

Abstract
Ca2+ imaging and (perforated) patch recording were used to analyze the mechanism of GABA- and glycine-induced depolarizations in lumbar motoneurons of spinal cord slices from fetal rats. In fura-2 ester-loaded cells, the agonist-induced depolarizations increased [Ca2+]i by up to 100 nm. The GABA- and glycine-evoked [Ca2+]i transients were suppressed by bicuculline and strychnine, respectively. Their magnitude decreased by ∼50% between embryonic days 15.5 and 19.5. The [Ca2+]i increases were abolished by Ca2+-free superfusate and attenuated by ∼65% by nifedipine, showing that the responses were mediated by voltage-activated Ca2+ channels. The [Ca2+]i rises were potentiated by >300% immediately after removal of Cl− from the superfusate but recovered to values of 50–200% of control during repeated agonist administration in Cl−-free saline. Bumetanide gradually suppressed the [Ca2+]i increases by >75%. Subsequent removal of Cl− reconstituted the responses and increased, upon repeated agonist application, the peak [Ca2+]i rises to values above control. Removal of HCO3− from the Cl−-free (bumetanide-containing) superfusate reversibly abolished both the agonist-induced [Ca2+]i rises and depolarizations that were reestablished by formate anions. In Cl−-containing superfusate, removal of HCO3− decreased both the peak and duration of the agonist-evoked membrane depolarization and [Ca2+]i response. Our findings show that HCO3− efflux has a major contribution to depolarizations mediated by GABAA and glycine receptor-coupled anion channels in prenatal neurons. We hypothesize that the HCO3−-dependent depolarizing component, which is likely to produce an intracellular acidosis, might play an important role during the early postnatal period when the Cl−-dependent component gradually shifts to hyperpolarization.
The principal hyperpolarizing and thus inhibitory neurotransmitters GABA and glycine exert a depolarizing action during development of neuronal structures (Ben-Ari et al., 1989; Reichling et al., 1994; Obrietan and van den Pol, 1995). In the immature hippocampus, depolarizing GABAergic IPSPs inhibit synaptic responses of CA3 pyramidal neurons (Psarropoulou and Descombes, 1999; Palva et al., 2000), but periodic GABA release produces a “giant” neuronal depolarization and action potential discharge (Ben-Ari et al., 1989). The rise in the concentration of free intracellular Ca2+([Ca2+]i) associated with the GABA-induced depolarization (Leinekugel et al., 1995; Garaschuk et al., 1998) might be implicated in trophic or hebbian modulation of developing synapses and activity-dependent formation of the hippocampal network (Cherubini et al., 1991; Leinekugel et al., 1999).
In motoneurons that are among the earliest neurons to differentiate within the brain, activity-related rises of [Ca2+]i are also supposed to have a trophic effect. Suppression of neurite outgrowth in developing motoneurons (Owen and Bird, 1997; Metzger et al., 1998) appears to be causally related with an increase of [Ca2+]iattributable to activation of Ca2+-permeable glutamate receptors (Metzger et al., 2000). Accordingly, maturation of motoneurons of cultured lumbar spinal cord is retarded upon blockade of glutamatergic neurotransmission (Xie and Ziskind-Conhaim, 1995). One week before birth, the isolated spinal cord of rats generates rhythmic nerve activity that is impaired by blockers of glycine and GABAA receptors (Nishimaru et al., 1996). It was found previously that GABA and glycine depolarize lumbar motoneurons that provide the output of this rhythmically active network in the fetus (Obata et al., 1978; Wu et al., 1992; Gao and Ziskind-Conhaim, 1995). The latter studies indicated a major role of Cl− ions in this depolarization. However, it is yet not clear whether efflux of HCO3− through the receptor-coupled anion pore (Bormann et al., 1987; Fatima-Shad and Barry, 1993) contributes to the GABA- and glycine-evoked responses in the fetal motoneurons as shown for a variety of neurons in postnatal nervous tissue (Kaila and Voipio, 1987; Kaila, 1994; Perkins and Wong, 1996; Backus et al., 1998). In particular, the extent is not known yet to which [Ca2+]iis elevated during such depolarization of fetal motoneurons and whether the expected [Ca2+]i rise depends on HCO3−.
We have analyzed the effects of exogenous GABA and glycine on [Ca2+]i in functionally identified lumbar motoneurons of acutely isolated, fura-2 ester-loaded lumbar spinal cord slices from embryonic rats. In addition, membrane potential (Vm) of the motoneurons was recorded with gramicidin-perforated and whole-cell patch-clamp techniques. The results show that the magnitude of the depolarization caused by GABA or glycine is sufficient to evoke a robust rise of [Ca2+]i secondary to Ca2+ influx through voltage-activated Ca2+ channels. Efflux of HCO3− through the anion pore has a significant contribution to the depolarization and the resulting [Ca2+]irise. It is hypothesized that expected similar [Ca2+]i transients associated with rhythmic activation of glycine and GABAA receptors are involved in control of neuronal survival and outgrowth of dendrites and axons within the motor network of the lumbar spinal cord in the fetus.
MATERIALS AND METHODS
Preparation of spinal cord slices. The experiments were performed on lumbar motoneurons of transverse spinal cord slices that were acutely isolated from Wistar rats of embryonic day 15.5 (E15.5) to E19.5. Pregnant rats were anesthetized with ether and killed by cervical dislocation. The fetuses were removed by cesarean section and decapitated. The lumbar part of the spinal cord was dissected and transferred to ice-cold solution (for composition, see below). Afterward, the spinal cord was embedded into 2% agarose solution at 35°C (Gao and Ziskind-Conhaim, 1995) and subsequently cooled on ice. The agarose block, including the spinal cord, was glued to the stage of a vibratome (Vibracut; FTB, Weinheim, Germany). Transverse 300-μm-thick slices were cut from the spinal cord together with the surrounding agarose. Before transfer to the recording chamber, the slices were stored at 30°C. For additional details, see Ballanyi (1999).
