

Abstract
Microglia with increased expression of the macrophage colony-stimulating factor receptor (M-CSFR; c-fms) are found surrounding plaques in Alzheimer's disease (AD) and in mouse models for AD and after ischemic or traumatic brain injury. Increased expression of M-CSFR causes microglia to adopt an activated state that results in proliferation, release of cytokines, and enhanced phagocytosis. To determine whether M-CSFR-induced microglial activation affects neuronal survival, we assembled a coculture system consisting of BV-2 microglia transfected to overexpress the M-CSFR and hippocampal organotypic slices treated with NMDA. Twenty-four hours after assembly of the coculture, microglia overexpressing M-CSFR proliferated at a higher rate than nontransfected control cells and exhibited enhanced migration toward NMDA-injured hippocampal cultures. Surprisingly, coculture with c-fms-transfected microglia resulted in a dramatic reduction in NMDA-induced neurotoxicity. Similar results were observed when cocultures were treated with the teratogen cyclophosphamide. Biolistic overexpression of M-CSFR on microglia endogenous to the organotypic culture also rescued neurons from excitotoxicity. Furthermore, c-fms-transfected microglia increased neuronal expression of macrophage colony-stimulating factor (M-CSF), the M-CSFR, and neurotrophin receptors in the NMDA-treated slices, as determined with laser capture microdissection. In the coculture system, direct contact between the exogenous microglia and the slice was necessary for neuroprotection. Finally, blocking expression of the M-CSF ligand by exogenous c-fms-transfected microglia with a hammerhead ribozyme compromised their neuroprotective properties. These results demonstrate a protective role for microglia overexpressing M-CSFR in our coculture system and suggest under certain circumstances, activated microglia can help rather than harm neurons subjected to excitotoxic and teratogen-induced injury.
- microglia
- hippocampus
- organotypic
- M-CSF
- excitotoxicity
- neuroprotection
- ribozyme
- laser capture microdissection
Introduction
Microglia play an important role during injury and infection in the nervous system. After ischemic or traumatic injury and in demyelinating diseases, microglia remove cellular debris by phagocytosis (Carson, 2002; Danton and Dietrich, 2003). During CNS bacterial and viral infections, microglia recruit leukocytes, express major histocompatibility complex class II antigens, and present antigen as part of the T-cell immune response (Nelson et al., 2002). Microglia may also inhibit the growth of CNS parasites (Luder et al., 1999). All of these actions are potentially beneficial. However, microglia may have negative effects. There is substantial evidence that activated microglia produce potentially toxic cytokines, reactive oxygen and nitrogen species, prostaglandins, and other factors that may injure neurons already compromised by disease. For example, in Alzheimer's disease (AD), microglia activated by the amyloid β (Aβ) peptide express proinflammatory cytokines, complement, nitric oxide, and superoxide free radicals that contribute to neurodegeneration (McGeer and McGeer, 2002). Similarly, after stroke, activated microglia may accelerate the death of injured neurons by secreting neurotoxic compounds, as well as recruiting large numbers of potentially neurotoxic leukocytes into the brain (Danton and Dietrich, 2003). During CNS infection, microglial-initiated leukocyte invasion, although potentially beneficial at first, may become fulminant and hence play a negative role during CNS infection (Nau and Bruck, 2002). Activated microglia may actually promote the growth of certain CNS neoplasia (Graeber et al., 2002). Clearly, microglia may have both beneficial and detrimental effects in neurologic disease.
