

Abstract
GABAA receptors are the principal sites of fast synaptic inhibition in the brain. These receptors are hetero-pentamers that can be assembled from a number of subunit classes: α(1–6), β(1–3), γ(1–3), δ(1), ε, θ, and π, but the majority of receptor subtypes is believed, however, to be composed of α, β, and γ2 subunits. A major mechanism for modulating GABAA receptor function occurs via the phosphorylation of residues within the intracellular domains of receptor subunits by a range of serine/threonine and tyrosine kinases. However, how protein kinases are targeted to these receptors to facilitate functional modulation remains unknown. Here we demonstrate that the receptor for activated C kinase (RACK-1) and protein kinase C (PKC) bind to distinct sites on GABAA receptor β subunits. Although RACK-1 is not essential for PKC binding to GABAA receptor β subunits, it enhances the phosphorylation of serine 409, a residue critical for the phospho-dependent modulation of GABAAreceptor function in the β1 subunit by anchored PKC. Furthermore, RACK-1 also enhances GABAA receptor functional modulation in neurons by a PKC-dependent signaling pathway with the activation of muscarinic acetylcholine receptors (mAChRs). This PKC-dependent modulation of neuronal GABAA receptors was mirrored by an increase in the phosphorylation of GABAA receptor β subunits with the activation of mAChRs.
Our results suggest a central role for RACK-1 in potentiating PKC-dependent phosphorylation and functional modulation of GABAA receptors. Therefore, RACK-1 will enhance functional cross talk between GABAA receptors and G-protein-coupled receptors and therefore may have profound effects on neuronal excitability.
- GABAA receptor
- protein kinase C
- receptor for activated C kinase
- muscarinic receptor phosphorylation
- cross talk
- GST fusion protein
GABAAreceptors mediate the majority of fast inhibitory neurotransmission in the brain (Rabow et al., 1995). These receptors are hetero-pentamers that can be assembled from a wide range of receptor subunits: α(1–6), β(1–3), γ(1–3), δ, ε, and θ (Rabow et al., 1995; Davies et al., 1997; Bonnert et al., 1999). In the brain most GABAA receptor subtypes are believed to be composed of α, β, and γ2 subunits (Rabow et al., 1995).
The exogenous regulation of GABAA receptor function by benzodiazepines and barbiturates is well established (Rabow et al., 1995). In contrast, the endogenous mechanisms that control receptor function remain only partly characterized, with receptor phosphorylation as a mechanism proposed to be of major significance (Brandon et al., 2000). It is evident that the GABAA receptor β1–β3 and γ2 subunits are the substrates of a number of protein kinases, including cAMP-dependent protein kinase (PKA), protein kinase C (PKC), cGMP-dependent protein kinase, and Ca/calmodulin-dependent protein kinase II (CamKII) (Moss et al., 1992a,b; Krishek et al., 1994; McDonald and Moss, 1997;McDonald et al., 1998). GABAA receptor β subunits are all phosphorylated by PKC on highly conserved serine (S) residues (S409 in the β1 and β3 subunits and S410 in the β2 subunit) (Krishek et al., 1994; McDonald et al., 1998). Moreover, for recombinant GABAA receptors the phosphorylation of these residues has been correlated directly with functional modulation (Kellenberger et al., 1992; Krishek et al., 1994; Lin et al., 1996). In addition, there is evidence from heterologous expression systems that PKC activity can modify receptor trafficking, an effect that appears to be independent of direct receptor phosphorylation (Moss and Smart, 2001). In neurons the receptor β subunits also are phosphorylated by PKC-dependent signaling pathways, and there is extensive evidence that the activation of PKC can modify neuronal GABAA receptor function (Browning et al., 1990;Brunig et al., 1999; Poisbeau et al., 1999; Brandon et al., 2000b).
Although progress has been made in identifying which GABAA receptor subunits are protein kinase substrates, presently little is understood about how kinases are targeted to these receptors to facilitate subunit-specific phosphorylation. Here we have examined the functional significance of the direct but independent interactions between the receptor for activated C kinase (RACK-1) and PKC-βII/α isoforms with GABAA receptors (Brandon et al., 1999). RACK-1 is essential for PKC-mediated phosphorylation of GABAA receptor subunits and functional modulation of the receptor by PKC-dependent signaling pathways after the activation of muscarinic acetylcholine receptors (mAChRs). Although RACK-1 is believed to function primarily as a “shuttle or anchoring protein,” facilitating the targeting of the α and βII PKC isoforms to their substrates (Chang et al., 1998; Liliental et al., 1998; Ron et al., 1999; Yarwood et al., 1999; Jaken and Parker, 2000), our results suggests a new insight into the role of RACK-1, acting to potentiate the catalytic activity of PKC anchored at GABAA receptors without affecting interaction with receptor subunits. This study demonstrates a new role for RACK-1 in modulating GABAA receptor phosphorylation and the functional modulation by PKC-dependent cell signaling pathways.
MATERIALS AND METHODS
Antibodies. The following primary antibodies were used for immunoblotting: a mouse monoclonal to RACK-1 (1:1000 dilution; Transduction Labs, Lexington, KY), a rabbit polyclonal to PKC isoforms α, β, and γ (1:500 dilution; Upstate Biotechnology, Lake Placid, NY), an antibody specific for the PKC-βII isoform (Brandon et al., 1999), or a rabbit polyclonal antiserum against the intracellular domain of the GABAA receptor (Tretter et al., 1997; Brandon et al., 1999). The following primary antibodies were used for immunoprecipitation: a mouse monoclonal antibody against the 9E10 epitope (9E10 clone, 5 μg/ml; a kind gift of G. Evan, Ludwig Institute, London, UK) (Connolly et al., 1996) and a rabbit polyclonal antibody to GABAA receptor β1/β3 subunits (Moss et al., 1992a,b; Krishek et al., 1994). To evaluate GABAA receptor phosphorylation, we used a phospho-specific antiserum that recognizes only phosphorylated serine residues 408 and 409 within the murine GABAA receptor β3 subunit (UCL 39; 1:50) (Jovanovic et al., 2001).
