

Abstract
Nonsteroid anti-inflammatory drugs (NSAIDs) are major drugs against inflammation and pain. They are well known inhibitors of cyclooxygenases (COXs). However, many studies indicate that they may also act on other targets. Acidosis is observed in inflammatory conditions such as chronic joint inflammation, in tumors and after ischemia, and greatly contributes to pain and hyperalgesia. Administration of NSAIDs reduces low-pH-induced pain. The acid sensitivity of nociceptors is associated with activation of H+-gated ion channels. Several of these, cloned recently, correspond to the acid-sensing ion channels (ASICs) and others to the vanilloid receptor family. This paper shows (1) that ASIC mRNAs are present in many small sensory neurons along with substance P and isolectin B4 and that, in case of inflammation, ASIC1a appears in some larger Aβ fibers, (2) that NSAIDs prevent the large increase of ASIC expression in sensory neurons induced by inflammation, and (3) that NSAIDs such as aspirin, diclofenac, and flurbiprofen directly inhibit ASIC currents on sensory neurons and when cloned ASICs are heterologously expressed. These results suggest that the combined capacity to block COXs and inhibit both inflammation-induced expression and activity of ASICs present in nociceptors is an important factor in the action of NSAIDs against pain.
Nonsteroid anti-inflammatory drugs (NSAIDs) have been generally considered as inhibitors of cyclooxygenases (COXs) (Walker, 1995; Vane and Botting, 1998). Their anti-inflammatory and analgesic action is thought to be mainly mediated via this inhibition. However, data have been accumulating through the years suggesting that NSAIDs also probably act on other targets to counteract pain. One recent result in this regard is that COX-1- and COX-2-deficient mice still show sensitivity to the analgesic action of NSAIDs (Ballou et al., 2000). On the other hand, administration of NSAIDs reduces both cutaneous (Steen et al., 1996) and corneal (Chen et al., 1997) pain induced by exposure to acidic pH in the absence of inflammation.
Tissue acidosis is a dominant factor in inflammation and in tumors and after ischemia (Reeh and Steen, 1996; Helmlinger et al., 1997) and has an important contribution in pain and hyperalgesia (Steen and Reeh, 1993; Steen et al., 1995). This is attributable to direct excitation of nociceptive sensory neurons by H+-gated currents (Krishtal and Pidoplichko, 1981a; Bevan and Yeats, 1991). Several channels corresponding to these currents have been cloned recently and belong to the acid-sensing ion channel (ASIC) family (Waldmann et al., 1996,1997a,b; Lingueglia et al., 1997; Chen et al., 1998) and to the vanilloid receptor family (Caterina et al., 1997; Tominaga et al., 1998) (for review, see Kress and Zeilhofer, 1999). Different ASIC isoforms have been identified and characterized: ASIC1a (Waldmann et al., 1997a) and its splice variant ASIC1b (Chen et al., 1998), both present in dorsal root ganglion (DRG) neurons; ASIC2a (Waldmann et al., 1996) and ASIC2b (Lingueglia et al., 1997), the latter being the more abundant in DRGs and the former being only present in large DRG neurons (Garcia-Añoveros et al., 2001); and ASIC3 (Waldmann et al., 1997b), abundant in peripheral sensory neurons and thought to mediate cardiac ischemic pain (Sutherland et al., 2001). This paper examines the relationships between ASICs, nociceptors, and inflammatory pain and shows that NSAIDs inhibit the activity of this class of channels in a COX-independent way, as well as their inflammation-induced expression.
MATERIALS AND METHODS
In situ hybridization. Sections (12 μm thick) of L4–L5 DRGs from normal or inflamed adult rats were fixed and hybridized (15 hr at 52°C) in 12.5% formamide, 4× SSC, 2.5× Denhardt's solution, 250 μg/ml herring sperm DNA, 125 μg/ml yeast tRNA, and 5 ng/μl oligonucleotides labeled with biotin-21-dUTP by terminal transferase. For fluorescent labeling, detection was performed with the ELF-97 mRNA In situ Hybridization kit (Molecular Probes, Eugene, OR) based on streptavidin–alkaline phosphatase–biotinylated probe interaction and the substrate ELF-97, which yields green fluorescent precipitates. For diaminobenzidine labeling, detection was performed with Dako (High Wycombe, UK) GenPoint with two cycles of amplification. Counterstain used hematoxylin. Probes were as follows: ASIC1a, CATTCTTGGAGACTTGGCTAAAGCGGAAC; ASIC1b, GGGTCATCACTCTCATCCAGTCCTAGCAT; ASIC2b, CCCAAACGGTCCATGAAGGCAGC; and ASIC3, CTGTTCCAGAAATACCCCAGGAC. Results were confirmed with a second set of probes. Control experiments were performed with ASIC1a sense oligonucleotide (CACAGATGGCTGATGAAAAGCAG), and background was assessed without probe. Each experiment was done on at least three animals.
