

Abstract
The cellular and network mechanisms of the transition of brief interictal discharges to prolonged seizures are a crucial issue in epilepsy. Here we used hippocampal slices exposed to ACSF containing 0 Mg2+ to explore mechanisms for the transition to prolonged (3–42 sec) seizure-like (“ictal”) discharges. Epileptiform activity, evoked by Shaffer collateral stimulation, triggered prolonged bursts in CA1, in 50–60% of slices, from both adult and young (postnatal day 13–21) rats. In these cases the first component of the CA1 epileptiform burst was followed by a train of population spikes at frequencies in the γ band and above (30–120 Hz, reminiscent of tetanically evoked γ oscillations). The γ burst in turn could be followed by slower repetitive “tertiary” bursts. Intracellular recordings from CA1 during the γ phase revealed long depolarizations, action potentials rising from brief apparent hyperpolarizations, and a drop of input resistance. The CA1 γ rhythm was completely blocked by bicuculline (10–50 μm), by ethoxyzolamide (100 μm), and strongly attenuated in hyperosmolar perfusate (50 mm sucrose). Subsequent tertiary bursts were also blocked by bicuculline, ethoxyzolamide, and in hyperosmolar perfusate. In all these cases intracellular recordings from CA3 revealed only short depolarizations. We conclude that under epileptogenic conditions, γ band oscillations arise from GABAAergic depolarizations and that this activity may lead to the generation of ictal discharges.
Cellular and network mechanisms of epileptiform discharges lasting a few hundred milliseconds, resembling interictal discharges, are understood in detail, largely because of experiments in vitro (Traub and Wong, 1982; Hablitz, 1987;Mody et al., 1987; Tancredi et al., 1990; Köhling et al., 1994;Gloveli et al., 1995). In the hippocampus they result from the interplay of intrinsic currents and synaptic interconnections in CA3 (Traub and Wong, 1982; Miles and Wong, 1986; Traub et al., 1994,1996a). Prolonged seizure-like (>2 sec; ictal) activity rarely occurs in adult slices (Anderson et al., 1986; Rafiq et al., 1993; Stasheff et al., 1993a; Traub et al., 1996a; Borck and Jefferys, 1999), but more often in juvenile tissue (Hablitz, 1987; Swann et al., 1993; Gloveli et al., 1995). Most reports on ictal activity in slices implicate prolonged, glutamatergic depolarizations variously depending on NMDA receptors [0 Mg2+ and electrographic “seizures” (Rafiq et al., 1993; Stasheff et al., 1993b; Traub et al., 1994)], AMPA receptors [4-aminopyridine (Traub et al., 1995)], or combined AMPA, NMDA and metabotropic glutamate receptors (mGluRs) [GABAA antagonists (Swann et al., 1993; Traub et al., 1996a; Merlin, 1999; Borck and Jefferys, 1999)].
Epileptic activity is often attributed to imbalanced glutamatergic excitation and GABAergic inhibition. However, GABAergic transmission remains effective in some epilepsy models and in epileptogenic human tissue (Prince and Wilder, 1967; Elger and Speckmann, 1983; Tancredi et al., 1990; Michelson and Lothman, 1992; Benardo, 1993; Westerhoff et al., 1995b; Esclapez et al., 1997; Köhling et al., 1998a). Functional GABAergic transmission does not necessarily mean inhibition. GABA can depolarize under tetanic stimulation, neuronal trauma, GABA uptake block, 4-aminopyridine, and during ontogenesis (Ben-Ari et al., 1989; Perreault and Avoli, 1992; Grover et al., 1993; Van den Pol et al., 1996; Kaila et al., 1997; Davies and Shakesby, 1999). This helps explain the susceptibility of juvenile tissue to ictal activity (Luhmann and Prince, 1991; Swann et al., 1992; Sutor and Luhmann, 1995), until close to maturity (Köhling et al., 1998b). Given that GABA can excite, the question is whether it does so under pathological, epileptogenic, conditions (Perreault and Avoli, 1992;Stasheff et al., 1993b; Higashima et al., 1996; Perkins and Wong, 1996).
γ frequency (30–100 Hz) oscillations are associated with cognition (Traub et al., 1999; Singer, 1999). One experimental form of γ oscillation is triggered in hippocampal slices, especially in CA1, by tetanic stimulation (Traub et al., 1996b). The main source of the prolonged depolarization during this γ oscillation was initially identified as mGluRs (Whittington et al., 1997), but several studies now implicate GABAergic depolarization (Grover et al., 1993; Kaila et al., 1997; Bracci et al., 1999; Cobb et al., 1999; Vreugdenhil et al., 1999). Ephaptic (field) effects provide the tight synchronization of neuronal firing into population spikes (Bracci et al., 1999). Here we put forward the hypotheses that similar discharges occur on the tail of epileptiform bursts induced by Mg2+withdrawal, and we explore the possibility that such oscillations support the transition to ictal activity in adult and juvenile hippocampal slices.
MATERIALS AND METHODS
Transverse hippocampal slices (450 μm, n = 89) were prepared from adult (>30 d postnatal; 90–350 gm;n = 34) and juvenile (9–21 d postnatal; 21–63 gm;n = 31) male Wistar or Sprague Dawley rats (anesthetized with ketamine and medetomidine). No strain differences were found in this study, so experiments from both strains were pooled. Slices were maintained in an interface-type chamber at 32–34°C in gassed (5% CO2 and 95% O2) artificial CSF (ACSF) containing (in mm): NaCl 125, NaHCO3 26, CaCl22, KCl 3, NaH2PO4 1.25, MgCl2 1, and glucose 10.