Expression constructs. Glutathione S-transferase (GST) fusion protein constructs of the GABAA β1 subunit were made by PCR amplification (Smith and Johnson, 1988;Brandon et al., 1999). A GST fusion protein of RACK-1 was constructed with the entire coding sequence of RACK-1 from a rat RACK-1 cDNA (Ron et al., 1994). The RACK-1 cDNA was a kind gift of D. Mochly-Rosen (University of California at San Francisco, San Francisco, CA). The 9E10-tagged GABAA receptor α1 and β1 expression constructs in pRK5 have been described previously (Krishek et al., 1994; Connolly et al., 1996).
GST protein “pull down” assays. Brains from adult Sprague Dawley rats were prepared in lysis buffer [10 mmtriethanolamine, pH 7.6, 1% Nonidet P-40, 0.5% deoxycholate, and (in mm) 150 NaCl, 5 EGTA, 5 EDTA, 50 NaF, 1 Na-orthovanadate, 100 PMSF plus 10 μg/ml leupeptin, pepstatin, antipain, and aprotinin] (Brandon et al., 1999). Insoluble material was removed by centrifugation. Lysates were exposed to GST fusion proteins on glutathione-agarose beads at 4°C for 2 hr. The beads were washed twice in 0.4% NP-40 and (in mm) 500 NaCl, 10 triethanolamine, pH 7.6, 5 EGTA, 5 EDTA, 1 Na-orthovanadate, and 1 PMSF and then twice in the same buffer supplemented with 150 mmNaCl. Proteins bound to the beads were separated by SDS-PAGE and analyzed by Western blotting or by an in vitro kinase assay and autoradiography. For the peptide competition assays after incubation with brain extract the beads were washed twice in pull down buffer, and peptides were added to a 100 nm final concentration (Billups et al., 2000). After rotation for 30 min at 4°C the beads were washed again with lysis buffer and then resuspended in SDS-PAGE sample buffer. Proteins bound to the beads were separated by SDS-PAGE and analyzed by Western blotting. The RACK-1 binding site competitive peptide had the sequence RHGVPGKGRI, whereas the scrambled control peptide had the sequence RRGGGIKPVH.
In vitro kinase assays and PKA phosphorylation. To analyze the phosphorylation of GABAA receptor subunits by associating protein kinases, we washed beads from pull down assays or immunoprecipitations twice in kinase buffer [containing (in mm) 20 Tris, pH 7.4, 20 MgCl2, 1 EDTA, 1 EGTA, 1 ouabain, 1 Na-orthovanadate, 0.1 DTT, 2 MnCl2] and resuspended them finally in 50 μl of kinase buffer containing 3–30 μCi32γP-ATP (Amersham Biosciences, Piscataway, NJ). The reactions were incubated at 30°C for 15 min. Beads were pelleted, and bound material was separated by SDS-PAGE. Phosphorylation of the β1 subunit was quantified by the use of a Bio-Rad PhosphorImager (Hercules, CA). For the PKA phosphorylation experiments (Moss et al., 1992a,b) fusion proteins were phosphorylated for 10 min at 30°C with purified PKA (Promega, Madison, WI); after extensive washing, they were exposed to brain extracts.
Filter overlay binding assays. Filter overlay assays were performed as described previously. GST fusion proteins of the GABAA receptor β1 subunit and GST alone were separated by SDS-PAGE, transferred to nitrocellulose membrane, and subjected to a guanidine denaturing–renaturing process in overlay buffer (10 mm HEPES, pH 7.5, 70 mm KCl, 5 mm EDTA). The filter was blocked in overlay buffer supplemented with 5% milk and then incubated with32P-labeled GST-RACK-1 overnight at 4°C. After extensive washing in overlay buffer, the bound32P-labeled GST-RACK-1 was detected by autoradiography.
Cell transfection and immunoprecipitation. Human embryonic kidney (HEK) 293 cells were transfected with the indicated constructs, left for 24 hr, and solubilized with a buffer containing 2% Triton X-100 (Krishek et al., 1994; Brandon et al., 1999). Brain lysates were prepared from adult rat brain under similar conditions (Brandon et al., 1999). Then GABAA receptors were isolated by using anti-β1/β3 sera coupled to protein A-Sepharose (Moss et al., 1992a,b; Krishek et al., 1994). Precipitated proteins were separated by SDS-PAGE and analyzed by immunoblotting. In some cases HEK 293 cells were labeled metabolically with [35S]methionine for 3 hr before immunoprecipitation; then the labeled receptor subunits were visualized by fluorography.
Patch-clamp electrophysiology. Whole-cell GABA currents were recorded from single HEK 293 cells by a List EPC7 amplifier. Patch electrodes (resistance, 2–6 MΩ) were filled with a solution containing (in mm): 140 KCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 11 EGTA, and 2 adenosine triphosphate, pH 7.2. Cells were superfused continuously with a Krebs' solution containing (in mm): 140 NaCl, 4.7 KCl, 1.2 MgCl2, 2.5 CaCl2, 10 HEPES, and 11 glucose, pH 7.4. Recordings were performed at 25°C, 24–48 hr after transfection voltage clamping at −40 mV (filtering at 5 kHz; −3 dB, sixth pole Bessel; 36 dB/octave). Drugs and Krebs' solution were applied rapidly to HEK 293 cells via a modified U tube. All drugs were dissolved in Krebs' solution. The RACK-1 peptides, GST fusion proteins, and PKC inhibitor peptide were dissolved directly in the patch pipette solution, whereas a stock solution of phorbol 12-myristate, 13-acetate (PMA; 0.5 mg/ml) was made in DMSO and diluted with a patch pipette solution (0.06% final DMSO concentration, which was inactive).
HEK 293 and neuronal cultures. HEK 293 cells were grown in DMEM supplemented with 10% fetal calf serum, 2 mmglutamine, 100 U/ml penicillin G, and 100 μg/ml streptomycin at 37°C in 95% air/5% CO2. Cells were electroporated with plasmids containing wild-type or mutant GABAA receptor subunit cDNAs together with a reporter plasmid expressing jellyfish green fluorescent protein. Rat superior cervical ganglion and cortical neurons were cultured as described previously (Krishek et al., 1994).