Substance P immunochemistry and isolectin B4 binding. Afterin situ hybridization, slides were treated with anti-substance P (SP) antibody (dilution of 1:100; Sigma, St. Louis, MO) and revealed with anti-IgG Texas Red-coupled antibodies (Jackson ImmunoResearch, West Grove, PA). For isolectin B4 (IB4) binding, FITC-labeled IB4 was applied at 12.5 μg/ml. IB4-positive cells appeared green, which was changed to red with Adobe PhotoShop 5.0 (Adobe Systems, San Jose, CA) before superposition with ASIC-labeled pictures. Cell proportions and profiles were calculated by measuring individually and counting cells.
Inflammation experiments. Right hindpaws of anesthetized male Wistar rats (7–9 weeks) were inflamed by a 50 μl injection of complete Freund's adjuvant (Stein et al., 1988). L4–L5 DRGs were removed at day 2 on both sides, the left ganglia serving as negative controls. The anti-inflammatory drug was infused intraperitoneally when the injection of adjuvant was given and was repeated the next day. The drugs used were (in mg/kg): 8 diclofenac, 4 ibuprofen, 40 salicylate, 40 aspirin, 20 nimesulide, 8 nordihydroguaiaretic acid (NDGA), 0.1 dexamethasone (all from Sigma), 20 Zileuton (kindly provided by Abbott Labs, Irving, TX), or 20 MK-886 (kindly provided by Merck-Frosst, Dorval, Quebec, Canada).
Reverse transcription-PCR experiments. Total RNA (2 μg) was reverse transcripted (First-strand cDNA synthesis kit; Amersham Pharmacia Biotech, Arlington Heights, IL). One-twentieth was used per PCR condition. Primers used were (5′–3′, sense/antisense): ASIC1a (Waldmann et al., 1997a), CACAGATGGCTGATGAAAAGCAG/CATGGTAACAGCATTGCAGGTGC; ASIC1b (Chen et al., 1998), ATGCCGTGCGGTTGTCCC/same as ASIC1a; ASIC2a (Waldmann et al., 1996), TCAACCTACAGATTCCCGACCCG/CGAGTCCCATCTCTGAGGAC-CGG; ASIC2b (Lingueglia et al., 1997), CTGCCTTCATGGACCGTTTG/same as ASIC2a; ASIC3 (Waldmann et al., 1997b), CCCAGACCCAGACCCAGCCCTCC/CTGTTCCAGAAATACCCCAGGAC; and β-actin (Nudel et al., 1983), GTGCCCATCTATGAGGGTTACGCG/GGAACCGCTCATTGCCGATAGTG. Analysis was performed after scanning autoradiograms of dot blots and/or ethidium bromide-stained agarose gel pictures (both gave the same results) with the NIH Image program. Results were normalized with actin signals. Calcitonin gene-related peptide mRNA level measurement was systematically performed by reverse transcription (RT)-PCR to assess the efficiency of the inflammatory treatment (Donaldson et al., 1992) with the primers TCTGAAGTTCTCCCCTTTCCTGG/GAAGGGTTTCAGTACCAAGAATG (Amara et al., 1985). Specific VR1 expression was measured using AGACAGACAGCCTGAAGCAGTTT/CTTGTCACGAACTTGGTGTTGTC (Caterina et al., 1997), VRL1 expression with TGCCGCCGCTACACCTTGGCTTC/GCTCCTGCTGGCTGGGAGCAGAA (Caterina et al., 1999), and VR5′sv expression using CCTCTTGGTGGAGAATGGAGCAG/same as VR1 (Schumacher et al., 2000).