Field and membrane potential recordings were obtained from CA3 and CA1 strata pyramidalia with blunt glass micropipettes (1–2 MΩ) filled with ACSF placed onto the slice surface and sharp microelectrodes (50–80 MΩ) filled with 2 m potassium methylsulphate, using a DC-coupled custom-made field potential amplifier and an Axoclamp 2B amplifier in bridge mode, respectively. In some experiments, three extracellular field potential electrodes were placed along the CA1 stratum pyramidale (interelectrode distances 300–400 μm) to investigate the spatial extent of oscillations within this subfield. Intracellular recordings were accepted provided the resting membrane potential was at, or negative to, −55 mV; mean resting levels were −67.2 ± 1.1 and −73.7 ± 3.7 mV (adult CA1 and CA3,n = 5 each), and −65.5 ± 2.1 and −64.5 ± 0.5 mV (juvenile CA1 and CA3, n = 11 and 5, respectively). A bipolar Nichrome wire stimulating electrode was used to stimulate Schaffer collaterals. Its position was approximately equidistant (400–500 μm) from both field potential recording electrodes. Stimulation intensity was set at 2× the intensity required to yield maximal population spikes (range, 30–100 V for 0.2 msec duration stimuli). Single or double (100 msec interstimulus interval) stimuli were delivered at fixed intervals of 10 min; 30 min for those slices in which spreading depression occurred. Before drug application, at least four of such stimulations were made to ensure that the response was uniform and stable. In some experiments, tetanic stimuli (20 at 100 Hz) were applied under control conditions to test whether slices generated oscillatory behavior.
Epileptogenic conditions were established by omitting Mg2+ from the perfusate. Instantaneous frequency of oscillations was calculated from the interval between the negative peaks of consecutive population spikes at different times after the stimulus. Drugs used were the GABAAreceptor antagonist bicuculline (50 μm); the membrane-permeable carbonic anhydrase blocker ethoxyzolamide (EZA; 100 μm) (Autere et al., 1999), and gap junction blockers (Perez-Velazquez et al., 1994; Ishimatsu and Williams, 1996; Draguhn et al., 1998) halothane (10 mm) and carbenoxolone (100 μm; added from stock solution dissolved in DMSO to yield a final concentration of 0.1%). Whenever DMSO was used as solvent, 0.1% DMSO was added as a control and had no effect on the activity. Osmolality changes were induced by addition of sucrose (50 mm) or distilled water (10%) to ACSF. All values are expressed as means ± SE.
RESULTS
Oscillations in the γ frequency range occur in the wake of epileptiform field potentials
Withdrawal of Mg2+ from the perfusate resulted in typical spontaneous epileptiform field potentials in adult hippocampal slices, consisting of a primary burst often followed by two to nine afterdischarges or secondary bursts, both in CA3 and CA1, as previously described by several groups (Traub et al., 1994; Whittington et al., 1995). In 9 of 25 slices, an additional field potential discharge could be observed in the wake of these epileptiform bursts, which was restricted to the CA1 subfield (Fig.1A). This discharge consisted of a barrage of population spikes reminiscent of γ oscillations elicited by tetanic stimulation (Fig.1B; Bracci et al., 1999). Pooling data from nine slices from different animals, ranging in age from P35 to P55, the instantaneous frequency of this oscillation was ∼70 Hz, rising to ∼90 Hz and then tailing off to ∼60 Hz (Fig. 1C), thus lying in the γ frequency band (30–100 Hz; Bracci et al., 1999). In one slice, such oscillations also appeared spontaneously and independently of epileptiform field potentials (data not shown). The oscillations typically lasted for >1 sec (1.6 ± 0.3 sec,n = 9), generally far outlasting the epileptiform burst in CA3 (Fig. 1). Intracellular recordings from CA1 pyramidal neurons revealed that they were associated with prolonged depolarizations of 10–25 mV, lasting 1–2 sec (n = 4; Fig. 1). These were sometimes sufficient to trigger action potentials synchronized with the population spikes, rising without any visible EPSP (Fig. 1). Pyramidal neurons in CA3 remained unaffected by these phenomena (data not shown).

γ frequency oscillations occur spontaneously under epileptogenic conditions in the CA1 subfield. A, Epileptiform field discharge induced by Mg2+withdrawal in an adult hippocampal slice. Field potential (fp) and membrane potential (ic) recordings from CA1 and CA3 strata pyramidale. In those 9 of 25 slices with γ oscillations on the tail of spontaneous epileptic bursts, γ occurred every second or third event, one of which is shown here.B, Same recording as in A on an expanded time scale and taken from the trace as defined by the dot.C, Plot of instantaneous frequency of oscillations obtained from nine slices. Data from a single representative oscillation are included for each slice.