Solutions and superfusion system. After transfer and immobilization of individual slices with a net, the recording chamber (volume of 3 ml) was superfused at 30°C with oxygenated standard solution (flow rate of 5 ml/min) of the following composition (in mm): 118 NaCl, 3 KCl, 1 MgCl2, 1.5 CaCl2, 25 NaHCO3, 1.2 NaH2PO4, and 10d-glucose. The pH was adjusted to 7.4 by gassing with 95% O2 and 5% CO2. Nominally CO2/HCO3−-free solution (Voipio and Ballanyi, 1997), pH-buffered with HEPES, was equilibrated with 100% O2 and contained (in mm): 118 NaCl, 3 KCl, 1.5 CaCl2, 1 MgCl2, 25 HEPES, and 10 d-glucose (pH adjusted to 7.4 with NaOH). Ca2+-free standard solution contained 1 mm EGTA, and MgCl2was elevated to 5 mm. In Cl−-free superfusate, gluconate− salts were used instead of Cl− salts of the constituents of the standard solution. Because of Ca2+chelation by gluconate−, Ca2+ concentration was increased to 12 mm (Kenyon and Gibbons, 1977). In formate−-containing Cl−-free solution, 10 mm Na-gluconate was substituted by 10 mm Na-formate (pH adjusted to 7.4 with NaOH).
Drugs purchased from Sigma (Munich, Germany), Tocris-RBI/Biotrend (Köln, Germany), and Calbiochem (Bad Soden, Germany) were added to the superfusate from stock solutions. The following stock solutions (in standard saline) were used: glutamate (100 mm), bicuculline (10 mm), strychnine (10 mm), muscimol (5 mm), and tetrodotoxin (TTX) (1 mm) were dissolved in H2O. Nifedipine (50 mm) and bumetanide (10 mm) were dissolved in dimethylsulfoxide (DMSO). GABA, glycine, and baclofen were added in final concentration to the standard solution. Agarose was dissolved in standard solution by heating to 100°C. Fura-2 AM, fura-2 pentapotassium salt (fura-2), and pluronic acid were obtained from Molecular Probes (Eugene, OR).
Fluorometric measurements of [Ca2+]i.[Ca2+]i was fluorometrically measured using an upright microscope [Axioscope (digital imaging system) or Standard-16 (photomultiplier system);Zeiss, Oberkochen, Germany] that was equipped with epifluorescence optics, a monochromator (Polychrome II; T.I.L.L. Photonics, Planegg, Germany), and either a CCD camera (T.I.L.L. Photonics) or a photomultiplier (Luigs und Neumann, Ratingen, Germany). For fura-2 excitation, cells were exposed to alternating wavelengths (360 and 380 nm), whereas emission was measured at 515 nm. Images were acquired at a frequency of 1 Hz, and exposure time was 20 msec. Fluorescence ratios of the fura-2 signals were converted into [Ca2+]i by using the following equation: [Ca2+]i =K(R −Rmin)/(Rmax− R) (Grynkiewicz et al., 1985) in which R is the fluorescence ratio (360/380 nm) and K is the effective dissociation constant of fura-2. In vivo calibrations to determine Rmin,Rmax, and K were performed according to the method described by Neher (1989). Briefly, whole-cell measurements were performed with three different pipette solutions (in mm): (1) 130 KCl, 1 MgCl2, 10 bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA), 10 HEPES, and 1 Na2ATP (low calcium; Rmin); (2) 130 KCl, 1 MgCl2, 3 CaCl2, 4 K4BAPTA, 10 HEPES, and 1 Na2ATP [intermediate calcium; 300 nm, according to aKD of 107 nm for BAPTA (Tsien, 1980)]; and (3) 130 KCl, 1 MgCl2, 10 CaCl2, 10 HEPES, and 1 Na2ATP (high calcium;Rmax). To each solution 100 μm fura-2 was added. The resulting intracellular fluorescence ratios were calculated according to the above equation. K was calculated as K = 300 nm (Rmax −R)/(R −Rmin). Cells were loaded with fura-2 by incubation of slices in standard solution containing 10 μm fura-2 AM, 5 μmpluronic F-127, and 0.1% albumin (bovine) for 30 min at 37°C.
Electrophysiological recording and stimulation. Gramicidin perforated-patch or whole-cell recordings ofVm were performed on superficial lumbar motoneurons under visual control using an EPC-9 patch-clamp amplifier (Heka, Lambrecht, Germany). Patch pipettes were obtained from borosilicate glass capillaries (GC 150TF; Clark Electromedical Instruments, Pangbourne, UK) using a horizontal electrode puller (Zeitz, München, Germany). The standard patch (low Cl−) pipette solution contained (in mm): 140 potassium gluconate, 1 MgCl2, 10 HEPES, and 1 Na2ATP, pH 7.3–7.4 adjusted with KOH. For perforated-patch recordings, a pipette solution containing 5 μg/ml gramicidin (Ebihara et al., 1995; Kyrozis and Reichling, 1995;Brockhaus and Ballanyi, 1998) was prepared from two stock solutions. Stock I contained 2 mg of gramicidin in 1 ml of DMSO and was freshly made every 4 hr. Stock II contained 140 mm KCl and 10 mm HEPES, pH adjusted to 7.4 with KOH. Stocks I and II were mixed at a ratio of 1:10, and this mixture was diluted 1:40 or 1:20 with the standard pipette solution. The pipettes were prefilled with 2 μl of standard pipette solution. The resistance of the patch electrodes ranged from 4 to 6 MΩ. After seal formation, series resistance decreased over 10–30 min to 40–100 MΩ to reveal aVm of at least −40 mV (mean of −45.6 ± 3.6 mV; n = 6) and overshooting spike discharge. Spontaneous or intended rupture of the perforated patch was evident by a negative change in the apparent restingVm by ∼10 mV (Brockhaus and Ballanyi, 1998). Furthermore, a depolarizing response to bath-applied GABA or glycine turned into a hyperpolarization. ThatVm is indeed more negative than −45 mV as measured with perforated-patch recordings in the present study is indicated by previous findings using whole-cell (Metzger et al., 2000) and microelectrode (Ziskind-Conhaim, 1988) recording techniques that revealed a resting Vm of between −60 and −50 mV. Because of the apparent similarities of resting potentials upon whole-cell or intracellular microelectrode recording, liquid junction potentials do not appear to cause a major voltage error and were thus not corrected for (Onimaru et al., 1996; Brockhaus and Ballanyi, 1998). Membrane input resistance was measured by application of hyperpolarizing current pulses (5–50 pA, 500 msec).