Macrophage colony-stimulating factor (M-CSF) is a hematopoietic cytokine that is expressed in brain by neurons and glia. M-CSF induces microglial proliferation and activation and results in expression of a variety of proinflammatory effectors. Interestingly, although M-CSF activates microglia, this cytokine has also been shown to have neuroprotective properties both in vivo and in cell culture (Berezovskaya et al., 1996; Fedoroff et al., 1997; Bruccoleri and Harry, 2000; Vincent et al., 2002a). The M-CSF receptor (M-CSFR) encoded by the proto-oncogene c-fms is upregulated on microglia after traumatic and ischemic brain injury and in AD and in transgenic mouse models for AD (Raivich et al., 1998; Wang et al., 1999; Murphy et al., 2000). We showed previously that increased microglial expression of the M-CSFR on cultured microglia and on microglia in organotypic hippocampal cultures induces microglial production of proinflammatory cytokines and nitric oxide, as well as promoting phagocytosis of Aβ (Mitrasinovic and Murphy, 2002, 2003; Mitrasinovic et al., 2004). In the present study, we sought to determine the effects of microglial overexpression of c-fms on the survival of neurons subjected to excitotoxic injury and to injury induced by the teratogen cyclophosphamide (CP). Increased microglial M-CSFR expression has been linked to CP neurotoxicity (Hao et al., 2002). Because the endotoxin lipopolysaccharide (LPS) has been used to stimulate microglial activation and microglial-induced neurotoxicity (Kim and Ko, 1998; Wang et al., 2002; Xie et al., 2004), we compared the effect of microglia overexpressing the M-CSFR on neuronal survival to that of microglia treated with LPS.
Materials and Methods
Microglial-hippocampal cocultures. The coculture system consisted of exogenous BV-2 mouse microglial cells and rat organotypic hippocampal cultures, as described previously (Mitrasinovic et al., 2001). BV-2 cells maintain many of the phenotypic features of primary microglial cells [for relevant citations, see Mitrasinovic et al. (2004)]. Cells were cultured as described previously (Mitrasinovic et al., 2001) and were used for experiments at a passage number of <15. Hippocampal organotypic slice cultures of 400 μm in thickness were prepared from 7-d-old neonatal rats and cultured on Millicell-CM membranes (Millipore, Bedford, MA) in six-well dishes, as described previously (Mitrasinovic et al., 2001). Hippocampal cultures were maintained for 2 weeks before experimentation to reduce microglial activation secondary to the explant procedure. A one-level coculture system was assembled as described previously (Mitrasinovic et al., 2001) by adding BV-2 cells onto the Millicell membrane supporting the hippocampal cultures (Fig. 1 A). The initial seeding density of BV-2 cells was 4 × 103 cells/cm2. In the one-level system, BV-2 cells were in direct contact with the organotypic culture. A two-level coculture system contained the same number of exogenous BV-2 microglia, but cells were seeded underneath the insert and adhered to the bottom of the tissue-culture plate (Fig. 1 B). In the two-level system, BV-2 cells were not in direct contact with the organotypic culture.

A, Schematic diagram of one-level coculture system. Hcs, Hippocampal cultures. B, Two-level coculture system. C, RNA secondary sequence of the anti-M-CSF hammerhead ribozyme (Rz). Cleavage occurs after nucleotide 266 (M-CSF mRNA sequence NM 007778), as indicated by the arrow.
Transfections. Microglial transfections were performed using a Lipofectamine Plus (Invitrogen, San Diego, CA) protocol essentially as described previously (Mitrasinovic et al., 2001). Briefly, BV-2 cells were grown to 65% confluency (∼5 × 104 cells) in six-well culture plates and treated with the plasmid complex containing 0.2 μg of simian virus 40-promoted c-fms (pTK1) plasmid or the pZeoSV control plasmid (lacking c-fms cDNA) preincubated with the Lipofectamine Plus reagent. Another control condition was BV-2 cells treated with transfection medium containing Lipofectamine Plus alone. We showed previously that transfection of BV-2 cells with the pTK1 c-fms plasmid results in an ∼75-fold increase in M-CSFR mRNA, as well as increased M-CSFR protein, and a 2.2-fold increase in microglial expression of the ligand, M-CSF (Mitrasinovic et al., 2001). Transfection efficiency is ∼90% (data not shown). To compare the effects of c-fms transfection with that of endotoxin, a classical stimulus for microglial activation that can induce microglial injury to monotypic neuronal cultures (Kim and Ko, 1998; Wang et al., 2002; Xie et al., 2004), some BV-2 cells were treated with 5 μg/ml LPS (Sigma, St. Louis, MO). After 24 h of transfection or LPS treatment, cells were detached from the wells by mechanical pipetting, quantified, and used for assembly of the organotypic coculture.