RESULTS
PKC and RACK-1 bind to distinct domains on the GABAAreceptor β1 subunit
We have demonstrated previously that RACK-1 and activated PKC-βII bind directly but independently to GABAA receptor β subunits (Brandon et al., 1999). To map the binding sites of RACK-1 and PKC in these proteins, we produced GST fusion protein constructs encoding regions of the major intracellular domain of the GABAA receptor β1 subunit (residues 302–426; GST-β1) (Moss et al., 1992a,b;Brandon et al., 1999). To identify the residues responsible for the binding of RACK-1, we incubated these proteins with recombinant RACK-1 expressed as a GST fusion protein in gel overlay assays. Using this approach, we were able to establish that RACK-1 binds to the C-terminal half of the β1 subunit intracellular domain between residues 395 and 404 (residues RHGVPGKGRI) (Fig.1A,B). The binding site for RACK-1 lies immediately upstream of Serine 409 (S409), which is a known substrate of PKC in addition to PKA and is of critical importance for GABAA receptor functional modulation with the PKC activation (Moss et al., 1992a,b, 1995; Krishek et al., 1994). However, the binding of RACK-1 to the intracellular domain of the β1 subunit is independent of S409, as shown by the positive interaction with the 366–404 construct, which does not contain this residue (Fig. 1A,B).

RACK-1 and PKC bind to differing sites on the GABAA receptor β1 subunit. A, The RACK-1 binding site was identified by using filter overlay assays. Shown is 2 μg of a series of GST fusion protein deletion constructs encoding the residues; residues 302–426 (GST-β1; lane 1), 302–365 (lane 2), 366–394 (lane 3), 395–426 (lane 4), 366–404 (lane 5), and 366–415 (lane 6) of the GABAA receptor β1 subunit intracellular domain or GST alone (lane 7) were separated by SDS-PAGE. Then duplicate gels were transferred to a nitrocellulose membrane and incubated with [32P]-labeled RACK-1 (right) or were stained with Coomassie blue to demonstrate equal loading of GST fusion proteins (left). The binding of RACK-1 was quantified with a PhosphorImager.B, The relative binding of PKC and RACK-1 to truncations of the β1 subunit intracellular domains as derived from overlay and pull down assays. +++, Binding similar (>95%) to control (GSTβ3 302–426); ++, 60% binding relative to control; —, no detectable binding. Similar results were obtained in four separate experiments.C, The PKC binding site was identified with GST pull down assays. GST fusion protein deletion constructs encoding residues 302–426 (GST-β1; lane 1), 302–365 (lane 2), 366–394 (lane 3), 395–426 (lane 4), 366–404 (lane 5), and 366–415 (lane 6) of the GABAA β1 subunit intracellular domain or GST alone (lane 7) were incubated with adult rat brain extracts, washed, and separated by SDS-PAGE and immunoblotting with anti-PKC-βII.Lane 8 represents 10% of the total brain extract that was exposed to the fusion proteins. The data are representative of three independent experiments. D, PKC associates with the β1 subunit intracellular domain independently of S409. GST fusion proteins encoding the entire intracellular domain of the β1 subunit (lane 1) or the mutant fusion protein GST-β1S409A (lane 2) or GST alone (lane 3) were incubated with adult rat brain extracts, washed, and separated by SDS-PAGE. Immunoblotting with anti-PKC-βII was performed to detect associating PKC. Lane 4represents 10% of the total brain extract that was exposed to the fusion proteins. The data are representative of three independent experiments. E, Schematic diagram of proposed distinct binding sites for RACK-1 (residues 395–404) and PKC (residues 405–415).
To identify the binding site for PKC on the intracellular domain of the GABAA β1 subunit, we used GST-affinity purification (pull down) assays from brain lysates. This methodology was used because we were unable to produce large amounts of the PKC-βII isoform (Brandon et al., 1999) required for use in gel overlay assays. PKC bound to a number of differing GABAA receptor β1 subunit constructs. This allowed us to deduce that the PKC was binding to the region between regions 405 and 415 (IRRRASQLKVKI) within the intracellular domain of the β1 subunit, straddling the phospho-acceptor site S409 (Fig.1B,C). This site is immediately downstream of where we propose RACK-1 binds and agrees with the observation that the two molecules can bind independently of each other (Brandon et al., 1999). Serine 409 is not absolutely critical for this interaction, because PKC still binds to the mutant fusion protein where S409 has been mutated to an alanine residue (GST-β1S409A) (Fig.1D) (Brandon et al., 1999). We have demonstrated previously that PKC-βII and RACK-1 interact directly in neurons andin vitro (Brandon et al., 1999), consistent with observations in other systems (Ron et al., 1999; Jaken and Parker, 2000).
Together, our observations suggest that PKC and RACK-1 bind independently to distinct residues within the GABAA receptor β1 subunit (Fig.1E). Therefore, RACK-1 may facilitate the targeting of PKC activity to its substrate S409 in this protein or play a role in regulating PKC activity at the GABAA receptor (Ron et al., 1999; Jaken and Parker, 2000).
RACK-1 facilitates PKC-mediated phosphorylation of the GABAA receptor β1 subunit
To examine further the role of RACK-1 binding to GABAA receptors, we attempted to block this interaction by using a synthetic peptide corresponding to the RACK-1 binding site (Pep-GC; RHGVPGKGRI) that we identified within the intracellular domain of the receptor β1 subunit (Fig. 1). GST-β1 was exposed to rat brain extract and then incubated with either the Pep-GC or a scrambled version of the same peptide (Pep-SC; RRGGGIKPVH). Inclusion of the RACK-1 peptide Pep-GC at 50 nm clearly abolished the binding of RACK-1 to GST-β1 (Fig.2A) without affecting the level of PKC binding to GST-β1 (Fig. 2A). The scrambled peptide did not alter the binding of either RACK-1 or PKC to GST-β1 (Fig. 2A).