For functional identification of lumbar motoneurons, a suction electrode was positioned close to the ventrolateral surface of the spinal cord slices in the vicinity of ventral nerve rootlets. As described in detail in our previous report (Metzger et al., 2000), single pulse stimulation via the suction electrode evoked an antidromic action potential (Ziskind-Conhaim, 1988; O'Donovan et al., 1994;Lev-Tov and O'Donovan, 1995). In the present study, tetanic antidromic stimulation (50 Hz, 10–60 V, 2 sec; single pulse duration of 100 μsec) evoked a [Ca2+]i increase of between 50 and 130 nm that was abolished by 0.5 μm TTX (see Fig. 1). The agonists GABA and glycine were administered to the cells via bath application because this procedure was shown to evoke a similar lumbar nerve burst as that occurring spontaneously in preparations from E13.5–E18.5 rats (Nishimaru et al., 1996).
Data analysis. Electrophysiological and microfluorometrical signals were sampled into a Macintosh PowerPC (Apple Computers Inc., Cupertino, CA) using Pulse/Pulsefit and X-Chart/Fura extension from Heka. Current and voltage signals were also digitized (VR-100A; Instrutech, Elmont, NY) and recorded on a video cassette recorder. Statistical significance of differences was assessed by ANOVA, followed by Dunnet's test using the GraphPad Prism software (GraphPad Software Inc., San Diego, CA). Values are given as a mean ± SEM.
RESULTS
Age-dependence of agonist-induced [Ca2+]i rises in identified lumbar motoneurons
In an initial experimental approach, lumbar spinal cord slices from E17.5 rats were loaded with fura-2 AM to study whether GABA (200 μm) and glycine (1 mm) elevate [Ca2+]i (see Fig.3). Bath application of GABA (Figs.1A,2A) or glycine (Figs.1B, 2A) for 20 sec led to a rise of [Ca2+]i with a peak value that was by between 15 and 90 nmhigher than resting level 36.0 ± 2.7 nm(n = 126) (for statistics, see Fig.2B). The [Ca2+]i increase caused by these amino acids was not changed by TTX (0.5 μm), which abolished the response to antidromic stimulation (Fig. 1). This indicates that the responses are attributable to direct activation of postsynaptic receptors. To assess putative developmental changes in the [Ca2+]i rises, the responses to GABA and glycine were also tested in preparations from E15.5 and E19.5 fetal rats (Fig. 2A). This revealed that the mean [Ca2+]i rises induced by both GABA and glycine declined with age. The peak value of the responses in preparations from E19.5 rats was <50% of those from E15.5 rats (Fig. 2B). Furthermore, the average response to GABA appeared to be larger than that to glycine, in particular at E15.5 and E17.5 (Fig. 2B). The [Ca2+]i transients attributable to the “inhibitory” neurotransmitters were compared with the peak [Ca2+]i increase evoked by exposure to glutamate (100 μm) for 20 sec. At E17.5, the mean glutamate-evoked [Ca2+]i rise was approximately fivefold larger than those caused by GABA or glycine. The glutamate responses were smaller in preparations from E15.5 and E19.5 rats (for statistics, see Fig. 2B).
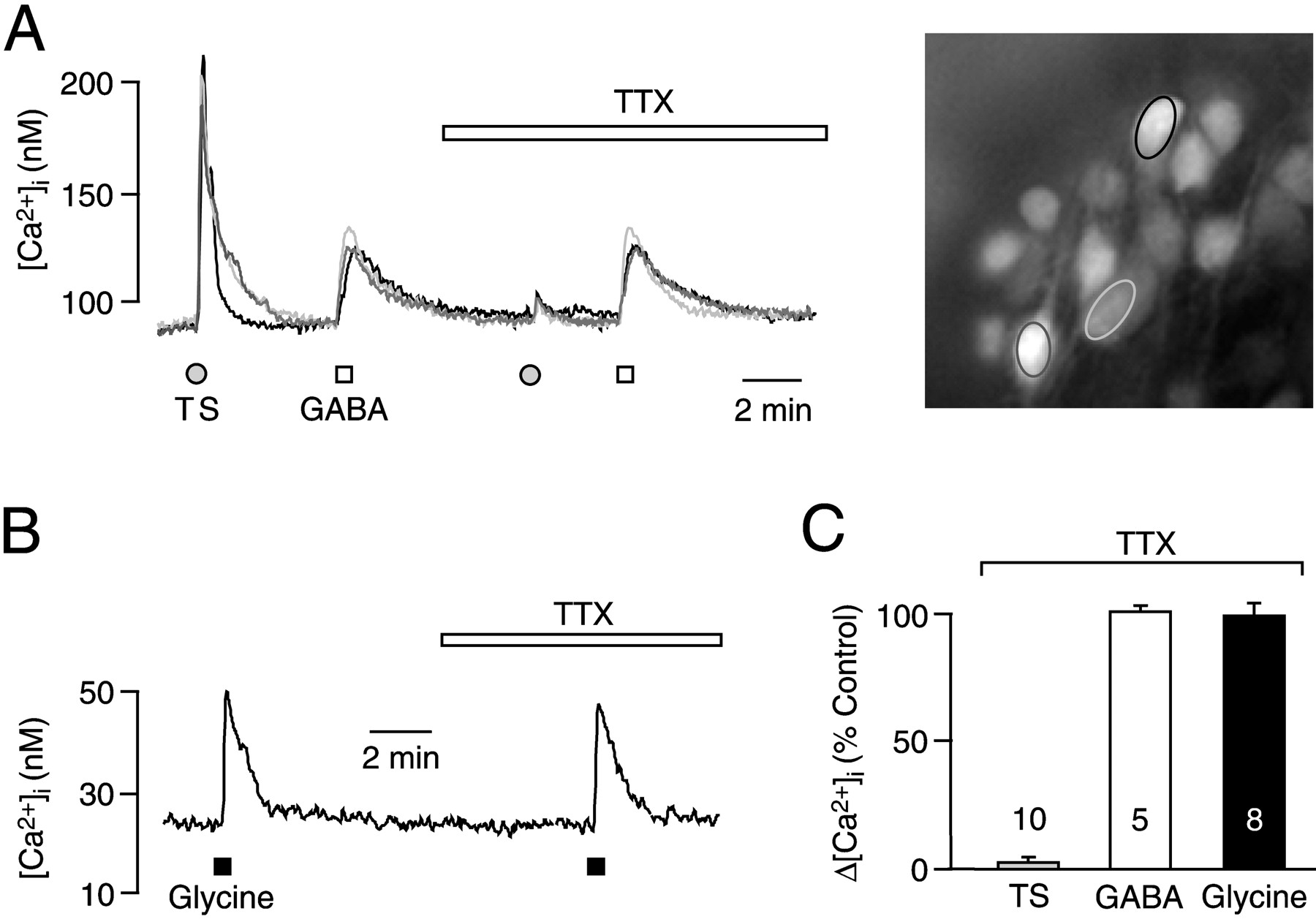