γ oscillations can precede seizure-like discharges
The prolonged depolarization observed in the wake of spontaneous epileptiform bursts in CA1 should provide an additional excitatory drive to CA1 neurons, and could, in principle, promote the transition from interictal burst to ictaform or seizure-like discharges (Swann et al., 1993; Traub et al., 1996a). This prompted us to search for a possible link between these two phenomena. For this purpose, we used single or paired electrical stimuli to the Schaffer collaterals which induced γ oscillations reliably in low Mg2+ (Fig.2). In adult slices, evoked γ oscillations had an average duration of 1.9 ± 0.18 sec and average population spike amplitude of 2.42 ± 0.51 mV (n = 18). The evoked oscillations again were always restricted to CA1 and were accompanied by prolonged (6.3 ± 2.3 sec; n = 3) neuronal depolarizations in CA1, but only short paroxysmal depolarization shifts lasting <1 sec in CA3 (Fig.2A). Spontaneous interictal epileptiform field potentials usually were interrupted by a long interval after each prolonged oscillatory discharge (Fig. 2A2). Most importantly, however, in 45% of the slices, prolonged afterdischarges or seizure-like bursts ensued after the oscillations (see also tetanically evoked γ in Traub et al., 1999, their Fig. 9.4). In adult rats the γ field potential oscillations and the seizure-like bursts both were always restricted to CA1.

Stimulus-induced γ frequency oscillations can precede seizure-like events under epileptogenic conditions. Field potential (fp) and membrane potential (ic) recordings from CA1 and CA3 strata pyramidale.A, Adult hippocampal slices reveal typical oscillation induced by paired stimuli (insets) in CA1, but not in CA3. A1, The oscillation is associated with a prolonged depolarization of a CA1 neuron, and, in this example, with prolonged afterdischarges or a seizure-like event in CA1, but not CA3. The region in the dashed box is expanded below, into thelarge dashed box, to show the early γ component and the onset of the epileptiform afterdischarges (*; action potentials are truncated on the ic trace). A2, CA3 neurons only show short depolarizations during the CA1 γ oscillation. In this experiment, no ictal activity followed the oscillation, although interictal bursts were present (calibrations same asA1). B, Juvenile hippocampal slices reveal typical oscillation (insets) in CA1, which is again not apparent in CA3. B1, The oscillation (from a P21 rat) is associated with a prolonged depolarization of a CA1 neuron and with prolonged afterdischarges or a seizure-like event in CA1, but not CA3. B2, CA3 neurons only show short depolarizations during γ oscillation in CA1, in spite of their involvement in the later ictal activity (from a P16 rat; calibrations same asB1). Note that ictal activity followed the oscillation in both CA1 and CA3 only in juvenile slices.
Juvenile tissue is known to be more susceptible to ictaform activity (Hablitz, 1987; Swann et al., 1993; Gloveli et al., 1995), so we extended our study to slices from neonatal rats of postnatal days 9–21 (P9–P21). Surprisingly, in slices from animals younger than P13 (n = 12), no γ oscillations could be elicited at all, either under epileptogenic conditions or with tetanic stimulation under control conditions. A recent report describes γ and high-frequency rhythms in relatively thick, submerged neonatal slices (Palva et al., 2000), but probably it is a different rhythm from the present in that it was spontaneous rather than evoked and was much weaker. Spontaneous interictal discharges did occur in juvenile tissue of all age groups, albeit more rarely than in adult tissue (25% of the slices). In preparations from P13 onward, however, single or paired stimuli evoked oscillations in all slices (n = 41), with features similar to those found in adult preparations (Fig.2B). Moreover, in 60% of the >P13 slices, seizure-like discharges could be observed in CA1, 73% of which, unlike adult preparations, also extended into CA3 (Fig.2B2). Apart from these seizure-like discharges, spreading depressions occurred in some slices. These could either start after the oscillations, or more frequently, after a seizure-like event. Spreading depression could occur in one hippocampal subfield independently of the other; it appeared in CA3 in 35% of the slices, and in CA1 in only 21% of the cases (compare Figs. 9, 10).
A typical field potential oscillation in a juvenile slice preparation is shown in more detail in Figure 3. After a double stimulus, the oscillation started 50–100 msec after the population spike (Fig. 3A). In CA3, only antidromic population spikes (Fig. 3A) or an epileptiform field potential (Fig. 4) could be observed. The oscillation typically lasted 1–2 sec (1.60 ± 0.32 sec;n = 41) and usually consisted of negative-going population spikes with an average amplitude of 4.6 ± 0.5 mV (Fig.3A). In Figure 3C, the average instantaneous frequency of the oscillation is plotted against time. As the graph demonstrates, the oscillations initially were in the γ frequency band (30–120 Hz) and then slowed to the β band (10–30 Hz), as previously described for tetanically evoked γ oscillations (Bracci et al., 1999). During these responses CA1 pyramidal cells experienced prolonged depolarizations, which usually outlasted the field oscillation. On average, this lasted 3.3 ± 1.6 sec (n = 5) in cases when no ictaform activity or spreading depressions followed. Action potentials synchronous with the population spikes were observed in five of eight neurons (Fig. 3B). Brief negative deflections often preceded the action potentials; in both tetanically induced oscillations and low-Ca2+ field bursts such deflections have been interpreted as evidence of field or ephaptic effects, because when the local extracellular field was subtracted they were revealed as net transmembrane depolarizations (Fig. 3B;Haas and Jefferys, 1984; Taylor and Dudek, 1984;Bracci et al., 1999). Such negativities could be observed, synchronous with the field potential deflections, and independently of action potentials in six of eight neurons; frequent synaptic potentials also occurred. A 50–70% drop in input resistance could be observed, which waned within 2–4 sec, in parallel with the prolonged depolarization (Fig. 4). Reduction of input resistance was a key factor in the identification of depolarizing GABA, rather than mGluRs, as the source of tetanically evoked depolarization (Kaila et al., 1997; Taira et al., 1997; Bracci et al., 1999; Cobb et al., 1999; Smirnov et al., 1999; Vreugdenhil et al., 1999).