Electrophysiology. Ion currents were recorded at room temperature on ASIC3-, ASIC1a-, or ASIC2a-transfected COS cells or on DRG neuron primary cultures using whole-cell or outside-out patch clamp and analyzed with Serf freeware (www.bram.org/serf/). Cells were voltage clamped at −60 mV. The pipette solution contained (in mm): 120 KCl, 30 NaCl, 2 MgCl2, 5 EGTA, and 10 HEPES, pH 7.2. The bath solution contained (in mm): 140 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, and 10 HEPES, pH 7.3. Solutions used for pH shifts were pH 5 for ASIC1a-transfected COS cells, pH 4 for ASIC3-transfected cells, and a more pathophysiological pH 6 for DRG cells, all giving comparable currents. These solutions were buffered with 10 mm MES plus 10 mmglycine instead of HEPES. After transient pH drops, NSAID solution was preapplied extracellularly for 10 sec before and during new pH changes.
Primary culture experiments. Dorsal root ganglion cells were prepared from Wistar adult male (5–7 weeks) and newborn rats by 0.1% collagenase dissociation and plating on collagen-coated P35 dishes in DMEM plus 5% fetal calf serum.
RESULTS
ASIC transcripts are present in small DRG neurons, and their levels are increased by inflammation
Using a double-labeling technique combining in situhybridization and histochemistry, we were able to localize ASIC transcripts in small sensory neurons i.e., nociceptors (Fig.1A). ASIC1a and ASIC3 are present in many SP- and IB4-positive neurons. SP and IB4 are specific markers of the two groups of unmyelinated nociceptors (C fibers), the neuropeptidergic fibers and the nonpeptidergic fibers, respectively (Snider and McMahon, 1998). ASIC1a is expressed in 62 ± 9% of SP-positive and 41 ± 4% of IB4-positive cells. Some SP-positive (40 ± 8%) and IB4-positive (48 ± 13%) cells do not express ASIC1a. ASIC3 is expressed in 50 ± 7% of SP-positive and 43 ± 9% of IB4-positive cells. Some SP-positive (48 ± 13%) and IB4-positive (39 ± 15%) cells do not express ASIC3. Similar labeling patterns were observed for ASIC1b and for ASIC2b (data not shown), which are expressed in SP- and IB4-positive and -negative neurons in the same proportions as the other ASICs (i.e., 40–50%).

Expression of ASIC mRNA in DRG nociceptors in normal and inflamed conditions. A, Colocalization (yellow) on DRG of ASIC1a or ASIC3 (green) with SP or IB4 (red).Arrows indicate characteristic cells:arrowhead indicates a double-labeled cell;pointing-up arrow indicates an ASIC-labeled cell; andpointing-down arrow indicates an SP- or IB4-labeled cell. B, Histograms represent the ASIC1a- and ASIC3-labeled cell proportion according to cell cross-sectional areas in normal (green) and inflamed (red) conditions. n = 3 animals for ASIC1a (control, n = 221 cells; inflamed,n = 210), and n = 4 animals for ASIC3 (control, n = 185 cells; inflamed,n = 214).
In normal conditions, ASIC1a mRNA is mainly expressed in cells with cross-sectional areas between 200 and 600 μm2 (mean area, 522 ± 36 μm2) that correspond to cell diameters between 15 and 30 μm (Fig. 1B). In inflammatory conditions, this area increases significantly to 722 ± 39 μm2 (p < 0.001). This increase can be explained by the appearance of a population of ASIC1a-positive cells with areas of 600–1200 μm2 (i.e., 30–40 μm in diameter). The area of cells coexpressing the ASIC1a transcript and SP is increased from 514 ± 47 μm2 in normal conditions to 664 ± 30 μm2 in inflamed conditions (p < 0.006) and the number of coexpressing cells from 40 ± 4 to 62 ± 7% (p < 0.03). No difference in ASIC3 distribution has been observed between normal (705 ± 43 μm2) and inflammatory (653 ± 33 μm2) conditions (Fig.1B). There is no significant difference for ASIC1b (560 ± 28 vs 556 ± 33 μm2) and ASIC2b (677 ± 35 vs 729 ± 44 μm2) either (data not shown).
