IV. After peripheral inflammation, an increase in SP synthesis by small DRG neurons that normally synthesize SP (Donnerer et al., 1993; Galeazza et al., 1995;Abbadie et al., 1996) as well as SP synthesis by large DRG neurons has been reported (Neumann et al., 1996). Release of SP by Aβ fibers could explain the SPR internalization observed in laminae III-IV after non-noxious stimulation in long-term inflammatory pain (Abbadie et al., 1997b). However, 3 hr after carrageenan injection, there would appear to be insufficient time for de novo synthesis and transport of SP from primary afferent cell body to the terminals in the spinal cord. These data suggest that the SPR internalization that is observed in lamina I after normally innocuous stimulation and the increased SPR internalization that is observed in laminae I-II and III-IV neurons after noxious stimulation is caused primarily by peripheral sensitization that manifests itself as a greater release and diffusion of SP from primary afferent neurons that already expressed SP. This increase in SP release from primary afferent fibers could thus lead to SP diffusing significantly greater distances, resulting in a switch from primarily synaptic to volume neurotransmission (Agnati et al., 1995; Zoli et al., 1998). Because SP would diffuse and interact with SPRs at both synaptic and extrasynaptic sites this increased release of SP from primary afferent fibers could also explain why there is significantly greater SPR internalization in lamina I SPR neurons after noxious stimulation under inflammatory conditions. Based on these observations, we suggest that this neurochemical signature of short-term inflammation is characterized by a lack of spontaneous SP release from primary afferents as reflected by the lack of ongoing SPR internalization, a lack of SPR upregulation, and a switch from synaptic to volume transmission so that there is an increase in both the number and the location of SPR-IR spinal neurons that are activated in response to innocuous or noxious stimuli.
Long-term inflammatory pain
CFA-induced unilateral inflammation and adjuvant-induced polyarthritis are two of the most commonly used models of long-term inflammatory pain. They elicit peak symptoms at 3 and 21 d, respectively. Similarities in the neurochemical signature of short- and long-term inflammatory pain include the lack of ongoing SPR internalization in basal unstimulated condition and an increase in the number and location of the spinal neurons that showed SPR internalization in response to either normally non-noxious or noxious stimuli.
The major difference in the spinal cords of animals with short- versus long-term inflammation is that in long-, but not short-term inflammation, there is a significant upregulation of the SPR on neurons in lamina I of the spinal cord. The increase in SPR mRNA observed in the spinal cord several days after peripheral inflammation has been reported to be blocked by morphine or SPR antagonists (Noguchi et al., 1988; McCarson and Krause, 1994, 1995, 1996), suggesting that SP release and/or SPR activation is necessary for SPR upregulation. However, in both chronic inflammatory pain, which is associated with an increase in SP in primary afferents (Lembeck et al., 1981; Donaldson et al., 1992), and in nerve injury, which is associated with a decrease in SP in primary afferents (Noguchi et al., 1989; Garrison et al., 1993), there is an increase in SPR immunoreactivity in lamina I of the spinal dorsal horn (Abbadie et al., 1996). Additionally, whereas cAMP has been reported to be involved in the regulation of SPR expression, SPR activation leads to the production of inositol phosphates, suggesting that SP is not the major regulator of SPR expression. These findings suggest that while SP could contribute to SPR upregulation, other neurotransmitters, acting directly on SPR-IR neurons or indirectly via the release of yet unknown factors, must be involved.
If there is a significant upregulation of the SPR in lamina I neurons in long-term inflammation, does it alter the response properties of these neurons? Several electrophysiological studies have shown that the response of spinal cord neurons to peripheral stimuli increases in an inflammatory pain state (Hylden et al., 1989; Haley et al., 1990;Simone et al., 1991; Dougherty et al., 1992; Stanfa et al., 1992; Urban et al., 1993; Neugebauer et al., 1994). This increased responsiveness is hypothesized to be largely mediated by a facilitated transmission through the NMDA receptor. SPR activation leads to the generation of diacyl glycerol and inositol triphosphate, inducing an increase in intracellular calcium and a synergistic facilitation of the activity of the protein kinase C. In turn, protein kinase C induces phosphorylation of the NMDA receptors, counteracting the magnesium block and allowing NMDA receptors to operate at a more negative potentials (for review, see Urban et al., 1994; Yaksh et al., 1995; Urban and Gebhart, 1998;Millan, 1999). These data suggest that SPR activation enhances NMDA receptor-mediated events and that the co-joint activation of SPR and NMDA receptors leads to increased neuronal excitability. The SPR upregulation observed in long-term inflammatory pain states may therefore contribute to the central sensitization observed in long-term inflammatory pain.
Conclusions
Previous experimental and clinical studies have suggested that there are distinctive differences between acute and chronic pain, including the shift from the sensitization of primary afferent neurons to a sensitization of spinal cord neurons. What is unique about the present approach is the ability to visualize and quantify neurochemical changes at the single and intracellular level as a pain “moves” from the acute to the long-term state. These results suggest that SPR internalization might serve as a marker of the contribution of ongoing primary afferent input to acute/persistent pain states. Using a similar approach to understand the changes that other neurotransmitter/receptor systems undergo as a pain moves from the acute to the chronic state should provide significant insight into the mechanisms involved in the generation and maintenance of chronic pain and may lead to novel therapies to control different pain states.
Footnotes
This study was supported by the National Institutes of Health, National Institute of Neurological Diseases and Stroke Grant 23970, National Institute on Drug Abuse Grant 11986, National Institutes of Health Training Grant DE07288, a Department of Veterans Affairs Merit Review, the Spinal Cord Society, and the Association Francaise pour la Recherche Therapeutique.
Correspondence should be addressed to Dr. Patrick W. Mantyh, Neurosystems Center, 18–208 Moos Tower, 515 Delaware Street, Minneapolis, MN 55455.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}