γ frequency population spikes are associated with transient membrane potential negativities in CA1.A, This cell, in a slice from a juvenile (P15) rat, fired infrequently during the evoked γ oscillation in Mg2+-free ACSF [field potential (fp) and membrane potential (ic) recordings from CA1 and CA3 strata pyramidale]. B, Same recording as in A on an expanded time scale and taken from the trace as defined by the dot reveals transient intracellular negativities during each population spike and preceding the one action potential shown. C, Plot of instantaneous frequency of oscillations obtained from 41 slices. Data from a single representative oscillation were included for each slice.

Neuronal input resistance drops during prolonged depolarization associated with field oscillation in CA1. Field potential (fp) and membrane potential (ic) recordings from CA1 and CA3 strata pyramidale. Input resistance of a CA1 neuron determined by −0.3 nA current injection (200 msec, 3 Hz).
Prolonged depolarizations develop gradually with Mg2+ washout
Several changes in synaptic activity occur after washout of Mg2+. NMDA receptor-mediated currents increase (Traub et al., 1994) because of removal of the well documented voltage-dependent block of the receptor by Mg2+ (Mayer et al., 1984; Nowak et al., 1984). In addition GABAA receptor-mediated inhibition erodes gradually (Whittington et al., 1995). In view of these findings, we investigated the temporal development of membrane potential changes with paired stimuli and progressive washout of Mg2+ in a cohort of P13–P21 rats.
CA1 neurons typically showed a 0.5–2 sec hyperpolarization after evoked EPSPs and action potentials (Fig.5A). After 30 min of washing out Mg2+, the hyperpolarization evoked by the first stimulus became smaller and, after the second, converted to a depolarization (Fig. 5A), a sequence of events found in all neurons after 45 min washout (Fig. 5B). In the illustrated neuron no hyperpolarization remained after 60 min washout; it was replaced by a prolonged depolarization, which was associated with prominent γ oscillations in the field potential (Fig. 5A). This effect was even more pronounced after 120 min washout, when the maximal depolarization typically reached 15–20 mV (Fig.5B). Qualitatively and quantitatively similar findings were also seen in adult preparations (>P21; data not shown). CA3 neurons under these conditions only show membrane potential changes comparable with those found for CA1 neurons in control ACSF (data not shown). The development of the prolonged depolarization correlated with the occurrence of ictaform activity. After <90 min washout of Mg2+, seizure-like discharges could be observed in only 22% of cases; after >90 min washout, ictal activity occurred in 60% of cases.

Prolonged depolarization associated with γ oscillations develops gradually with Mg2+ washout.A, Typical membrane potential changes of a juvenile CA1 neuron in response to paired (100 msec interstimulus interval) electrical stimuli to Schaffer collaterals in control ACSF (black line), after 30 min (dark gray line), and 60 min Mg2+-withdrawal (light gray line). Field (fp) and membrane potential (ic; action potentials are truncated) recordings in CA1 and CA3 strata pyramidale. B, Membrane potential changes at different time points of juvenile CA1 neurons (n = 16) in response to paired stimuli in control ACSF, after 45 and 120 min Mg2+ withdrawal. Time 0 msec denotes the end of the second stimulus.
The gradual development of γ oscillations under epileptogenic conditions is also mirrored in a spatial spread of oscillatory field discharges. In three experiments, the spatial extent of oscillations within CA1 was judged with three field potential electrodes positioned at either end and in the middle of the CA1 region. In normal ACSF, double stimuli elicited no oscillation. Tetanic stimuli, however, led to γ oscillations of nearly equal amplitude at all three locations (Fig. 6). Single population spikes appeared to be initiated first at the site closest to the stimulation electrode (Fig. 6). By contrast, after 60 min Mg2+ withdrawal, a paired stimulus now generated large-amplitude oscillations, which were typically absent at the site closest to the stimulation electrode and were initiated at the subicular end of CA1 (Fig. 6).

γ oscillations are initiated at the subicular end of CA1 under epileptogenic conditions. Field potential recordings from CA1. Typical γ oscillation in CA1 induced by tetanic stimulation (100 Hz, 200 msec) of Schaffer collaterals (black line) in a juvenile (P17) hippocampal slice. Oscillations of nearly equal amplitude occur in the CA1 area most proximal to the stimulation electrode (CA1 c) at the border of CA1 and CA2, in the middle of the CA1 subfield (CA1 b), and at its subicular end (CA1 a). Single spikes first arise at CA1c. By contrast, paired stimulus-induced γ oscillation in Mg2+-free ACSF in the same slice is generated only in CA1 a and b, and is initiated at the subicular end (gray line).