The mRNA levels of the different ASICs mainly expressed in DRGs are highly increased 1–2 d after Freund's adjuvant-induced inflammation (Fig.2A,B). The highest increase was found for ASIC1a, ASIC2b, and ASIC3 (from sixfold to 15-fold) by semiquantitative RT-PCR, the only feasible technique because the amount of RNA in individual DRGs is low. ASIC2a mRNA that we find either undetectable or present at a low level in control DRG cells remains at a low level after inflammation. Indeed, ASIC2a is present in large mechanosensitive neurons (Garcia-Añoveros et al., 2001) that have no nociceptive role.

In vivo studies of the expression of ASIC mRNA levels in inflamed conditions and after action of NSAIDs.A, In situ hybridization experiments on DRG with ASIC1a and ASIC3 probes. It presents L4 DRG on the inflamed side of the animal (middle), contralateral non-inflamed DRG (left), and DRG from inflamed animal treated with aspirin (right). Arrowheads show examples of unlabeled (white) and labeled (black) cells (on 5 animals at least). B shows semiquantitative RT-PCR results. Top, Representative experiments showing RT-PCR in normal (−) and inflamed (+) conditions (−NSAID) and after treatment with NSAID (+NSAID, here aspirin). Bottom, Induction factors (mean ± SEM) of mRNA levels by inflammation (Freund) and after inflammation plus treatment with dexamethasone or NSAIDs (n = 3–10 animals per experiment). Band densities are measured and normalized to that of actin (act) in the same preparation, and the induction factor corresponds to the ratio of the normalized densities in treated over untreated conditions.
The transcript levels of the different vanilloid receptors were measured on the same samples. No difference was observed in VR1 transcript level between normal and inflamed conditions, contrary to ASICs (Fig. 2B). No changes were observed for the splice variant VR5′sv (induction factor of 0.8 ± 0.1) or for VRL1 (induction factor of 0.9 ± 0.3).
If inflammatory conditions trigger a significant increase in mRNA levels for some of the ASICs, anti-inflammatory corticoids such as dexamethasone completely abolish this increase of expression for all of the increased ASICs (Fig. 2B). This increase is also suppressed by NSAIDs such as aspirin, diclofenac, and ibuprofen (Fig.2A,B) and also salicylic acid, nimesulide, NDGA, Zileuton, or MK-886 (data not shown). None of these compounds have an effect on ASIC expression in normal animals. The action of these compounds on ASIC mRNA expression is probably attributable to the lowering of inflammatory mediators with a return to a more normal “non-inflamed” phenotype.
ASIC-type currents are sensitive to NSAIDs in DRG neurons
Under our experimental conditions, DRG neurons express three main types of H+-induced currents (Escoubas et al., 2000) as it has been described for trigeminal ganglion sensory neurons (Krishtal and Pidoplichko, 1981b). Sustained type 1 shows a small rapid transient current, followed by a sustained current that remains open as long as the acid stimulus is applied. Slow-inactivating type 2 is a transient current with a slow inactivation rate. Fast-inactivating type 3 has a large transient phase with a fast rate of inactivation, followed by a sustained phase, relatively small compared with the peak current amplitude. Salicylic acid or aspirin (500 μm) inhibits the transient current of type 1 and type 2 responses and the sustained current of type 1 and type 3 responses (Fig.3A,B), all in a reversible manner (data not shown). The fast component of type 3 response is not altered at the concentrations used. The type 2 response is reversibly inhibited by flurbiprofen (Fig. 3C), and this flurbiprofen-inhibited current is sensitive to 10 nm psalmotoxin-1 (PcTX1), a spider toxin that has been shown to be specific for ASIC1a (Escoubas et al., 2000) (Fig.3C). Acetaminophen (500 μm) or 200 μm tolmetin, piroxicam, or etodolac have no effect on H+-gated currents.

Action of NSAIDs on DRG cells in primary culture. Salicylic acid (A) and aspirin (B) inhibit H+-induced currents; inset in A shows a 2.5× magnification of the inhibition. C, Flurbiprofen (500 μm) reversibly inhibits the psalmotoxin-1-sensitive current (n = 9). D, Capsaicin-activated currents are insensitive to salicylic acid.E, Current-clamp experiments show that aspirin reduces acid-induced spiking on DRG neurons.
DRG neurons also contain another type of pH-sensitive current, the capsaicin-induced current (Bevan and Yeats, 1991). This current is sensitive to neither salicylic acid (Fig. 3D) nor flurbiprofen, aspirin, and diclofenac, at pH 7.3 or 6.0.