Increases of [Ca2+]i in lumbar motoneurons of rat embryonic spinal cord slices. A, Tetanic antidromic stimulation (TS; 50 V, 50 Hz, 2 sec) of the ipsilateral ventral nerve rootlet evokes a noticeable rise of [Ca2+]i that is abolished by 0.5 μm TTX. In contrast, the more moderate [Ca2+]i increase in response to bath application (20 sec) of 200 μm GABA is not affected by TTX. Traces correspond to somatic measurements in the regions of interest indicated in the micrograph. B, In a different spinal cord slice, TTX has no effect on the [Ca2+]i increase upon bath application (20 sec) of glycine (1 mm). C, Statistical analysis of the effects of TTX on [Ca2+]i rises caused by TS, GABA, and glycine. Numbers indicate the population of measured cells. Means ± SEM.

Age-dependence of agonist-induced [Ca2+]i increases. A, In a spinal cord slice from an E15.5 rat, bath-applied (20 sec) GABA (200 μm), glycine (1 mm), and glutamate (100 μm) elevate motoneuronal [Ca2+]i by between 25 and 100 nm (top trace). Middle traceexemplifies that the response to glutamate is greatly potentiated in a slice from an E17.5 rat, whereas the [Ca2+]i increases caused by GABA and glycine are smaller than at E15.5. Bottom traceillustrates that the GABA- and the glycine-induced [Ca2+]i rises are even less pronounced in a preparation from an E19.5 rat. Also, the response to glutamate is smaller than at E17.5. B, Statistical analysis of the agonist-evoked [Ca2+]i responses. Note the different scale for the glutamate effects.
Pharmacology and origin of the GABA- and glycine-mediated [Ca2+]i rises
The above results showed that GABA and glycine produce a robust rise of [Ca2+]i in motoneurons of fetal rats and that the magnitude of these [Ca2+]i transients decreases toward birth. To avoid age-dependent scatter of data, the pharmacology and ionic mechanism of these [Ca2+]i transients were determined at one particular age. Although the responses were smaller than those revealed at E15.5, preparations from E17.5 rats were chosen for this purpose because these were more stable with regards to long-term (>30 min) measurements of [Ca2+]i. For dose–response relationships, the cells of individual slices were exposed consecutively to different concentrations of the agonists. As illustrated in Figure 3, A andC, recovery to baseline after washout of the agonists occurred within several minutes and, thus, recording periods often exceeded 1 hr. Within time periods of up to 2 hr, no “rundown” of the peak of the [Ca2+]i transients was observed during repeated application of agonists. The dose–response relationships showed that the threshold for the GABA-induced [Ca2+]i rise was between 5 and 10 μm and that saturation occurred at concentrations >200 μm (Fig.3A; for statistics, see Fig. 3B). The concentration threshold for the glycine-induced [Ca2+]i increase was ∼50 μm, and the response saturated at concentrations >250 μm (Fig. 3C; for statistics, see Fig. 3D).

Dose relationship of agonist-evoked [Ca2+]i increases. A, Original recording of [Ca2+]i rises caused by bath application (20 sec) of GABA. B, Statistical analysis reveals that the response to GABA saturates at concentrations >200 μm. C, Original recording of [Ca2+]i rises in response to bath application (20 sec) of glycine. D, Statistical analysis reveals that the response to glycine saturates at concentrations >250 μm.