Disrupting RACK-1 binding to the GABAA β1 subunit receptor reduces PKC phosphorylation of the receptor. A, Pep-GC prevents RACK-1 binding to the intracellular domain of the GABAA receptor β1 subunit. GST-β1 was exposed to adult rat brain extracts and then alone (lane 1), with 50 nm Pep-SC (lane 2), or with 50 nm Pep-GC (lane 3). Lane 4 represents 10% of the input that was used. Bound material was blotted with an anti-PKC-βII antibody (top) or anti-RACK-1 (bottom). B, GST-β1 fusion protein was phosphorylated by associated PKC activity on S409. GST-β1 (lanes 1–4) or GST-β1S409A(lane 5) was exposed to adult rat brain extracts. After extensive washing, the fusion proteins were subjected to kinase assays alone (lanes 1, 4, 5) or in the presence of either 0.1 μm (lane 2) or 0.01 μm(lane 3) PKC inhibitor peptide (PKC19–36).C, Pep-GC reduced the phosphorylation of GST-β1. GST-β1 was exposed to brain lysate and then incubated with 50 nm Pep-GC (lane 2), 50 nm Pep-SC (lane 3), or buffer alone (lane 1). After extensive washing, the bound material was subjected to in vitro kinase assays. The reaction products were separated by SDS-PAGE and analyzed by autoradiography (top). Also shown is a Coomassie stain of the fusion proteins to demonstrate equal loading (bottom). The data are representative of four independent experiments. D, The changes in phosphorylation of the β1 subunit in B were quantitated by the use of a PhosphorImager. The effects of Pep-GC and Pep-SC were calculated as the change relative to the untreated sample. Pep-GC produced a 27 ± 2.1% decrease in phosphorylation of GST-β1 compared with control (p > 0.001; Student's t test; n = 4).E, Binding of PKC to GST-β1 was modulated via phosphorylation of S409. GST-β1 (lanes 1, 2) or GST alone (lanes 3, 4) was subjected to in vitro kinase assays with purified PKA with (+) or without (−) ATP before exposure to adult rat brain. The final stoichiometry of phosphorylation of the GST-β1 in these experiments was 0.8 ± 0.05 mol/mol. Samples were washed and separated by SDS-PAGE. Immunoblotting with an anti-PKC-βII antibody was used to detect the association of PKC. The data are representative of five independent experiments. The average binding of PKC to phosphorylated GST-β1 was decreased by 90 ± 5% of control (p > 0.001; Student's t test; n = 5).
To measure whether PKC associated with GST-β1 is catalytically active, we used in vitro kinase assays. This approach revealed that GST-β1 was phosphorylated in these assays and that phosphorylation was blocked completely by a specific PKC inhibitor (Fig. 2B), confirming our immunoblotting approaches (Fig. 2A). Moreover, the sole site of phosphorylation for PKC in GST-β1 is S409 (Moss et al., 1992a,b; Krishek et al., 1994; Brandon et al., 1999) because GST-β1S409A was not phosphorylated significantly in this assay (Fig. 2B), but PKC can still bind to this protein (Fig. 1D). Together, these results confirm that activated PKC interacts with the intracellular domain of receptor β subunits in agreement with previous observations (Moss et al., 1992a,b; Krishek et al., 1994; Brandon et al., 1999). To test the importance of RACK-1 in facilitating PKC-mediated phosphorylation of GST-β1, we exposed material associating with this protein from brain extracts to Pep-GC or the control peptide Pep-SC. After extensive washing, the effects of these agents on PKC-mediated phosphorylation of GST-β1 were analyzed. Pep-GC produced a highly significant 30 ± 2.1% decrease compared with control (p > 0.001; Student's t test;n = 4) in the PKC-mediated phosphorylation of GST-β1 (Fig. 2C,D). In contrast, Pep-SC was without effect on PKC-dependent phosphorylation of GST-β1 (Fig.2B,C).
Together, our results reveal that Pep-GC specifically blocks the binding of RACK-1 to GST-β1 without having any effect on the level of PKC binding. Moreover, the loss of RACK-1 binding correlates with a reduction in the activity of PKC bound to the GABAA receptor. Therefore, our results demonstrate that RACK-1 and PKC bind to differing but adjacent sites on the GABAA receptor β1 subunit and suggest that the role of RACK-1 at GABAA receptors is not simply to target PKC activity to these proteins (Ron et al., 1999;Jaken and Parker, 2000) but to potentiate receptor phosphorylation by associated PKC.
PKC, but not RACK-1, binding to the GABAA receptor β1 subunit is modulated via the phosphorylation of the PKC binding site
Inhibiting RACK-1 binding to the intracellular domain of the GABAA receptor β1 subunit does not appear to modify PKC binding. Although the binding of PKC to the β1 subunit residues 405–415 did not require the presence of S409 (Fig.1B,C), the possibility that phosphorylation of this residue may modulate PKC binding still remains. Therefore, we tested this possibility by using fusion proteins that had been phosphorylatedin vitro by PKA before exposure to neuronal lysates. We were unable to test the effect of PKC phosphorylation, because purified PKC would bind to the fusion proteins and interfere with additional analysis. Therefore, GST-β1 was phosphorylated via PKA, which also phosphorylates S409 (Moss et al., 1992a,b), without binding to GABAA receptor β subunits (N. J. Brandon and S. J. Moss, unpublished observation), allowing us to investigate the phospho-dependence of PKC binding to GST-β1.
Phosphorylation of GST-β1 on S409 to a final stoichiometry of 0.8 ± 0.05 mol of phosphate/mol of protein (n = 4) by PKA significantly reduces the level of PKC binding in an ATP-dependent manner (Fig. 2E) by 90 ± 5% (p > 0.001; Student's t test;n = 5). In contrast, the binding of RACK-1 to the β1 subunit intracellular domain was independent of the phosphorylation status of S409 (data not shown). Therefore, it appears evident that PKC and RACK-1 binding to GABAA receptors can be regulated differentially via the phosphorylation of S409 within the binding site for PKC on the β1 subunit.
Complexes of RACK-1, PKC, and GABAA receptors are found in neurons and HEK 293 cells
To examine the association PKC and RACK-1 with GABAA receptors of defined subunit composition and functional properties, we used transient expression of recombinant receptors in HEK 293 cells (Pritchett et al., 1989; Rabow et al., 1995;Connolly et al., 1996). HEK 293 cells expressing GABAA receptors composed of α1/β1 subunits were labeled with [35S]methionine and immunoprecipitated with anti-β1/β3. This antiserum precipitated two prominent bands of 52 and 58 kDa (Fig.3A) that were not present in untransfected cells or precipitated with control IgG and thus represent the α1 and β1 subunits, respectively, as demonstrated previously (Moss et al., 1992a,b; Krishek et al., 1994). Precipitated material also was tested for the presence of RACK-1 and PKC. Endogenous RACK-1 and PKC clearly coimmunoprecipitated with anti-β1/β3 (Fig.3B), but not with control IgG as measured by immunoblotting. Identical results were obtained when immunoprecipitation was performed with antisera against the α1 subunit.