After exposure to pH 6, DRG neurons depolarize and trains of action potentials are generated. Aspirin (500 μm) suppresses this acid-induced repetitive activity, and this inhibition is reversible (Fig. 3E). Diclofenac (200 μm), 500 μm salicylic acid, or 500 μm flurbiprofen have the same effect. This shows that H+-induced currents are able to trigger action potentials on sensory neurons and that the inhibition of these currents by NSAIDs prevents the electrical activity attributable to acidification of the extracellular medium.
ASICs expressed in COS cells are directly inhibited by NSAIDs
Because flurbiprofen selectively inhibits the PcTX1-sensitive current (Fig. 3C), we tested its inhibitory effect on ASIC1a channels expressed in COS cells (Fig.4A,B). It blocked the channel activity with an IC50 of 349 ± 40 μm (Fig. 4C). Its analog ibuprofen has the same effect.

Action of NSAIDs on ASIC1a transfected in COS cells. Flurbiprofen (A) or ibuprofen (B) inhibit the current in a dose-dependent manner (C) (5–10 cells per data point).
Aspirin (n = 9) or salicylic acid (n = 12) (500 μm) do not inhibit ASIC1a. This result suggests that the native type 2 responses in DRGs, which have kinetics of inactivation similar to ASIC1a (Waldmann and Lazdunski, 1998), can be attributable not only to homomeric ASIC1a channels (PcTX1-sensitive currents) but also to other types of isoforms compositions that lead to PcTX1-insensitive currents. Piroxicam, tolmetin, etodolac, nimesulide, or naproxen (all at 200 μm) or 500 μm indomethacin have no effect on ASIC1a activity.
ASIC1b and ASIC2a are unaltered by 500 μm aspirin, salicylic acid, or flurbiprofen or 200 μm diclofenac (data not shown).
ASIC3 generates a biphasic current with a transient fast-inactivating phase followed by a sustained phase (Waldmann et al., 1997b). Salicylic acid (IC50 of 260 ± 21 μm), aspirin, or diclofenac (IC50 of 92 ± 19 μm) inhibit the sustained current component of ASIC3 but not the transient component (Fig.5A–C). Piroxicam, etodolac, nimesulide, naproxen (all at 200 μm) or 500 μm indomethacin or acetaminophen have no effect on ASIC3 components (data not shown).

Action of NSAIDs on ASIC3 expressed in COS cells. Salicylic acid, aspirin, and diclofenac inhibit the current in whole-cell (A–C) and outside-out (D) patches. Dose dependence of the inhibition of the sustained component by salicylic acid (E) or diclofenac (F). Five to 10 cells per data point.
When outside-out patches are excised from ASIC3-transfected COS cells, extracellular application of aspirin (Fig. 5D) or salicylic acid (data not shown) inhibits channel activity. This seems to indicate that the interaction of aspirin with the ASIC3 channel is direct and does not involve intracellular messengers. It is unlikely that a COX is excised along with the channel and acts as the target for the observed NSAID action on ASIC3 because COS cells do not express COX enzymes (O'Neill et al., 1994) and some NSAIDs specific for COX-1 do not inhibit the pH-induced current.
ASIC3/ASIC2b heteromultimers (Lingueglia et al., 1997) are also inhibited by 500 μm salicylic acid (66 ± 4%;n = 6) or 200 μm diclofenac (49 ± 5%; n = 9) in the same way as for ASIC3 (data not shown).
Thus, aspirin, salicylate, and diclofenac are effective on ASIC3 and ASIC2b/3 currents and none of the other NSAIDs tested are. Ibuprofen and flurbiprofen are the only active drugs against ASIC1a currents. All of the other NSAIDs tested are without significant effect.
DISCUSSION
ASIC isoforms are present in nociceptors and are increased by inflammation
Nociceptive fibers can be divided into several groups, the largest being formed by the C fibers. These nociceptors have a small cell body diameter (15–30 μm), unlike other sensory neurons (Aβ fibers, 30–50 μm; large Aα fibers, >50 μm) (Harper and Lawson, 1985). The precise localization of ASICs in DRG neuron subtypes was still not very well known. One study had reported that ASIC1 was partially coexpressed with substance P in small DRG neurons (Olson et al., 1998) and another that ASIC1a and ASIC1b were localized in peripherin-positive and -negative neurons (Chen et al., 1998).