Excitotoxicity. Neuronal toxicity was induced with NMDA (Tocris Cookson, Ballwin, MO), introduced into hippocampal growth medium at final concentrations of 5 or 100 μm at the time of the coculture assembly. Cocultures were treated with NMDA for 24 h, except where indicated otherwise in the figures. Pilot studies showed that c-fms-transfected microglia proliferated rapidly in coculture with NMDA-treated slice cultures. To test whether protection against NMDA toxicity conferred by c-fms-transfected BV-2 cells in coculture might be merely a result of increased cell numbers, we assembled cocultures containing the same density of wild-type BV-2 cells as found in c-fms-transfected cocultures after 24 h of NMDA treatment (6.0 × 104 cells/cm2). These were compared with cocultures containing the standard starting density of BV-2 cells (4 × 103 cells/cm2).
Teratogen-induced neurotoxicity. It was reported that conditioned medium from neurons treated with the neurotoxic teratogen compound CP induces microglial expression of the M-CSFR, and it was hypothesized that this promotes microglial neurotoxicity (Hao et al., 2002). To test directly the role of increased microglial M-CSFR in CP-induced neurotoxicity, cyclophosphamide monohydrate (Sigma) was dissolved in nuclease-free water (Promega, Madison, WI) at a concentration of 10 μg/ml and was added to the coculture at a final concentration of 1 μg/ml for 30 min at 37°C after assembly of the coculture system (Hao et al., 2001, 2002). Seeding density and coculture conditions were the same as described above for NMDA experiments.
Propidium iodide labeling. At the end of neurotoxin treatment, 2.5 μl of propidium iodide solution (PI; 1 mg/ml; Molecular Probes, Eugene, OR) was added to the growth medium (1.2 ml), and coculture plates were returned to the incubator for an additional 30 min. Labeling reactions were stopped by removing the growth medium, after which sections were washed two times with PBS buffer and then fixed with 4% paraformaldehyde for 20 min at room temperature before finally being washed four times with PBS. The Millicell membrane was cut from the insert and mounted on a glass slide using the SlowFade Antifade kit (Molecular Probes). Microscopy was performed on an Axioskop 2FS microscope (Carl Zeiss, Jena, Germany) in the cyanine 3 channel with a 10× objective. After image acquisition, quantitative analysis was performed with MetaMorph 5.0 software (Universal Imaging Corporation, West Chester, PA). PI fluorescence was quantified as integral regional density, which corresponds to the intensity of the PI emitted fluorescence per unit area after subtraction of the background fluorescence. All comparative measurements were taken using identical acquisition settings and mercury lamp intensity. Mercury lamp intensity was preset based on the highest intensity image to prevent pixel saturation.
FluoroJade staining. After 24 h of coculture, hippocampal slices with BV-2 cells were fixed in 4% paraformaldehyde, washed three times with PBS, and cryoprotected at 4°C in 20% sucrose before freezing. Cultures were then sectioned on a cryostat at 15 μm and mounted on glass slides. FluoroJade staining was performed following the protocol of Schmued et al. (1997). Slides were first air dried for 30 min, then treated with 100% ethanol for 3 min, 70% ethanol for 1 min, and washed with distilled water (dH2O) for 1 min. Subsequently, sections were incubated with 0.06% potassium permanganate for 15 min with gentle shaking and then washed with dH2O for 1 min. FluoroJade (Chemicon, Temecula, CA) solution was prepared as 0.001% in dH2O containing 0.1% acetic acid. Slides were incubated with the FluoroJade solution for 30 min at room temperature and then washed three times with distilled H2O. After air drying for 30 min, slides were rapidly immersed in xylene and then coverslipped before confocal microscopy.