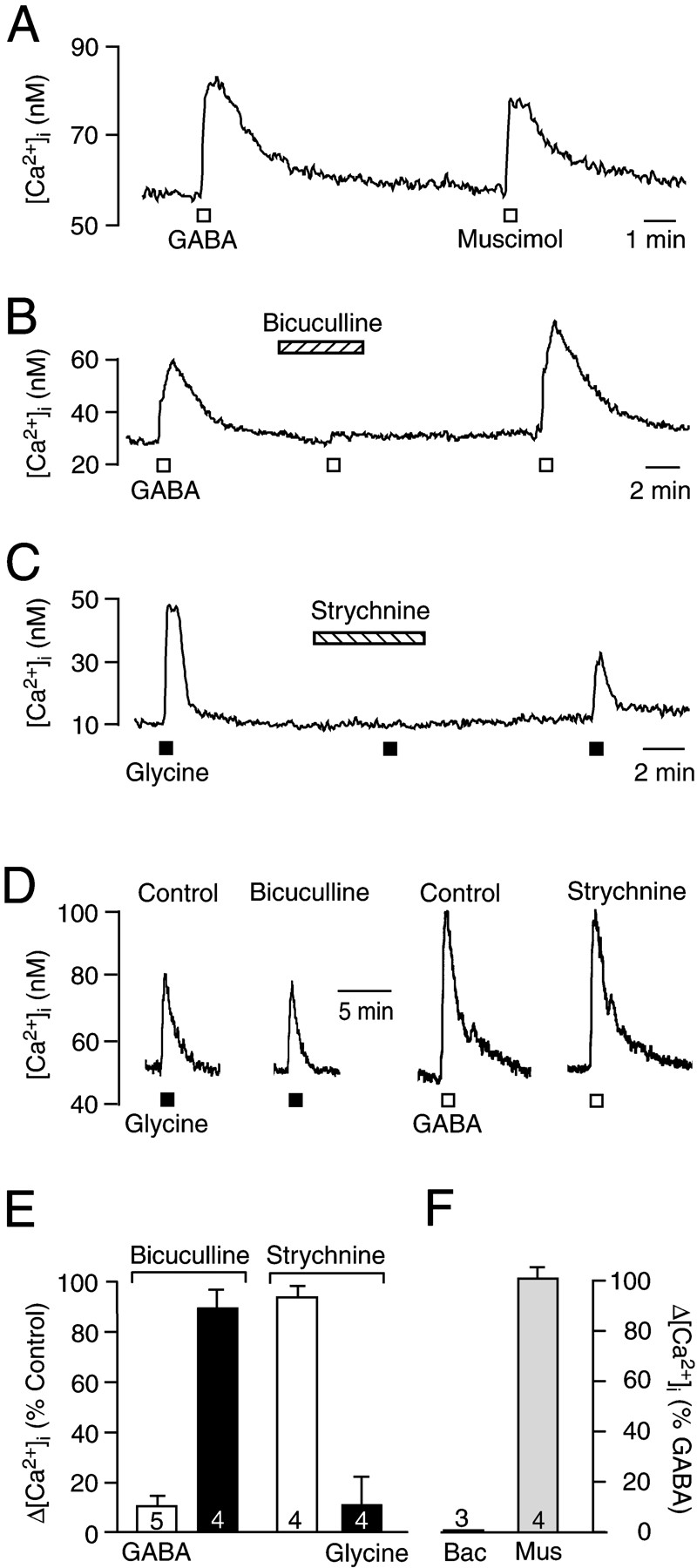
For further pharmacological analysis, GABA and glycine were applied at a concentration of 200 μm and 1 mm, respectively. Bath application of the GABAAreceptor agonist muscimol (10 μm, 20 sec) elevated [Ca2+]i by 26.6 ± 5.3 nm (n = 4), whereas the GABAB receptor agonist baclofen (50 μm, 20 sec) had no effect (n = 3) (Fig.4A,F). The GABAA receptor antagonist bicuculline (50–100 μm) abolished the GABA-induced [Ca2+]i elevation (Fig. 4B) but had no major effect on the response to glycine (Fig. 4D; for statistics, see Fig.4E). The glycine receptor blocker strychnine (10 μm) suppressed the response to glycine (Fig.4C), but the drug had almost no effect on the response to GABA (Fig. 4D; for statistics, see Fig.4E). The origin of the GABA- and glycine-mediated [Ca2+]i rise was elaborated by exposure to Ca2+-free superfusate and to the L-type Ca2+ channel blocker nifedipine. Preincubation of the slices in Ca2+-free superfusate induced a fall of [Ca2+]i baseline by between 4 and 35 nm. Under these conditions, the [Ca2+]i rise in response to GABA (Fig. 5A) or glycine (Fig. 5B) was abolished (for statistics, see Fig.5D). Nifedipine (50 μm) reduced the response to GABA and glycine to 29.6 ± 8.2 (n = 5) and 40.0 ± 8.7% (n = 4) of control, respectively (Fig. 5C,D).

Pharmacology of agonist-induced [Ca2+]i increases. A, Bath-applied (20 sec) muscimol (10 μm) mimics the response to GABA (200 μm). B, Bicuculline (100 μm) reversibly abolishes the GABA-evoked [Ca2+]i rise. C, The response to glycine (1 mm) is suppressed by strychnine (10 μm). D, Bicuculline does not evoke a major suppression of the [Ca2+]i increase caused by glycine, whereas strychnine does not influence the response to GABA. E, Statistical analysis of the percentage antagonist-induced reduction of the GABA and glycine responses.F, Statistical analysis of the response to baclofen and muscimol, represented as the percentage of the peak [Ca2+]i rise elicited by GABA.

Mediation of anion channel-induced [Ca2+]i increases by voltage-activated Ca2+ channels. A, The [Ca2+]i transient caused by bath-applied GABA (200 μm) is suppressed in nominally Ca2+-free superfusate. B, Ca2+-free saline also abolishes the [Ca2+]i rise in response to glycine (1 mm). C, The Ca2+ channel antagonist nifedipine (50 μm) reduces the glycine-evoked [Ca2+]i response. D, Statistical analysis of the blocking effects of Ca2+-free saline and nifedipine on agonist-induced [Ca2+]i rises.
Ionic mechanism of the GABA- and glycine-mediated [Ca2+]i rises
The above results indicated that the [Ca2+]i rises associated with activation of GABAA or glycine receptor-coupled anion channels are caused by depolarization-induced opening of voltage-activated Ca2+channels. In the following, results are described that aimed to analyze the ionic mechanism of such agonist-evoked depolarizations. For example, in mature peripheral neurons (Gallagher et al., 1983; Ballanyi and Grafe, 1985) or immature central neurons (Ben-Ari et al., 1989; Ito and Cherubini, 1991), it was demonstrated that Cl− efflux through the anion pore constitutes a major component of GABA- or glycine-associated depolarizations. To study the contribution of Cl− to agonist-evoked [Ca2+]i increases in the fetal motoneurons, the preparations were superfused with nominally Cl−-free solution. The [Ca2+]i rises in response to GABA (Fig.6A) or glycine (Fig.6B) were potentiated up to 10-fold when the agonists were applied within the first 1–2 min after introduction of the Cl−-free superfusate (for statistics, see Fig. 6D). The [Ca2+]i transients decreased in amplitude upon consecutive application in Cl−-free saline, but the peak response could remain larger than under control (Fig.6C,D). As also evident from the recording of Figure 6C, recovery from the agonist-induced [Ca2+]i transients in Cl−-free solution was incomplete in ∼30% of cases.