Role of field effect (ephaptic) interactions
Small negative membrane potential fluctuations, coinciding exactly with the negative peaks of oscillatory field potentials (Fig. 3), have been interpreted as a sign of ephaptic interactions and were found to be of critical importance for the generation of synchronous tetanically evoked γ oscillations (Bracci et al., 1999). Such field effects are most prominent when the extracellular resistance is relatively high (Korn and Faber, 1980; Jefferys, 1995; Vigmond et al., 1997). Consequently, field effects can be manipulated by changing the osmolality of the extracellular fluid. Hyperosmolar solutions make cells shrink and widen the extracellular space and thus decrease its resistance; conversely, hypo-osmolar solutions induce cell swelling and an increase of extracellular resistance. Hyperosmolar ACSF reversibly blocked all oscillatory activity and subsequent epileptiform afterdischarges (n = 3; Fig.7), as already reported for tetanically induced oscillations and nonsynaptically mediated 0 Ca2+ epilepsy (Dudek et al., 1990; Bracci et al., 1999). The neuronal depolarization, as demonstrated in the neuron shown in Figure 7, was still present, but had a much shorter duration (∼200 msec) than the prolonged depolarization before the manipulation of osmolality. Hypo-osmolar ACSF increased the amplitude of oscillations and prolonged the ictal discharges (data not shown;n = 1).

γ frequency oscillations and seizure-like events are abolished by expansion of the extracellular space. A, γ oscillation and subsequent ictal discharge, evoked by paired stimuli, in Mg2+-free ACSF in a juvenile (P17) hippocampal slice before and after perfusion with hyperosmolar (30 mm sucrose added) ACSF. Field potential (fp) and membrane potential (ic) recordings from CA1 and CA3 strata pyramidale.B, Same recording as in A on an expanded time scale and taken from the trace as defined by the dotted rectangle during the γ oscillation.
Oscillations and prolonged depolarizations are GABA mediated and provide a possible mechanism for the generation of seizure-like events
We tested whether the γ oscillations observed under epileptogenic conditions depend on GABA, similarly to tetanically induced γ oscillations (Bracci et al., 1999), and whether the associated ictal events were affected by manipulation of the inhibitory system. Blockade by bicuculline and ethoxyzolamide provided evidence that the ∼15–20 mV, >3 sec depolarization evoked by tetanic stimulation resulted from a massive release of GABA (Kaila et al., 1997; Taira et al., 1997; Bracci et al., 1999; Cobb et al., 1999;Smirnov et al., 1999; Vreugdenhil et al., 1999).
The membrane-permeable carbonic anhydrase blocker EZA, leads to a drop of HCO3− availability, and hence reduces its contribution to the depolarizing GABAA response (Kaila et al., 1997; Taira et al., 1997; Autere et al., 1999). In the present study, application of EZA (100 μm;n = 3) for 30 min greatly decreased the γ oscillation and blocked seizure-like events (Fig.8).

γ frequency oscillations and seizure-like events are abolished by permeable carbonic anhydrase blocker ethoxyzolamide (100 μm). Typical γ oscillation and subsequent ictal discharge, evoked by paired stimuli, in Mg2+-free ACSF in a juvenile (P15) hippocampal slice before and after perfusion with ethoxyzolamide. Field potential recordings (fp) from CA1 and CA3 strata pyramidale.
Blockade of GABAA receptors by bicuculline (10–50 μm; 30 min; n = 7) in this study had one of two distinct consequences. In four cases, all from P13–P21, in which CA3 was involved in the ictal activity, this activity persisted, whereas γ oscillations in CA1 were blocked. In these cases the ictal bursts were shorter and had a morphology different from those in the absence of bicuculline (Fig.9A). In the three other P13–P21 cases, ictal activity was restricted to CA1, and γ oscillations again were blocked, but in these cases the seizure-like events were abolished, whereas interictal-type discharges persisted (Fig. 9B). An experiment on an adult slice replicated the latter result; γ oscillations and ictal activity within CA1 were both blocked by bicuculline. These results suggest that ictogenesis depends on differential mechanisms in CA1 and CA3. In cases in which spreading depressions occurred after the ictal discharges, these were abolished by bicuculline (Fig. 9).

γ frequency oscillations are abolished by GABAA receptor blockade, whereas seizure-like events persist if they are generated in CA3. Field potentials in CA1 and CA3 strata pyramidale evoked by paired stimuli, separated by 100 msec, to the Shaffer collaterals. γ oscillation with following ictal discharges are seen either in both CA1 and CA3 (A, from a P18 rat) or only in CA1 (B, from a P19 rat) in Mg2+-free ACSF in juvenile hippocampal slices before and after perfusion with bicuculline (50 μm). Note that interictal activity is generated in B and that spreading depressions are abolished by bicuculline in both A andB.