The present results reveal that ASIC transcripts are present in small DRG neurons i.e., nociceptors. This is in accordance with electrophysiological data showing that currents associated with H+-activated channels, and particularly ASIC1a- and ASIC3-like currents, are found in half of the small DRG neurons (Krishtal and Pidoplichko, 1981a; Bevan and Yeats, 1991), consistent with a prominent role of these channels in acid perception. These H+-induced currents are recorded in part in IB4-positive cells, as well as in SP-positive cells (Petruska et al., 2000). These H+-sensitive currents, which are inhibited by amiloride, are distinct from the capsaicin-sensitive H+-induced currents because their characteristics are different and they are not present in the same population of cells (Petruska et al., 2000). The two families of channels probably have distinct roles in acid perception. One important example is ASIC3. It has been shown to be the main sensor of acid variations in the cardiac nociception system (Sutherland et al., 2001), and it is present in nociceptors that barely express a response to capsaicin.
In inflammatory conditions, the transcript levels of different ASIC isoforms are highly increased. This could signify a particular sensing role for these channels during inflammation. Moreover, the ASIC1a transcript appears in medium-sized cells in this condition. These newly ASIC1a-expressing cells probably could correspond to Aβ fibers. This is suggested by results of Neumann et al (1996), who have shown that inflammation causes a phenotypic switch of a population of Aβ neurons that then acquires a pain fiber resembling phenotype by newly expressing SP (Neumann et al., 1996). This switch could participate in the hypersensitivity observed in inflammatory pain. With the appearance of ASIC1a in newly recruited neurons, an inflammatory acid stimulus can thus activate more fibers, increasing the excitability of spinal cord neurons via the release of SP.
The capsaicin (vanilloid) receptor VR1 is a cation channel expressed by primary sensory neurons. It is believed to play an important role in pain sensation (Tominaga et al., 1998; Caterina et al., 2000; Davis et al., 2000). VR1 is activated by vanilloid compounds and heat but also by protons (Caterina et al., 1997; Tominaga et al., 1998). However, the VR1 H+-induced current is different from the currents generated by ASICs and is not present in the same categories of nociceptors (Petruska et al., 2000). Mice lacking the capsaicin receptor, in which the H+-induced capsaicin-sensitive VR1 current is abolished, have an unchanged proportion of H+-induced capsaicin-insensitive current (Caterina et al., 2000; Davis et al., 2000). The capsaicin-activated current is insensitive to NSAIDs, and no difference was observed in VR1 transcript level between normal and inflamed conditions. Thus, it seems that, at least in inflammatory conditions, ASICs may have a pronounced role in acid perception. These findings are consistent with the observation that capsaicin-induced allodynia is not sensitive to ibuprofen (Kilo et al., 1995) and that capsazepine, a VR1 antagonist, does not prevent nociceptor activation induced by a combination of inflammatory mediators and low pH (Habelt et al., 2000).
ASIC current subtypes in DRG neurons
A comparison of NSAIDs action on native H+-induced currents on DRG neurons (Fig.3) and on heterologously expressed ASICs (Figs. 4, 5) leads to new information concerning the molecular identity of native H+-induced currents. First, there are two types of slow-inactivating type 2 currents: those generated by homomeric ASIC1a channels, which are PcTX1 sensitive (Escoubas et al., 2000) and flurbiprofen sensitive (Fig. 4) and those generated by other molecular arrangements of ASIC subunits that lead to salicylate–diclofenac sensitivity. These arrangements may contain ASIC3 subunits, because ASIC3 is the only ASIC subunit to be sensitive to salicylate and diclofenac. Second, the type 3 current can be attributable to ASIC3 alone or in combination with ASIC2b (Waldmann and Lazdunski, 1998), because kinetics are similar and because, in all cases, there is a sustained phase that is sensitive to the same NSAIDs. Finally, type 1 responses could represent a more complex situation resulting from more than one current population. The type 1 response could be partly attributable to a PcTX1-insensitive type 2 current (the transient phase is inhibited by salicylate and diclofenac, like the type 2) associated with a sustained type 3-like component, because the plateau phase of the type 1 response is sensitive to the same compounds and has the same type of amplitude (several hundreds of picoamperes) as the plateau phase of type 3.