Biolistic transfections of the organotypic cultures. Hippocampal sections were prepared as described above and after 7 d were biolistically transfected with a CD11b-c-fms plasmid or a CD11b-enhanced green fluorescent protein (EGFP) construct or the pZeoSV control vector using a Helios Gene Gun system, as described previously (Mitrasinovic et al., 2004). The CD11b promoter specifically targets microglia endogenous to the organotypic hippocampal cultures and results in increased expression of microglial M-CSFR. After biolistic transfection, slices were treated with 100 μm NMDA and returned to the incubator. After 24 h, sections were treated with PI for neurotoxicity assessment.
Cell proliferation assay. The number of exogenous microglia in coculture was monitored at 24 and 48 h. BV-2 microglia were removed from the coculture by trypsinization for 4 min and then collected by centrifugation at 13,000 rpm for 4 min. Cells were resuspended in PBS buffer and quantified with an automatic particle counter (Beckman Coulter, Miami, FL) using 4 μm as the particle threshold as described previously (Mitrasinovic et al., 2001). Each sample was counted three times, and each treatment was repeated three times.
M-CSF-deficient microglia. An anti-M-CSF hammerhead ribozyme catalytic RNA oligonucleotide was designed with Mfold software (Zuker, 2003). The sequence of the ribozyme is shown in Figure 1C (custom synthesized; Dharmacon Corporation, Boulder, CO). BV-2 microglia were grown to 5 × 104 cells per well in a six-well tissue culture plate and treated with 2 μg of the anti-M-CSF RNA oligonucleotide incubated with 15 μl of Oligofectamine (Invitrogen) reagent at 37°C for 30 min. Cells were simultaneously cotransfected with the c-fms plasmid as described above. Control transfections were performed using an RNA oligonucleotide with the same sequence but synthesized in the reverse 5′ to 3′ orientation. After 24 h, cotransfected BV-2 microglia were used for coculture assembly. Subsequent treatments and outcome measures were identical to those described above.
RNA isolation, reverse transcription, and real-time quantitative PCR. Total RNA was extracted from BV-2 cells using Trizol (Invitrogen). Reverse transcription (RT) was performed with SuperScript reverse transcriptase (Invitrogen). To quantify M-CSF and c-fms mRNA in BV-2 cells, we used a real-time PCR assay with SYBR Green (Applied Biosystems, Foster City, CA), as described previously (Mitrasinovic et al., 2001).
Laser capture microdissection. Pure samples of hippocampal neurons from microglial-organotypic cocultures were obtained as described previously (Vincent et al., 2002b), with some modifications. Cultures were frozen on dry ice, sectioned at 15 μm thickness, and mounted on glass slides for membrane-based laser capture. To visualize the neuronal layer, sections were stained with hematoxylin as described. Individual neurons were subsequently captured using a Leica Microsystems (Bannockburn, IL) AS LMD laser microdissection system. An average of 30 neuronal cells was collected from each slice culture, evenly distributed throughout the CA1-CA3 neuronal layer. Total RNA was extracted from captured neurons using the Absolutely RNA Microprep kit (Stratagene, Cedar Creek, TX) and reverse transcribed using Sensiscript (Qiagen, Valencia, CA) as described previously (Vincent et al., 2002b). Real-time PCR with SYBR Green was performed as described previously (Mitrasinovic et al., 2001). All PCR was performed in quadruplicate. To confirm that neuronal cells had been captured, we tested for neurofilament heavy chain (NF-H) expression as described previously (Vincent et al., 2002b). To test for glial contamination, RNA samples were assessed with real-time RT-PCR for expression of the astrocyte markers GFAP and S100β and the microglial marker CD11b. We then tested for neuronal expression of M-CSF, M-CSFR, brain-derived neurotrophic factor (BDNF), and the BDNF receptor variants neurotrophic tyrosine kinase receptor type 2 (NTRK2) full-length and NTRK2 T1 (truncated). Primer sequences used for real-time SYBR Green PCR are presented as supplemental material (available at www.jneurosci.org). To determine expression of the full-length (M55291) and truncated (M55292) NTRK2 splice variants, sequences were aligned, and nonoverlapping domains were used to design primers for differentiation of isoforms. Neuronal RNA samples captured from cocultures containing control, LPS-treated, and c-fms-transfected BV-2 microglia were compared. Gene expression fold changes between experimental conditions were calculated using the standard curve method [user bulletin 2 (1997); Applied Biosystems]. To adjust for RNA loading, we quantified glyceraldehyde-3-phosphate dehydrogenase expression in each sample.