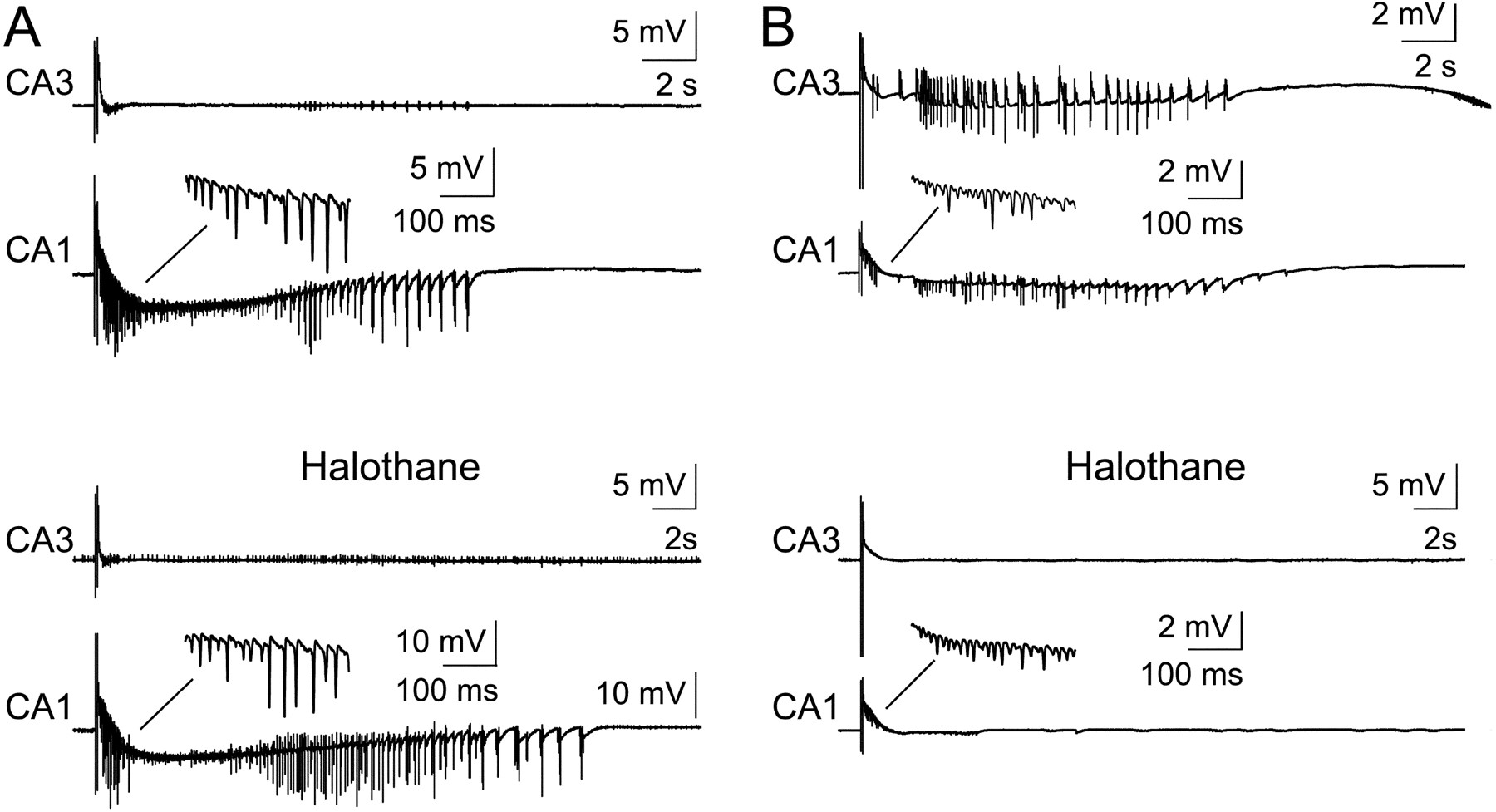
Gap junctional coupling has been implicated in shaping prolonged low-Mg2+ discharges that originate in CA3 (Köhling et al., 1999). Here we used the gap junction blockers halothane (10 mm;n = 5) and carbenoxolone (100 μm;n = 2) to determine whether they selectively affected either (1) oscillations or (2) seizure-like events. Again, the effect of halothane depended on which region generated ictal events. If CA1 alone showed prolonged discharges, halothane had no influence either on the γ oscillations or on the seizure-like event (n = 2; Fig.10A). In contrast, in slices with ictal activity in both subfields, both halothane and carbenoxolone blocked this activity but did not influence the appearance of γ oscillations (n = 3 and 2, respectively; Fig. 10B).

γ frequency oscillations remain unaffected, but seizure-like events are abolished by gap junction blockade if generated in CA3 but persist if they are generated in CA1 only. Field potential recordings from CA1 and CA3 strata pyramidale. Typical paired stimulus-induced γ oscillation and subsequent ictal discharges in CA1 only (A, P18 rat) and both in CA1 and CA3 (B, P19 rat) in Mg2+-free ACSF in a juvenile hippocampal slice before and after perfusion with halothane (10 mm).
CA1 and CA3 both can initiate seizure-like events
These pharmacological manipulations suggest that CA1 and CA3 possess different mechanisms for the generation of seizure-like discharges, which in CA1 depend on GABAergic depolarization, and in CA3 on gap junctions, presumably in conjunction with recurrent excitation (Traub et al., 1994). If this were so, one would expect that either region can initiate discharges and that the subfields should compete for the leading or pacemaking role in this process. We therefore analyzed all experiments (at P13–P21) in which both subfields showed ictal events for any indication of such “competition” by evaluating the time lag between each afterdischarge in the CA1 and CA3 recordings. Figure 11 gives a typical example of such a discharge. The expanded insets of Figure 11Bshow that, in this example, it is the CA1 region that led for the first 19 afterdischarges, with very variable latencies ranging from 5 to 36 msec with respect to CA3 (Fig. 11C). Only from the 20th afterdischarge on, CA3 consistently led with a less variable time lag of 5–8 msec (Fig. 11C). Such a behavior, i.e., an initial lead of CA1 with subsequent lead of CA3, was seen in 31% of the slices. In 57% of the preparations, CA3 always led, and in 12% there was a continuous change of lead between CA1 and CA3.