NSAID antinociception and direct inhibition of ASICs
NSAIDs are organic acids (Fig. 6) classified according to their chemical structure: salicylic acid derivatives (aspirin and salicylate), indole (indomethacin and etodolac) and heteroaryl (diclofenac and tolmetin) acetic acids, arylpropionic acids (ibuprofen, flurbiprofen, and naproxen), enolic acids (piroxicam), para-aminophenol derivatives (acetaminophen), and alkanones. NSAIDs show different potency against COX isoforms. Flurbiprofen, indomethacin, naproxen, and aspirin are more selective for COX-1, rofecoxib, nimesulide, and etodolac are more selective for COX-2, and ibuprofen, diclofenac, salicylate, tolmetin, and piroxicam are relatively equipotent on both COX isoforms (Warner et al., 1999). Acetaminophen is a weak inhibitor of COX unless it acts on another isoform (Simmons et al., 2000).
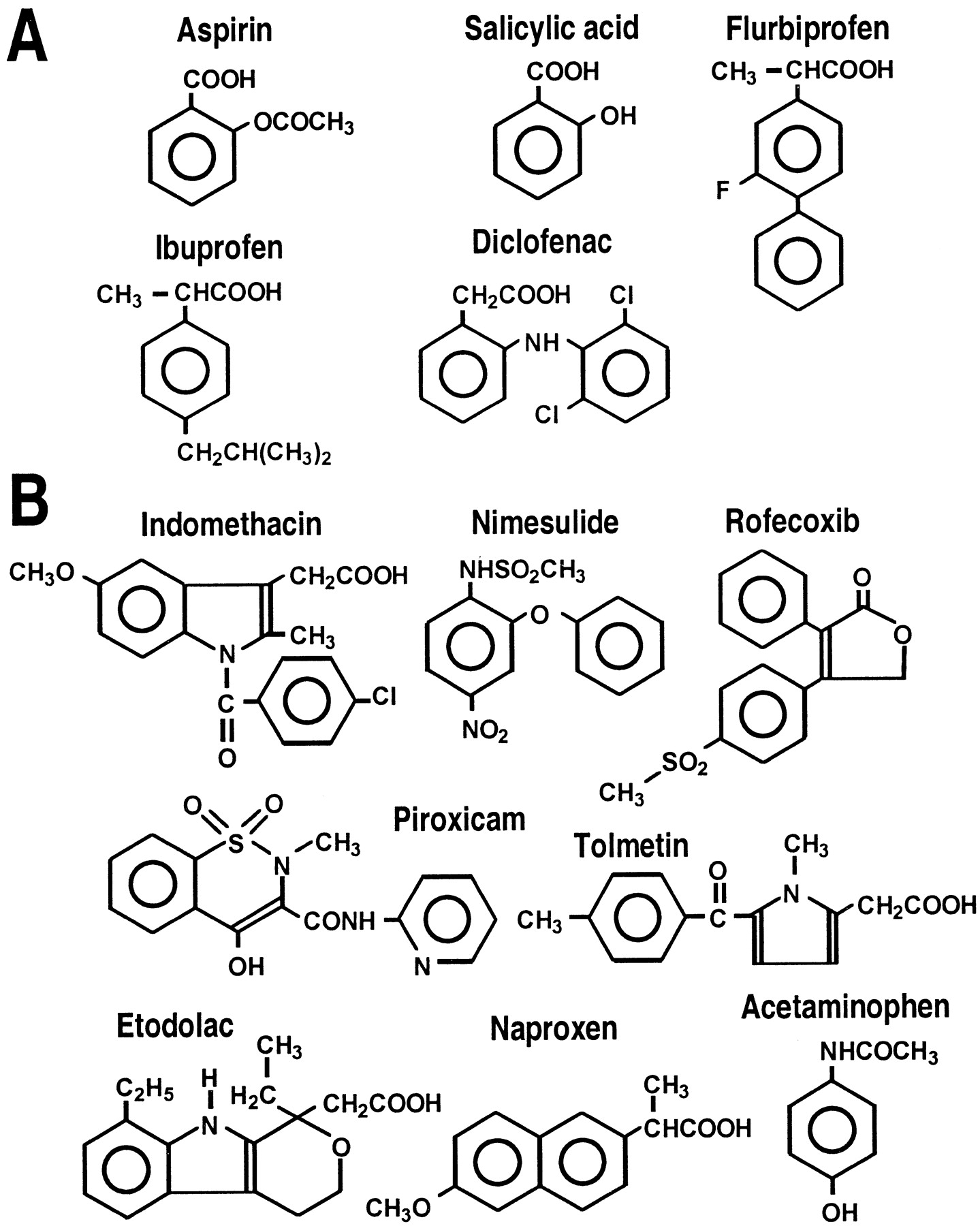
Chemical formulas of the NSAIDs tested on ASIC currents showing NSAIDs inhibiting ASIC activity (A) and those with no action on ASICs (B).