Statistical analyses. Data were analyzed using one- and two-way ANOVAs. Post hoc comparisons between means were performed using the Scheffé correction to control for type 1 error. All data points represent a minimum of three experimental replicates. For neuroprotection experiments, each data point represents a minimum of six replicates, each replicate consisting of data from three individual slice cultures.
Results
Microglia overexpressing the M-CSFR were found to be neuroprotective. Figure 2 illustrates the effects of overexpression of the M-CSFR by exogenous microglia on NMDA-induced neurotoxicity in organotypic hippocampal cultures. A two-way ANOVA revealed a significant interaction between the presence or absence of NMDA and microglial treatment/transfection condition (p < 0.0001). Cocultures containing microglia overexpressing the M-CSFR and treated with NMDA showed significantly less neurotoxicity (Scheffé-corrected p < 0.05) than hippocampal cultures treated with NMDA, NMDA-treated cultures grown with BV-2 cells transfected with the control plasmid, and NMDA-treated cocultures containing exogenous microglia pretreated with LPS. There were no significant differences in neurotoxicity among the other treatment conditions after NMDA treatment. Among cultures not treated with NMDA, there were no significant differences among treatment conditions.
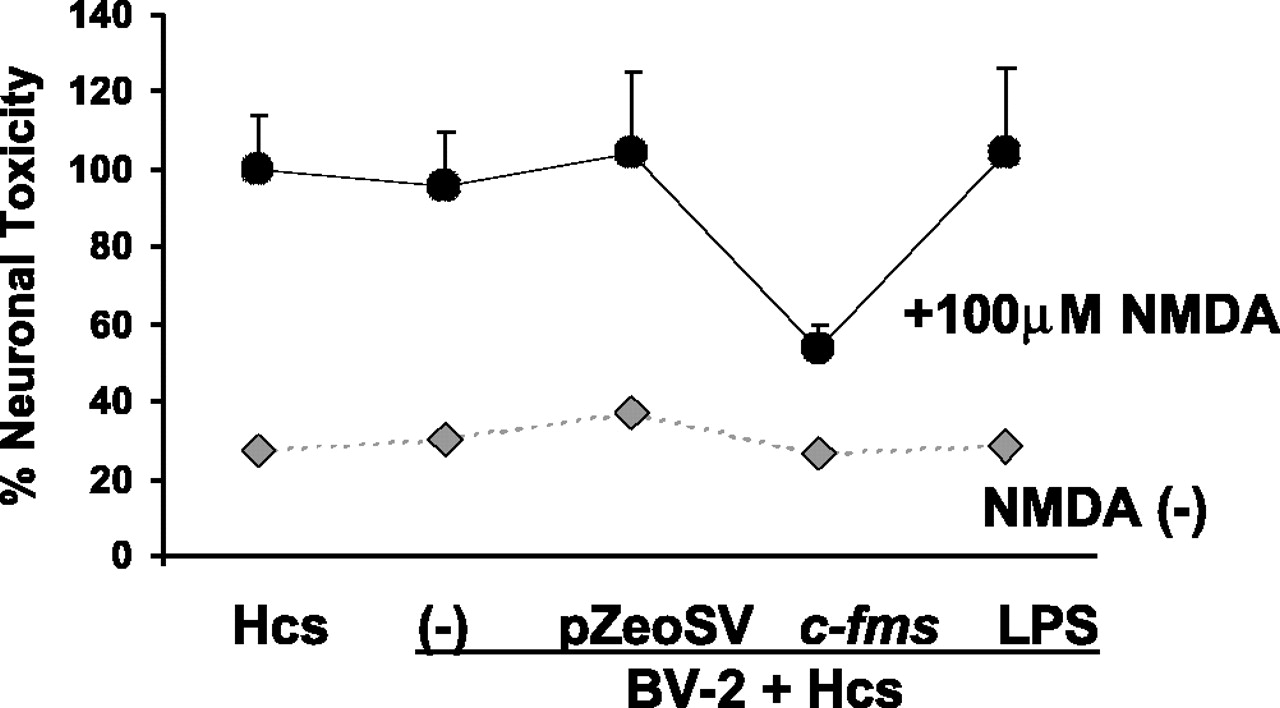
Neuroprotective effects of BV-2 microglia overexpressing M-CSFR in the microglial organotypic coculture system after NMDA treatment. The black data points represent NMDA-treated cocultures, whereas the gray data points represent cocultures not treated with NMDA. The y-axis represents PI staining calculated as a percentage of the staining seen hippocampal cultures (Hcs) alone (no microglia) treated with NMDA. This value is compared with a coculture system containing nontransfected microglia treated with transfection medium alone (-), microglia transfected with the control plasmid (pZeoSV), the c-fms plasmid (c-fms), or pretreated with LPS (LPS). Only c-fms-transfected exogenous microglia were neuroprotective. Error bars represent SD.