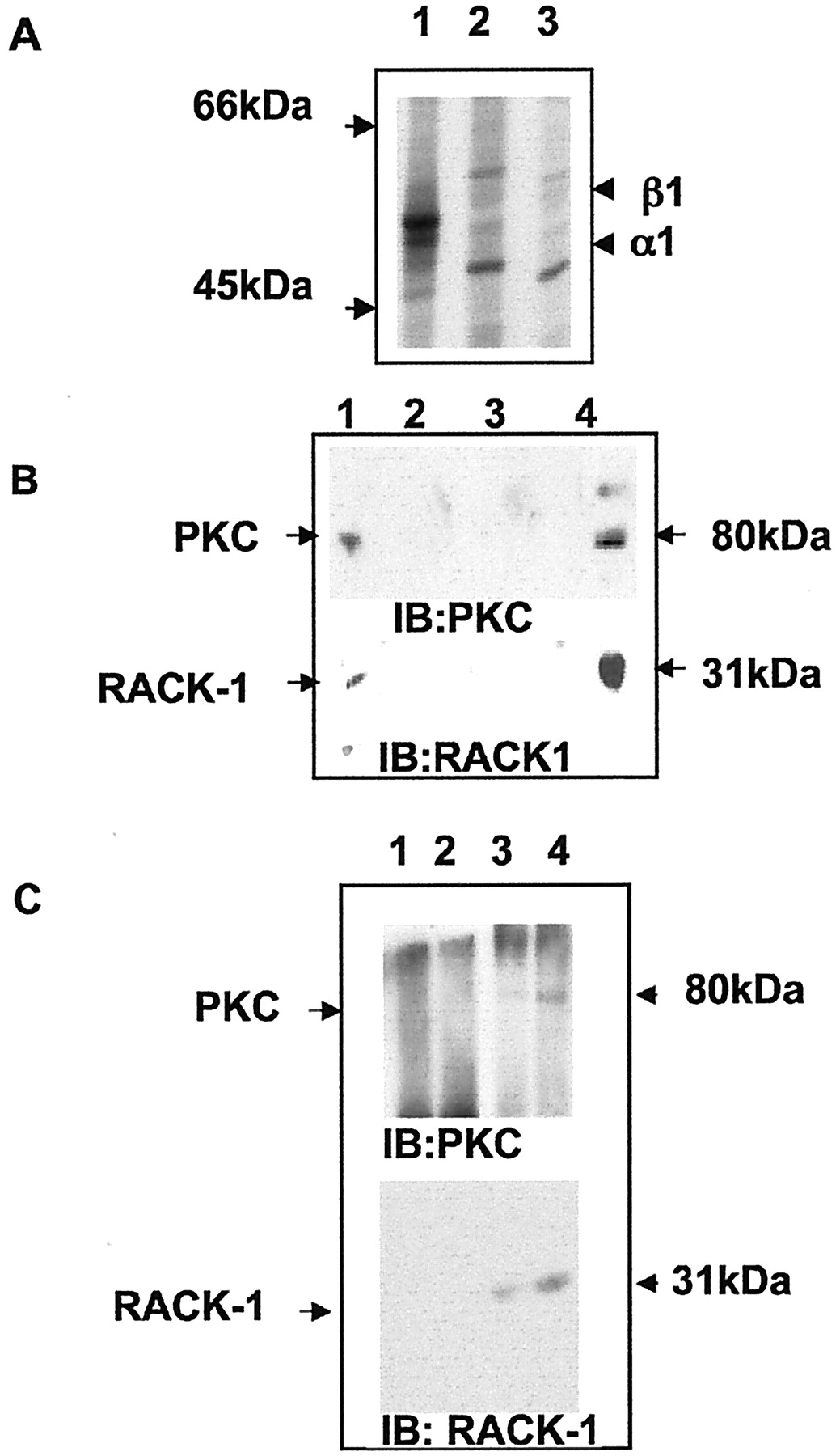
GABAA receptors form a complex with PKC and RACK-1 in HEK 293 cells and in the brain. A, Immunoprecipitation of GABAA receptors from HEK 293 cells. Transfected cells expressing the receptor α1/β1 subunits (lanes 1, 2) or control mock-transfected cells (lane 3) were labeled with [35S]methionine and lysed. Cell extracts then were immunoprecipitated with anti-β1/β3 antisera (lanes 1, 3) or control nonimmune IgG (lane 2). Precipitated material was separated by SDS-PAGE, and the receptors were visualized by autoradiography. B, Coimmunoprecipitation of RACK-1 and PKC with GABAA receptors from HEK 293 cells. HEK 293 cells expressing receptor α1/β1 subunits (lanes 1, 2) or mock-transfected cells (lane 3) were immunoprecipitated with anti-β1/β3 (lanes 1, 3) or nonimmune IgG (lane 2). Cell extracts were immunoblotted with an anti-PKC-βII antibody (top) or with an antibody specific for RACK-1 (bottom). Lane 4 represents 20% of the material that was used for the immunoprecipitations. C, PKC and RACK-1 form complexes with GABAA receptors in vivo. Adult rat brain extracts (1 mg of total protein, lanes 1, 3; 5 mg of total protein, lanes 2, 4) were subject to immunoprecipitation with either anti-GABAA β1/β3 antisera (lanes 3, 4) or nonimmune IgG (lanes 1, 2). Precipitated material was separated by SDS-PAGE; then immunoblotting was performed with an antibody against PKC-βII (top) or with anti-RACK-1 antibody (bottom).
To determine whether RACK-1 and PKC are associated with neuronal GABAA receptors, we again used immunoprecipitation, followed by Western blotting. PKC and RACK-1 coimmunoprecipitate with GABAA receptor β1/β3 subunits from adult rat brain with the use of antisera against the receptor β1/β3 subunits (Brandon et al., 1999), but not with control IgG (Fig. 3D). Therefore, these observations demonstrate that complexes of GABAA receptors RACK-1 and PKC are found in neurons and heterologous systems.