Seizure-like discharges can be initiated in either CA3 or CA1 subfields. A, Typical paired stimulus-induced γ oscillation and consecutive seizure-like discharge in Mg2+-free ACSF in a juvenile (P16) hippocampal slice. Field potential (fp) and membrane potential (ic) recordings from CA1 and CA3 strata pyramidale. B, Expanded traces of the recording inA at time points corresponding to the fourth (a), fifth (b), and 102nd (c) afterdischarge. Dotted linesindicate onset of afterdischarge, revealing that CA1 leads ina and b, whereas CA3 leads inc. C, Plot of the time lag between onset of field potential afterdischarges between CA1 and CA3; negative values indicate CA1 is leading. Each dot represents an afterdischarge of the event shown in A.
DISCUSSION
Processes governing γ oscillations under epileptogenic conditions
The central observation here is that trains of population spikes at γ frequencies, in CA1, prolong 0 Mg2+interictal events previously shown to be initiated in CA3 (Mody et al., 1987; Colom and Saggau, 1994; Köhling et al., 1994; Köhling et al., 1999). These γ trains closely resemble tetanically induced γ oscillations in hippocampal slices (Whittington et al., 1997). Our current view is that depolarizing actions of GABA play a major role, particularly in the vicinity of the stimulation electrode (Kaila et al., 1997; Bracci et al., 1999; Vreugdenhil et al., 1999). Several lines of evidence suggest that under epileptogenic conditions of the present study the γ oscillations and concomitant neuronal depolarizations observed were also mediated by a massive, depolarizing release of GABA. (1) The oscillations observed here appeared solely in CA1 and showed the same frequency dynamics as the tetanically induced oscillations, starting in the γ band and then shifting to the β band. (2) They were associated with a reduction of neuronal input resistance. In this context, experiments showing no γ oscillations at recording sites very close to the stimulating electrode (Fig. 6) can be interpreted as being attributable to the highest GABA levels causing neurons to fail to fire because of a massive loss of resistance, as proposed for tetanically induced oscillations (Vreugdenhil et al., 1999). (3) γ oscillations under epileptogenic conditions, and accompanying depolarizations, were blocked by bicuculline and severely attenuated by ethoxyzolamide.
The synchronizing drive, which provides for neuronal firing and the generation of oscillatory population spikes, appeared to be ephaptic under epileptogenic conditions. Relevant observations in the present paper parallel those for tetanically induced oscillations (Bracci et al., 1999). (1) Membrane potential negativities appeared in precise synchrony with the field population spikes. (2) Action potentials sometimes arose from these negativities, not all cells generated spikes, and some produced partial spikes (compare Figs. 3 and 4). (3) Manipulations of osmolality changed the activity in the predicted manner. The role of ephaptic interactions is further emphasized by the fact that in preparations younger than P13, no γ oscillations can be elicited. In juvenile tissue, the extracellular space is known to be wide, and thus field effects are less likely (Lehmenkühler et al., 1993).
Why do γ oscillations arise under epileptogenic conditions?
Here we show that under epileptogenic conditions, brief epileptiform bursts, either evoked by single or paired electrical stimuli, or in some cases spontaneous, were sufficient to evoke γ oscillations. In control ACSF, single or paired stimuli never suffice to elicit oscillations, and instead tetanic stimulation is necessary. The drive for the initiation of an oscillation thus appears to be synchronous, synaptically mediated activity, either evoked or spontaneous. The increase in neuronal excitability because of Mg2+ withdrawal certainly plays an important role. As others have pointed out, it is probably brought about by: loss of surface charge (not an essential requirement;Jefferys and Traub, 1998), increased NMDA-mediated synaptic currents, and reduced activity of the Mg2+ dependent Na+/K+ ATPase (Anderson et al., 1986; Mody et al., 1987; Tancredi et al., 1990; Traub et al., 1994). Synchronous bursting under these conditions is facilitated and spreads rapidly within the neuronal population via recurrent excitation (Traub et al., 1996a). Presumably the epileptiform burst replaces the external tetanic stimulus in driving GABAergic interneurons (Bracci et al., 1999) and activates interneurons by local circuits (Knowles and Schwartzkroin, 1981; Esclapez et al., 1997). The prolonged presence of GABA then causes a depolarizing shift of the GABAA receptor reversal potential because of one or more of: accumulation of [Cl−]i, accumulation of [K+]o, or spillover of GABA to receptors with a greater HCO3− permeability (Kaila et al., 1997; Taira et al., 1997; Perkins, 1999; Smirnov et al., 1999; Staley and Proctor, 1999).