Anion-dependence of agonist-evoked [Ca2+]i increases. A,B, Shortly after introduction of a nominally Cl−-free superfusate, the [Ca2+]i rise in response to bath-applied GABA (200 μm; A) or glycine (1 mm; B) is potentiated several-fold.C, HCO3−-free saline abolishes the [Ca2+]i transient evoked by GABA (200 μm) after its potentiation by Cl−-free superfusate. Subsequent addition of formate anions (10 mm) fully restores the response.C, Statistical analysis of the initial potentiation (left columns) and the steady-state (right columns) effect of Cl−-free superfusate on the agonist-induced [Ca2+]i increases represented as percentage of control. Note the differences in the scale of the ordinates.
The observation that the [Ca2+]i increases persisted in the absence of extracellular (and thus intracellular) Cl− (Ballanyi et al., 1987; Ballanyi and Schlue, 1990) indicated that another anion substitutes for Cl− to produce the GABA- or glycine-induced depolarizations. Receptor-coupled anion channels are permeable to several anions, including HCO3− (Bormann et al., 1987; Kaila and Voipio, 1987; Fatima-Shad and Barry, 1993; Staley et al., 1995). We thus investigated whether HCO3− efflux through the anion pore induces a [Ca2+]i rise in the absence of Cl−. After the agonist-induced [Ca2+]i rises have stabilized in Cl−-free saline, CO2/HCO3−was removed from the superfusate. This led to reversible reduction of the GABA- and glycine-evoked [Ca2+]i transients by >90% (Fig. 6C; for statistics, see Fig.6D). That no complete block was induced in ∼50% of preparations is likely to be related to the fact that the cells within the slices produce CO2, and thus HCO3−, while using aerobic metabolism (Voipio and Ballanyi, 1997). It was also tested whether the anion formate substitutes for Cl− and HCO3− to produce the agonist-induced Ca2+ signals. The GABAA receptor-permeant weak-acid anion formate that has an equilibrium potential set by the transmembrane pH gradient has been used as a substitute for Cl−and/or HCO3− in a variety of previous studies (Bormann et al., 1987; Mason et al., 1990;Lamsa and Kaila, 1997). As illustrated in Figure 6C, bath application of formate (10 mm) restituted the response to GABA in Cl−- and HCO3−-free saline (for statistics, see Fig. 6D). Next, the extent to which removal of HCO3− from the superfusate affects the evoked responses in the presence of Cl− was studied. As exemplified in Figure7A, the peak [Ca2+]i rise caused by GABA or glycine was reduced to 66.2 ± 6.8 (n = 5) and 62.6 ± 6.0% (n = 4) of control, respectively, in the absence of HCO3− (Fig.7C). Because recovery to [Ca2+]i baseline was considerably faster in all cells in the CO2/HCO3−-free superfusate, the overall rise of intracellular Ca2+ caused by GABA or glycine was reduced to ∼50% of control. In contrast, the glutamate-induced [Ca2+]i transient was not attenuated by the HEPES pH-buffered saline (Fig. 7B; for statistics, see Fig. 7C).

Attenuation of anion channel-mediated [Ca2+]i rises by HCO3−-free superfusate.A, CO2/HCO3−-free superfusate reduces both the peak amplitude and duration of [Ca2+]i rises elicited by glycine (1 mm) or GABA (200 μm). B, In a different slice, omission of CO2/HCO3− does not reduce the response to glutamate (100 μm).C, Statistical analysis of the effects of CO2/HCO3−-free saline on the peak of the [Ca2+]i rise represented as percentage of the control agonist response.
To elucidate the role of inwardly directed Na+/K+/2Cl−cotransport in the [Ca2+]i rises attributable to activation of receptor-coupled anion channels, the effects of the selective inhibitor bumetanide (Haas, 1994; Rohrbough and Spitzer, 1996) were analyzed. Bumetanide (100 μm) produced a consecutive decrease in the magnitude of the agonist-evoked Ca2+ transients. However, on average, 24.5 ± 6% (n = 5) of the GABA-induced [Ca2+]i increase and 21.0 ± 3.8% (n = 4) of the glycine-induced [Ca2+]i transient persisted even during exposure to the drug for time periods of >1 hr (Fig.8A,B). In motoneurons in which the response to GABA or glycine was strongly attenuated by bumetanide, Cl−-free saline initially potentiated the evoked [Ca2+]i rises in a similar way as observed in the absence of bumetanide (Fig.8C). Upon consecutive exposure to the agonists in bumetanide-containing, Cl−- free solution, the potentiated response decreased in magnitude but could remain larger than under control conditions (Fig. 8C). Under these conditions, HCO3−-free solution reduced the remaining responses to GABA (n = 3) or glycine (n = 3) by >90% (Fig. 8C).

Role of an inwardly directed Cl− pump in anion channel-mediated [Ca2+]i increases. A, The Na+/K+/2Cl−-cotransport blocker bumetanide (100 μm) attenuates the [Ca2+]i rise caused by glycine (1 mm). B, In a different preparation, bumetanide leads to a more moderate depression of the [Ca2+]i transient caused by GABA (200 μm). The remaining component is almost abolished by CO2/HCO3−-free superfusate. C, After bumetanide-related suppression of the GABA-induced [Ca2+]i rise, Cl−-free solution exerts a potentiating effect that is attenuated by CO2/HCO3−-free saline. Note that the bumetanide-dependent attenuation of the [Ca2+]i rise was not attributable to rundown because the response to the agonists remained stable during repeated application for time periods >1 hr (compare with Fig.3).