PKC modulation of GABAA receptors via direct phosphorylation of receptor β subunits is enhanced by RACK-1
The effects of RACK-1 on GABAA receptor modulation by PKC activity were examined first, by using patch-clamp recording, in HEK 293 cells expressing receptors composed of α1β1 subunits, which are modulated negatively by PKC activity (Krishek et al., 1994). This subunit combination was chosen because it represents the simplest combination of GABA-sensitive functional receptors that are modulated by direct PKC-mediated phosphorylation of a single biochemically defined phosphorylation site within the β1 subunit. Control GABA-activated currents were stable for 50 min after formation of the whole-cell recording mode. Activation of PKC with the phorbol ester PMA, externally perfused via the Krebs' solution, reduced the peak amplitude of the GABA-activated current by up to 46 ± 5% (Fig. 4A,E), which was mediated via the phosphorylation of S409 by PKC in the β1 subunit (Krishek et al., 1994). Control currents are well maintained in these cells, with no rundown evident after 50 min of recording (Krishek et al., 1994) To disrupt the interaction between RACK-1 and GABAA receptors, we included GST fusion proteins encoding parts of the intracellular domain of the receptor β1 subunit in the patch pipette solution to compete for RACK-1 binding. In the presence of the GST fusion protein 366–404, which contains the RACK-1 binding site, PKC-dependent regulation of GABAAreceptor function is blocked almost totally (Fig.4B,E). In contrast, fusion proteins that do not contain the RACK-1 binding site (either residues 302–365 or 366–394) (Fig. 4C,E and D,E) or GST alone (data not shown) were without effect. To analyze further the role of RACK-1 in PKC-dependent regulation of GABAA receptor function, we used the decapeptide that blocked the binding of RACK-1 to the receptor β1 subunit Pep-GC. Inclusion of Pep-GC peptide in the patch pipette significantly blocked the reduction in peak amplitude of the GABA-activated current after PKC activation with 500 nm PMA (Fig. 5), similar to our observations that used the GST-β1 366–404 fusion protein (Fig. 4B,E). In contrast, the control-scrambled peptide, Pep-SC, did not reduce PKC-dependent inhibition of GABA-activated currents (Fig. 5). As an additional control for any potential background effects of Pep-GC that are not related to the PKC-dependent phosphorylation of S409, we assessed the activity of this peptide in HEK 293 cells coexpressing the GABAA receptor α1 subunit and a mutant β1 subunit in which S409 had been replaced by an alanine residue (β1S409A; data not shown). In contrast to cells expressing wild-type receptors, PKC activation had no effect on the GABA-activated response in cells expressing receptors composed of α1β1S409A subunits. Moreover, Pep-GC had no significant functional effect on these receptors when included in the patch pipette, suggesting that for the wild-type receptor it was the prevention of RACK-1 binding to the β1 subunit that caused the reduction in GABA current inhibition after PKC activation (data not shown). Together, our results illustrate that RACK-1 binding to GABAA receptors composed of α1/β1 subunits facilitates the PKC-dependent regulation of receptor function via phosphorylation of S409 in the β1 subunit.

PKC modulation of GABAA receptor activation is dependent on RACK-1. GABA-activated currents were recorded under whole-cell voltage clamp at 25°C from single HEK 293 cells expressing α1β1 GABAA receptors at a holding potential of −50 mV. GABA (10 μm) was used to activate a near-maximal response and was applied for the durations indicated at 5, 10, 15, and 25 min after formation of the whole-cell recording mode (P + t min) either in the absence (solid lines) or presence (broken lines) of 100 nm PMA. The patch pipette solution contained the control or unsupplemented solution (A; also see Materials and Methods) or incorporated a GST fusion protein of the intracellular domain of the β1 subunit, including the RACK-1 binding site (120 μg/ml; B) or the GST fusion proteins, but not including the RACK-1 binding site, sequence 302–365 (C) and sequence 366–394 (D) at 200 μg/ml. The inhibition of peak GABA-activated currents caused by PKC activation for each protocol is indicated in E. The filled bar (mean ± SEM) represents the control 10 μm GABA-activated current at P + 1 min to which all subsequent responses to GABA in each cell were normalized. The open bar (+PMA) andshaded bars (patch pipette solution supplementation with Pep-GC, GST-320–365, or GST-366–394) represent current amplitudes measured at P + 30 min in n = 4–5 cells. Thehorizontal broken lines indicate, for reference, the mean inhibition induced by 100 nm PMA in control HEK 293 cells recorded with unsupplemented patch pipette solution (E, top). The time calibration is 5 sec, and the membrane current calibrations are 100 pA (A) and 300 pA (B–D). *Significantly different from control (p > 0.01; Student's ttest; n = 4).

Time dependence of RACK-1 peptides in modulating GABA-activated currents. HEK 293 cells expressing α1β1 GABAA receptor subunits were exposed to 0.5 μm PMA at 25°C after the formation of the whole-cell recording mode at −50 mV holding potential. The patch pipette solution was the control solution (open circles), or the solution was supplemented with 120 μg/ml RACK-1 binding site peptide (Pep-GC;filled circles) or 120 μg/ml of a scrambled version of the same peptide (Pep-SC; open squares). The GABA-activated currents were normalized to the initial currents recorded after formation of the whole-cell recording (= 1), and PMA was applied at P + 10 min. *Significantly different from cells treated with phorbol esters alone (p > 0.05; Student'st test; n = 4).
Regulation of GABAA receptor function by PKC signaling pathways in neurons is enhanced by RACK-1
To study the role of RACK-1 in the functional modulation of neuronal GABAA receptors, we used cultured superior cervical ganglion neurons (SCG). This preparation was studied because of its relative low diversity of neuronal cell types (Lees, 1986). Moreover, we have established that zinc-sensitive GABAA receptor function in these neurons can be inhibited by PKC activity, suggesting a significant presence of receptors containing the β1 subunits (Moss et al., 1992a,b;Krishek et al., 1994). Control whole-cell voltage-clamp recordings revealed that GABA-induced responses were stable for up to 30 min without any significant rundown in the response amplitude (Fig.6). Inclusion of 500 nm PMA in the Krebs' solution produced a large reduction in the response amplitude of, on average, 40% (Fig. 6), as described previously, an effect that can be blocked completely by specific PKC inhibitors (Krishek et al., 1994). To test the role of RACK-1 in the PKC-dependent modulation of receptor function, we made recordings from SCG neurons with either Pep-GC or Pep-SC included in the patch pipette. In the presence of Pep-GC the effect of PKC activity on GABA-activated currents was reduced significantly to an inhibition of 10% compared with that observed in the presence of PMA alone (Fig. 6). In contrast, the scrambled peptide Pep-SC had no effect on PKC-mediated inhibition of GABA-activated currents (Fig. 6). Therefore, our results illustrate that RACK-1 facilitates GABAA receptor functional modulation in SCG neurons by PKC activity.