The increased excitability under epileptogenic conditions also involves interneurons, which should thus fire more readily than under normal circumstances (Domann et al., 1991). There are reports that inhibition is weakened in 0 Mg2+. Whittington et al. (1995) report an “erosion of inhibition” measured in CA3. LeBeau and Alger (1998) found a transient reduction in IPSPs in CA1, although the relative contributions of Cl− and HCO3− to IPSPs in these Cl−-loaded neurons may be consistent with the results reported here. Others found substantial inhibition in this model. Tancredi et al. (1990) showed that neuronal GABA-mediated postburst hyperpolarizations occur and that IPSPs could be elicited during the generation of epileptiform bursts in hippocampal slices.Westerhoff et al. (1995b) reported that, even after single 0 Mg2+-induced epileptiform bursts, GABA-mediated Cl− currents occurred with average durations of 500 msec, considerably longer than normal. Thus, we think that a widespread release of GABA occurs under epileptogenic conditions, triggered by synaptic mechanisms during the initial epileptiform burst. This is likely to be exacerbated by the ability of prolonged Mg2+ omission to cause downregulation of the KCC2 K+/Cl−cotransporter selectively in CA1 (Rivera et al., 1999), an effect that is particularly remarkable because low Mg2+ allosterically stimulates K+/Cl−cotransport (Jennings, 1999).
GABA as a possible ictogenic mechanism
One question that remains is why GABA in some instances can be ictogenic and why ictal activity was even blocked by bicuculline, a widely used epileptogenic substance. For the appearance of seizure-like events, a prolonged neuronal excitation, extending beyond the primary epileptiform discharge, is required (Swann et al., 1993; Traub et al., 1996a; Borck and Jefferys, 1999). This prolonged excitation is generally attributed to enhanced glutamatergic transmission of various kinds: local synaptic networks, direct actions of the epileptogenic manipulation, potentiation of mGluRs, or indirectly by GABAB-mediated reduction of inhibition (Anderson et al., 1986; Rafiq et al., 1993; Stasheff et al., 1993b; Swann et al., 1993; Traub et al., 1994, 1995; Merlin and Wong, 1997; Merlin, 1999;Motalli et al., 1999). Three reports, dealing with electro- graphic discharges in vitro, have speculated that actions of GABA may play a role in seizure generation in hippocampus, without, however, providing evidence for prolonged depolarizations (Stasheff et al., 1993a,b; Higashima et al., 1996).
How does the GABA-mediated depolarization initiate a seizure-like event in CA1? In some instances, ictal discharges remained restricted to CA1; indeed, Tancredi et al. (1990) reported that the isolated CA1 subfield can sustain 0 Mg2+ seizure-like activity when triggered by a brief single stimulus. Several processes may lead to ictogenesis in CA1. (1) Prolonged exposure to low-Mg2+ can cause downregulation of the KCC2 K+/Cl−cotransporter protein in CA1, resulting in a depolarizing shift of the GABAA receptor reversal potential in CA1 (Rivera et al., 1999). However, epileptic bursts have also been reported in CA1 after γ-frequency discharges evoked by tetanic stimulation (I. M. Stanford and J. G. R. Jefferys, unpublished observations, reported in Fig. 9.4 of Traub et al., 1999); these epileptic bursts occurred in normal Mg2+, which argues that the downregulation of KCC2 is not a necessary requirement. (2) A rise in extracellular K+ during the γ oscillation would increase excitability for some time after the oscillation terminated (Vreugdenhil et al., 1999). (3) Ephaptic interactions, which synchronize population spikes, are more powerful in CA1 than in CA3, because of the relatively small extracellular space (McBain et al., 1990). (4) Fi-nally, the repetitive, synchronous firing of CA1 pyramidal cells may lead to posttetanic and long-term potentiation of the excitatory synapses between them (Traub et al., 1998), thus setting up a network prone to repetitive epileptic bursts through mechanisms analogous to those identified in CA3 (Traub and Wong, 1982; Traub et al., 1987, 1993, 1994). Long-lasting GABAA-dependent depolarizations have been described in the entorhinal cortex exposed to 4-aminopyridine (Avoli et al., 1996; Lopantsev and Avoli, 1998); however, they did not produce the rhythmic γ band population spikes, found in CA1 in 0 Mg2+, which set the scene for the transition to final, prolonged component of the ictal discharges reported here.
Seizure-like events also occurred in CA3, either initially or secondary to CA1. The latter case could be because of antidromic, ectopic spikes generated in CA3 axons, as reported for another model by Stasheff et al. (1993a). Subsequently, recurrent excitation, already well described in CA3 (Traub and Wong, 1982; Traub et al., 1987, 1993, 1994; Knowles et al., 1987), may be potentiated and enable CA3 to lead ictal discharges by itself (Fig. 11B,C).
In summary, we propose that at least two distinct mechanisms of ictogenesis coexist in hippocampal slices under epileptogenic conditions: (1) recurrent excitation in CA3 (Traub et al., 1994), possibly involving gap junctional coupling (Köhling et al., 1999), and (2) GABAergic depolarizations in CA1. The coexistence of these mechanisms can account for the diverging results with application of bicuculline. In the former, GABA has antiepileptic properties, and hence bicuculline prolongs discharges (Tancredi et al., 1990; Traub et al., 1994), in the latter, GABA is proepileptic and hence bicuculline blocks bursts. These findings may explain why some antiepileptic, putatively GABA-promoting drugs, have occasionally been found to be proconvulsant in clinical cases (Schapel and Chadwick, 1996; Elger et al., 1998).
Footnotes
We thank the Wellcome Trust for supporting this work.
Correspondence should be addressed to John G. R. Jefferys, Division of Neuroscience (Neurophysiology), The Medical School, The University of Birmingham, Birmingham B15 2TT, UK. E-mail:j.g.r.jefferys{at}bham.ac.uk.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}