GABA- and glycine-induced depolarizations
In a final series of experiments, the extent was measured to which GABA (200 μm) and glycine (1 mm) depolarized the fetal motoneurons of the acute slices. Because the response is dependent on Cl−, whole-cell recordings were not feasible. Accordingly, gramicidin perforated-patch recording was done in six cells. Figure9A shows a representative recording that revealed a depolarization by ∼20 mV (for statistics, see Fig. 9B) and a concomitant decrease of input resistance by >90% in response to GABA. After recovery, exposure to GABA was repeated in HCO3−-free solution, which showed that the amplitude and the duration of the response were reversibly reduced (Fig. 9A; for statistics, see Fig. 9B). A similar attenuating effect of removal of extracellular HCO3− was observed for the response of two cells to glycine (data not illustrated). Furthermore, it was investigated whether formate anions can substitute for Cl− and HCO3− to mediate the agonist-induced depolarization. Figure 9C shows that the GABA-induced depolarization was first potentiated by Cl−-free solution. Subsequent exposure to both Cl−- and HCO3−-free saline considerably attenuated the response that was restituted by 10 mm formate. Similar observations were made in three other motoneurons. Finally, it was studied whether the HCO3−-dependent component of the agonist-induced depolarizations can be revealed in cells that are whole-cell recorded with the standard low Cl− patch solution. As exemplified in Figure 10, GABA induced a fall of input resistance by >90% and a concomitant hyperpolarization of ∼20 mV. After recovery, the slice was exposed to Cl−-free solution for 5 min. Subsequent administration of GABA evoked a depolarization by ∼15 mV and a decrease of input resistance by ∼65%. This response was reversibly abolished upon removal of HCO3−. Similar observations were made in three additional neurons. In two of these cells, the HCO3−-dependent depolarizing component of the GABA response showed a rundown after several minutes of whole-cell recording.

Perforated-patch recording of anion channel-mediated depolarizations. A, HCO3−-free saline attenuates and shortens the depolarization in response to bath-applied GABA (200 μm). B, Statistical analysis of the depolarization induced by GABA and of the attenuating effect of CO2/HCO3−-free saline. C, Cl−-free superfusate increases the peak of the GABA depolarization. After introduction of HCO3−-free solution, the GABA depolarization is abolished but reappears upon addition of formate anions (10 mm).

Whole-cell recording of the HCO3−-dependent component of the response to GABA. After rupture of a gramicidin perforated patch (low Cl− patch solution), GABA (200 μm) evokes a hyperpolarization and decrease of input resistance. The response shifts to a depolarization in Cl−-free superfusate and is reversibly blocked by removal of HCO3−.
DISCUSSION
The effect of opening of receptor-coupled anion channels on [Ca2+]i andVm was analyzed with Ca2+ imaging and (perforated) patch clamp in developing lumbar motoneurons. We found that efflux of HCO3− (in addition to that of Cl−) has a noticeable contribution to GABA- or glycine-evoked depolarizations. The resulting Ca2+ channel-mediated [Ca2+]i rise might serve as a trophic signal.
Origin and pharmacology of the GABA- and glycine-induced [Ca2+]i rise
The blocking effects of Ca2+-free superfusate and nifedipine suggest that GABA- and glycine-induced [Ca2+]i rises are caused by depolarization-evoked Ca2+influx through voltage-activated Ca2+channels as shown previously for other immature nervous structures (Reichling et al., 1994; Leinekugel et al., 1995; Owens et al., 1996). The sensitivity to bicuculline and the observation that muscimol, but not baclofen, mimicked the GABA-induced [Ca2+]i rise consolidate that GABAA receptors are responsible for the GABA-induced depolarization of spinal neurons (Obata et al., 1978; Mandler et al., 1990; Wu et al., 1992). The finding that the depolarizing response to glycine was blocked by strychnine also agrees with earlier findings in perinatal motoneurons (Takahashi, 1984; Wu et al., 1992; Gao and Ziskind-Conhaim, 1995; Singer et al., 1998) and other developing central neurons (Ito and Cherubini, 1991; Ehrlich et al., 1999).
Ionic mechanism of the GABA- and glycine-induced [Ca2+]i rise
Previous studies have demonstrated that Cl− efflux is a major constituent of the depolarizing neuronal response to GABA or glycine in supraspinal structures (Ballanyi et al., 1984; Ben-Ari et al., 1989; Owens et al., 1996). That the response of fetal motoneurons in the present study depends on Cl− is evident from the observation that Cl−-free solution, which causes initially a positive shift of the Cl− equilibrium potential (ECl) (Ballanyi et al., 1987; Ballanyi and Schlue, 1990), greatly potentiated the agonist-evoked [Ca2+]i rise (Li et al., 1998). Our results are in accordance with findings on immature spinal neurons (Reichling et al., 1994; Wang et al., 1994; Serafini et al., 1995; Rohrbough and Spitzer, 1996), including identified motoneurons (Wu et al., 1992; Gao and Ziskind-Conhaim, 1995). It has been established for more than two decades that the prerequisite for a Cl−-dependent depolarizing GABA or glycine action is an inwardly directed Na+/K+/2Cl−cotransport (Nicoll, 1978; Gallagher et al., 1983), with NKCC-1 as the molecular identity (Haas, 1994; Rohrbough and Spitzer, 1996; Clayton et al., 1998; Kakazu et al., 1999). Measurement of intracellular Cl− activity has demonstrated that such a Cl− pump elevates intracellular Cl− to >30 mm and that intracellular Cl− decreases by several millimolar during GABA-induced membrane depolarization (Ballanyi et al., 1984; Ballanyi and Grafe, 1985). Several studies have shown that loop diuretics block the GABA- or glycine-evoked neuronal depolarizations by inhibition of Na+/K+/2Cl−cotransport (Ballanyi and Grafe, 1985; Misgeld et al., 1986; Jarolimek et al., 1999; Kakazu et al., 1999). Also in the present study, bumetanide strongly attenuated or even abolished the evoked [Ca2+]i rises. This shows that a (NKCC-1 type) Cl− pump is responsible for the Cl−-dependent component of the depolarization caused by receptor-coupled anion channels in fetal motoneurons.