Interaction of RACK-1 with PKC modulation of GABA-activated currents in sympathetic neurons. The bar graph represents GABA-activated currents recorded at 25°C from cultured rat sympathetic ganglionic neurons [10 d in vitro (10 DIV)] at −50 mV holding potential. Peak current amplitudes were measured to 5 μm GABA at P + 25 min after formation of the whole-cell recording mode and were normalized to the currents recorded initially on achieving the whole-cell mode. The neurons were subjected to the following conditions: GABA-activated currents were recorded with control pipette solution (filled bar) and also recorded from neurons exposed to 100 nm PMA by using control pipette solution (open bar) or by using a patch pipette solution supplemented either with 120 μg/ml RACK-1 binding site peptide (Pep-GC; shaded bar) or with 120 μg/ml of the scrambled version of the RACK-1 peptide (Pep-SC; hatched bar). *Significantly different from cells treated with phorbol esters alone (Student'st test; p > 0.05;n = 4).
To analyze further the involvement of RACK-1 in the modulation of GABAA receptors by PKC activity in a more physiological context, we examined the effects of mAChR activation on GABAA receptor function. SCG neurons predominately express the M1 mAChR subtype, which mediates many of its downstream effects via the activation of PKC (Lees, 1986; Brown et al., 1997; Haley et al., 1998). Activation of mAChRs with 1 μm muscarine in SCG neurons produced a significant decrease in GABA-mediated currents similar in onset and magnitude to those seen on the direct activation of PKC. To establish whether this effect was mediated by PKC activity, we made recordings with a patch pipette solution containing a specific peptide inhibitor of PKC (PKC19–36). This inhibitor completely blocked the effects of mAChR activation on GABA-mediated currents (10 ± 5% inhibition), demonstrating that mAChR modulation of GABAA receptor function is dependent on the activation of PKC (Fig. 7). To ascertain the role of RACK-1 in mAChR modulation of GABAAreceptor function, we included in the patch pipette solution Pep-GC, which blocks the binding of RACK-1 to the GABAAreceptor β1 subunit. Pep-GC significantly reduced the effects of mAChR activation on GABA-mediated currents to 20 ± 5% inhibition (Fig. 7). The scrambled peptide was without effect on muscarine-dependent inhibition of GABA-mediated currents.

mAChR activation modulates GABA-induced currents via a RACK-1-dependent mechanism. GABA-activated currents are represented as a bar graph recorded at 25°C from cultured sympathetic neurons at 6 DIV and at 30 min after the initiation of whole-cell recording at −50 mV holding potential. All currents were normalized to the response to 5 μm GABA recorded after formation of the whole-cell recording mode and represent the mean ± SEM. GABA-activated currents were recorded in control Krebs' solution with normal pipette solution (filled bar) and also after treatment with 1 μm muscarine (musc) to activate muscarinic acetylcholine receptors. The muscarine-treated cells were recorded with normal pipette solution (open bar) and after supplementation of the pipette solution with either Pep-GC (shaded bar) at 200 μg/ml or PKC inhibitor peptide (hatched bar) at 15 μg/ml. *Significantly different from cells treated with muscarine alone (Student's t test; p > 0.05;n = 4).
To determine whether mAChR activation can modulate GABAA receptor β subunit phosphorylation, we used a phospho-specific antiserum raised to bind specifically to di-phospho S408/S409 in the β3 subunit, both of which are phosphorylated by PKC activity in neurons (McDonald et al., 1998;Jovanovic et al., 2001). Furthermore, the β3 subunit, like the β1 subunit, can bind directly both RACK-1 and PKC (Brandon et al., 1999). For these studies we used cultured cortical neurons, because it is impossible to produce sufficient numbers of SCG neurons in culture for biochemical experiments. Activation of mAChRs in cortical neurons by 1 μm muscarine produced a sixfold enhancement in the phosphorylation state of S408/S409 residues in the β3 subunit compared with control untreated samples (Fig.8); the effects of muscarine were transient, and S408/S409 phosphorylation returned to basal levels after 30 min of treatment. Importantly, the enhanced phosphorylation of the β3 subunit by muscarine was blocked completely by calfostin, a specific PKC inhibitor. Therefore, it is apparent that GABAA receptors are phosphorylated on functionally relevant residues with the activation of mAChRs in neurons by a PKC-dependent process. The transient nature of mAChR-induced GABAA receptor phosphorylation is interesting and may arise from mAChR desensitization. However, the kinetics of receptor phosphorylation observed in cortical neurons cannot be compared directly with the electrophysiological experiments in SCG neurons because of the differences in the incubation temperature of 37 or 25°C used, respectively, in these experiments.

Muscarinic acetylcholine receptor activation facilitates PKC-dependent phosphorylation of GABAA receptor β subunits. A, The level of phosphorylation of GABAA receptor β3 subunit in cortical neurons (8 DIV) under basal conditions (lane 1) or treated with muscarine (1 μm) for 10 min (lane 2), 20 min (lane 3), or 30 min (lane 4) or with muscarine for 20 min in the presence of the PKC inhibitor calfostin C (500 nm; lane 5) was assessed by SDS-PAGE and immunoblotting with either P-β3 408/409 antibody (UCL39; 1:50; top blot) or β3 antibody (Tot-β3; 1:200;bottom blot), followed by incubation with125I-anti-rabbit secondary antibody. B, The level of β3 subunit phosphorylation on S408/S409 was quantitated with a PhosphorImager. The bar graph represents the levels of β3 subunit phosphorylation on S408/S409 calculated for each treatment as a percentage of control (untreated) normalized for total β3 subunit levels under the same experimental conditions. Similar results were seen in three separate experiments.
Together, our observations suggest a central role for RACK-1 in facilitating PKC-dependent phosphorylation and functional modulation of GABAA receptors with the activation of mAChRs in neurons.
DISCUSSION
Targeting of protein kinase activity to ion channels is emerging as a critical factor in controlling their functional modulation in addition to neuronal activity (Fraser and Scott, 1999; Levitan, 1999). In the case of nicotinic acetylcholine or ionotropic glutamate receptors, direct interactions of receptor subunits with protein kinases or interactions with scaffold molecules such as A kinase-anchoring proteins known to bind PKA holoenzyme are believed to facilitate the phosphorylation of these receptors (Swope and Huganir, 1993; Yu et al., 1997; Fraser and Scott, 1999; Levitan, 1999; Westphal et al., 1999). In contrast, the mechanisms that control the targeting of signaling molecules to GABAA receptors to facilitate subunit phosphorylation and receptor functional modulation remain unknown.