Comparing the NSAIDs that inhibit ASIC currents (Fig.6A) with those that do not (Fig.6B) leads to several observations. First, none of the used COX-2-specific drugs block ASIC activity, not even the highly COX-2-selective inhibitor rofecoxib (A. Baron and M. Lazdunski, unpublished observation). Second, only some of the COX-1-specific compounds and some NSAIDs that are not COX-isoform specific act on ASIC activity. Third, the constant motif in the ASIC-inhibiting NSAIDs is a carboxylic moiety and a benzene ring. Fourth, we can predict that aspirin, which acetylates the active site of COXs (Roth et al., 1983), does not acetylate the ASICs, because their inhibition is reversible and salicylate has the same potency as aspirin.
ASICs can thus be considered as new direct targets for NSAIDs. This is not opposite to the general view of the NSAIDs mode of action involving COXs as major targets. There has been much evidence showing that NSAIDs have other targets in addition to COXs that could mediate their analgesic actions. First, COX-2-deficient mice do not present significant differences in NSAIDs-sensitive nociception compared with normal animals (Ballou et al., 2000). Second, it has been shown often that NSAIDs have analgesic effects that are independent of their action on COXs; for example S- and R-flurbiprofen show comparable analgesic potency, although only the former inhibits COX activity (Brune et al., 1992), and diclofenac has an analgesic action that cannot be only explained by COX inhibition (Tonussi and Ferreira, 1994). In fact, a lack of correlation between the antinociceptive effects of NSAIDs and their anti-inflammatory activities has often been observed, suggesting that their analgesic properties cannot be attributable entirely to their anti-inflammatory effects (McCormack and Brune, 1991; Clarke et al., 1994; McCormack, 1994), and some COX inhibitors significantly reduce pain only when administrated at a dose 100-fold greater than necessary to inhibit COX-derived prostaglandin synthesis (Wallace, 1999).
Conclusions
One of the main conclusions of this work is that various NSAIDs are inhibitors of H+-gated channels in sensory neurons as well as cloned ASICs. The inhibition of ASICs occurs at values in the range of therapeutic doses of NSAIDs, because (1) concentrations of NSAIDs are high in inflamed areas in which they accumulate and slowly eliminate (Brune, 1977; Makela et al., 1981), (2) these compounds are often applied topically (e.g., diclofenac reaches 1–2 mm in the dermal tissue layers after skin application) (Muller et al., 1997), and (3) when given orally, high doses may be needed (e.g., aspirin and salicylate are often prescribed to reach 1–3 mm plasma concentrations) (Famaey and Paulus, 1992). Our results can also explain why topical applications of commercial solutions of NSAIDs (such as aspirin and ibuprofen) are able to relieve cutaneous pain induced by infusions of acidic solutions in humans (Steen et al., 1996). In addition to their direct effect on H+-gated channels, NSAIDs block inflammation and hence the large inflammation-induced increase of ASIC transcription. We propose that the two effects, i.e., direct channel block and inhibition of inflammation-induced ASIC expression, play an important role in the antinociceptive effects of NSAIDs in addition to their well known effects on COXs and more particularly in case of inflammation. These observations could lead to new therapeutic openings to treat pain.
Footnotes
This work was supported by the Centre National de la Recherche Scientifique, the Association Française contre les Myopathies, and the Association pour la Recherche sur le Cancer. We thank Dr. F. Kuper and Dr. A. Baron for fruitful discussion and M. Jodar and C. Widmann for expert technical assistance.
Correspondence should be addressed to Prof. Michel Lazdunski, Institut de Pharmacologie Moléculaire et Cellulaire, Centre National de la Recherche Scientifique Unité Mixte de Recherche 6097, 660 route des Lucioles, Sophia Antipolis, 06560 Valbonne, France. E-mail:ipmc{at}ipmc.cnrs.fr.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}