In a considerable portion of cells, the evoked [Ca2+]i rise persisted (although at reduced amplitude) after bumetanide inhibition of Na+/K+/2Cl−cotransport. Similarly, long-term superfusion of Cl−-free solution did not abolish the agonist-induced [Ca2+]i transient. Its peak amplitude could even remain larger than under control conditions. Removal of HCO3− from the superfusate almost abolished this remaining component of the agonist-evoked [Ca2+]i transient or depolarization, and formate anions effectively restituted the responses. These results are in accordance with findings from biophysical measurements that have established that the GABAA or glycine receptor–channel complex has a noticeable permeability not only to Cl−but also to a variety of other anions (Bormann et al., 1987; Kaila and Voipio, 1987; Mason et al., 1990; Fatima-Shad and Barry, 1993; Staley et al., 1995). Because EH+/HCO3− is close to 0 mV (Kaila, 1994; Lückermann et al., 1997; Kaila and Ransom, 1998), efflux of HCO3−through the anion pore causes a noticeable depolarization during activation of GABAA or glycine receptors (Kaila et al., 1993; Staley et al., 1995; Perkins and Wong, 1996; Backus et al., 1998; Dallwig et al., 1999; Frech et al., 1999). Also in physiological solution, efflux of HCO3− has a significant influence on the response of fetal motoneurons to GABA and glycine. This is indicated by the finding that both the peak amplitude and the duration of the agonist-evoked [Ca2+]i rise were considerably reduced in HCO3−-free solution (Frech et al., 1999). This attenuating effect on [Ca2+]i is not attributable to a possible effect of a HCO3−-dependent change in intracellular pH on intracellular Ca2+(for references, see Trapp et al., 1996; Kaila and Ransom, 1998). This is because the GABA-evoked depolarization as the primary source for the [Ca2+]i rise was diminished in a very similar way in both amplitude and duration (compare Figs. 7A, 9A). The magnitude of the GABA- or glycine-induced responses was considerably larger in Cl−-free solution than in the presence of bumetanide. Furthermore, removal of Cl−reincreased the peak of the responses after suppression by bumetanide. This suggests that a passive distribution of Cl− after block of the Cl− pump (which results in an ECl of approximately −55 mV, corresponding to resting Vm) (Ballanyi et al., 1984;Ballanyi and Grafe, 1985) provides a shunt of the depolarizing action of HCO3− efflux. In contrast, the response gets considerably closer to EH+/HCO3− in the absence of Cl− as a charge carrier.
Functional relevance of GABA- or glycine-induced [Ca2+]i transients
Rises of [Ca2+]i associated with activation of GABAA or glycine receptors appear to be involved in maturation of neuronal networks (Collins et al., 1991; Meier et al., 1991; Spitzer, 1994; Barker et al., 1998). As example, bicuculline-sensitive giant depolarizing potentials (“early network oscillations”) produce a rhythmic [Ca2+]i rise in the immature hippocampus (Leinekugel et al., 1995; Garaschuk et al., 1998). This might constitute a mechanism for controlling formation of glutamatergic synapses, which are proposed to be immature when the GABAergic system is already functional (Cherubini et al., 1991;Ben-Ari et al., 1997; Leinekugel et al., 1999; Psarropoulou and Descombes, 1999; Palva et al., 2000).
Also in the fetal spinal cord, the neuronal response to exogenous GABA and glycine appears earlier than that to glutamate (Mandler et al., 1990). At E16–E17, depolarizing dorsal root-evoked potentials are blocked by GABAA and glycine receptor antagonists, whereas at E19 strong excitatory (glutamatergic) synaptic inputs are formed (Wu et al., 1992). Furthermore, at E14.5–E15.5, glutamate receptor blockers fail to suppress glycine- and GABA-mediated spontaneous lumbar motoneuronal activity but abolish the rhythm at E17.5 (Nishimaru et al., 1996). Thus, it is possible that elevation of [Ca2+]i caused by periodic activation of glycine and GABAAreceptors is crucial for formation of synaptic connectivity within the lumbar motoneuronal network responsible for locomotion. It was reported that activation of the Ca2+-permeable AMPA receptor has a regulatory effect on outgrowth of dendrites of lumbar motoneurons (Metzger et al., 1998). Interestingly, the rise of [Ca2+]iattributable to these glutamate receptors is of similar modest magnitude (<100 nm) (Metzger et al., 2000) as that elicited by GABA and glycine in the present study. However, in the latter as well as in the present study, the magnitude of the agonist-induced [Ca2+]i rises might have been underestimated because the Ca2+ indicator fura-2 increases internal Ca2+ buffering as shown recently for lumbar motoneurons in situ (Palacek et al., 1999). Furthermore, receptor desensitization attributable to slow bath application of agonists as well as a change in intracellular Cl− during sustained opening of the anion channels might result in a noticeable attenuation of the peak of the depolarizations, thereby reducing Ca2+influx through Ca2+ channels (Ballanyi and Grafe, 1985; Huguenard and Alger, 1986).
The [Ca2+]iresponses to both GABA and glycine decreased in amplitude toward birth as reported previously for the depolarization of lumbar motoneurons evoked by the agents (Wu et al., 1992; Gao and Ziskind-Conhaim, 1995). This is not attributable to a decrease in the expression of voltage-activated Ca2+ channels, which rather increases (Gao and Ziskind-Conhaim, 1998). Accordingly, the amplitude of the glutamate-induced [Ca2+]i transient, which greatly depends on voltage-activated Ca2+ channels, (Metzger et al., 2000) increased between E15.5 and E17.5. This is consistent with findings on an age-dependence of glutamate-evoked motoneuronal depolarizations (Seno et al., 1984). It is probable that this developmental decrease in the magnitude of the GABA- and glycine-induced motoneuronal [Ca2+]i transients correlates with the time course of maturation of connectivity in the lumbar spinal cord and a concomitant increase in expression of an outwardly directed Cl− pump (KCC2) (Rivera et al., 1999).
During the developmental period at which EClapproaches resting Vm and then gets more negative (Rivera et al., 1999), HCO3− efflux might constitute the only component for a depolarizing GABA or glycine action. Previous work has established that receptor-activated efflux of HCO3− induces a prominent decrease of intracellular pH (Kaila and Voipio, 1987; Trapp et al., 1996; Lückermann et al., 1997). Future studies will illuminate whether GABA- or glycine-induced intracellular acidosis has a similar second-messenger role in neuronal maturation as assumed for Ca2+.
Footnotes
This study was supported by the Deutsche Forschungsgemeinschaft and the Hermann und Lilly-Schilling-Stiftung.
Correspondence should be addressed to Dr. Klaus Ballanyi, Physiologisches Institut, Universität Göttingen, Humboldtallee 23, D-37073 Göttingen, Germany. E-mail:kb{at}neuro-physiol.med.uni-goettingen.de.
Dr. Nishimaru's permanent address: Department of Physiology, Institute of Basic Medical Sciences, University of Tsukuba, Tsukuba, Ibaraki 305, Japan.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}