To address this issue, we have investigated the functional significance of the interactions between PKC isoforms and RACK-1 with GABAA receptors (Brandon et al., 1999). The function of RACK-1 is unclear, but this protein has been implicated in the subcellular targeting of α and βII PKC isoforms (Ron et al., 1999; Jaken and Parker, 2000). This is based on observations in cell lines in which the activation of PKC, via the dopamine D2 receptor, results in the translocation of both PKC and RACK-1 to the same sites in the plasma membrane (Ron et al., 1999). RACK-1 also can interact with a number of other proteins, including integrin β subunits, phosphodiesterase isoforms, and Src (Chang et al., 1998; Liliental and Chang, 1998; Yarwood et al., 1999). Here we have established that RACK-1 and PKC isoforms bind independently but directly to adjacent sites within the intracellular domain of the GABAA receptor β1 subunit. PKC bound to residues that incorporate the only PKC phosphorylation site in this protein, S409. Although binding affinity was not dependent on the identity of this phospho-acceptor residue (Moss et al., 1992a,b;Krishek et al., 1994), it was shown to decrease with the phosphorylation of S409, which implies that covalent modification of receptor structure in this intracellular domain is important for kinase association. In contrast, RACK-1 bound to residues N-terminal to the PKC binding site and was affected neither by the presence of S409 nor by its phosphorylation status, strongly suggesting that the interaction of RACK-1 and PKC with GABAA receptors may be regulated differentially. Importantly, the specific binding of RACK-1, but not PKC, to the β1 subunit could be blocked by using a peptide corresponding to the RACK-1 binding site within this protein. Therefore, our results reveal that RACK-1 and PKC bind independently to the GABAA receptor β1 subunit, demonstrating that RACK-1 does not simply facilitate the targeting of α and βII PKC isoforms to GABAA receptors. Interestingly, the residues that mediate RACK-1 and PKC binding are partially conserved in all receptor β subunits (Ymer et al., 1989; Rabow et al., 1995). Given that most GABAA receptor subtypes incorporate β subunit isoforms (Ymer et al., 1989; Benke et al., 1994; Rabow et al., 1995; Connolly et al., 1996), this suggests an intimate association of most populations of neuronal GABAA receptors with RACK-1 and PKC isoforms. Blocking RACK-1 binding significantly reduced PKC-mediated phosphorylation of the β1 subunit on S409 without affecting PKC binding and also the inhibitory effect of PKC on GABA-activated membrane currents recorded from both recombinant and neuronal GABAA receptors. This is consistent with a role for RACK-1 in potentiating the activity of PKC bound to the β1 subunit and in facilitating phosphorylation rather than simply playing a role in PKC anchoring, especially because PKC-βII and RACK-1 can bind in the absence of GABAA receptor subunits (Brandon et al., 1999; Ron et al., 1999; Jaken and Parker, 2000). The precise mechanism of PKC-dependent modulation on GABAA receptor function is complex, with effects on both receptor internalization and channel kinetics being reported (for review, see Brandon et al., 2000a). PKC can modulate GABAA receptor gating in neurons apparently without affecting cell surface stability for receptor subtypes containing β2/β3 subunits (Brandon et al., 2000a,b). In agreement with this, the activity of GABAA receptors expressed in Xenopus oocytes or HEK 293 cells are modulated by PKC activity via direct phosphorylation of intracellular serine residues within the receptor β and γ2 subunits (Brandon et al., 2000a; Moss and Smart, 2001). However, in recombinant systems PKC activity also appears to modulate GABAA receptor cell surface stability via a mechanism that is independent of direct receptor phosphorylation (Chapell et al., 1998; Brandon et al., 2000a).
RACK-1 is also of relevance to PKC regulation of neuronal GABAA receptors. In SCG neurons, blocking RACK-1 binding dramatically reduced the PKC modulation of GABAA receptor function (Krishek et al., 1994;Brandon et al., 2000a). We also established by using SCG neurons that GABAA receptor function could be modulated by the activation of mAChRs via a PKC-dependent pathway, which inhibited receptor function. Importantly, this endogenous G-protein-coupled receptor-based mechanism in SCG neurons for the regulation of GABAA receptor function was also dependent on RACK-1 binding to receptor subunits. Interestingly RACK-1 also has been implicated in PKC-dependent modulation of GABAAreceptor function by 5-HT2 receptor activation in cortical neurons (Feng et al., 2001). Interestingly, in cortical neurons mAChR activation enhances PKC-dependent phosphorylation of the GABAA receptor β3 subunit on residues S408/S409, key sites for receptor functional modulation (Brandon et al., 2000a), suggesting that mAChR modulation of GABAA receptor function may be attributable to direct modification of receptor phosphorylation. However, to confirm this awaits the production of mutated mouse lines in which the phosphorylation of individual subunits has been ablated via homologous recombination.
In addition to binding PKC and the GABAA receptor β subunits, RACK-1 is also able to bind the tyrosine kinase Src (Chang et al., 1998). Interestingly, GABAAreceptors are modulated via direct tyrosine phosphorylation by Src of sites primarily within the γ2 subunit, and this kinase has been shown recently to bind to receptor β and γ2 subunits (Moss et al., 1995;Brandon et al., 2001). Therefore, RACK-1 also may play a central role in targeting Src to GABAA receptors to facilitate receptor tyrosine phosphorylation (Moss et al., 1995; Brandon et al., 2001).
Our results have revealed an important role for RACK-1 in the phosphorylation and functional modulation of GABAA receptors with the activation of neurotransmitter receptors that signal via PKC-dependent pathways. Therefore, RACK-1 may facilitate functional cross talk between G-protein-coupled receptor-signaling pathways and GABAA receptors. Because GABAA receptors are critical mediators of synaptic inhibition, RACK-1/PKC-dependent phosphorylation of these receptors and its functional consequences may have profound effects on neuronal excitability.
Footnotes
This work was supported by the Wellcome Trust and the Medical Research Council.
Correspondence should be addressed to Dr. Stephen J. Moss, Medical Research Council, Laboratory of Molecular Cell Biology, University College London, Gower Street, London WC1E 6BT, UK. E-mail:steve.moss{at}ucl.ac.uk.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}