Examination of PI-stained sections revealed that the decrease in NMDA-induced neurotoxicity occurs within both the CA1 and CA3 regions of the hippocampus, as well as in the dentate gyrus (Fig. 3A). In PI-stained sections, BV-2 cells surrounding the organotypic culture never showed any signal (Fig. 3B; compare with phase-contrast images in Fig. 5A). PI fluorescence was limited to the neuronal layer, and no signal was observed in neuropil regions of the hippocampus despite the presence of numerous overlying BV-2 cells. To verify that PI signal was caused by nerve-cell injury and not caused by PI uptake by other cells endogenous to the organotypic culture, we examined FluoroJade as a highly specific marker for neuronal injury. Results showed strong FluoroJade staining in sections from hippocampal slices treated with NMDA and cultured with wild-type BV-2 cells (Fig. 3D) but little staining in slices cocultured with BV-2 cells overexpressing the M-CSFR (Fig. 3C).

PI labeling of neurotoxicity in the microglial-hippocampal coculture system. A, Left column, Hippocampal cultures (Hcs) treated with 5 or 100 μm NMDA, no BV-2 cells. Middle column, Hippocampal cultures treated with 5 or 100 μm NMDA and cocultured with BV-2 microglia transfected with c-fms construct. Right column, Hippocampal cultures treated with 5 or 100 μm NMDA and cocultured with BV-2 microglia treated with transfection medium alone (-). B, High-power view of the edge of microglial-hippocampal organotypic coculture treated with NMDA and containing wild-type BV-2 cells. The strong PI staining is in the pyramidal layer (py) of CA1. The stratum oriens (or) shows no staining, and the Millicell membrane (mem) adjacent to the slice, although containing numerous BV-2 cells, shows no PI signal. C, Confocal image of section from a microglial-organotypic coculture containing BV-2 cells transfected to overexpress the M-CSFR and treated with NMDA and stained with FluoroJade. There was no FluoroJade signal visible using acquisition settings identical to those in D. In C, the overall brightness was increased after image acquisition to make the CA3 and granule cell layer landmarks faintly visible. D, Confocal images of section from sister coculture stained with FluoroJade. The coculture contained wild-type BV-2 cells transfected to overexpress the M-CSFR and was treated with 100 μm NMDA. Note the strong neuronal staining in the CA3 and granule cell layers (gcl). Scale bar, 100 μm.

Microglial proliferation induced by c-fms transfection and NMDA treatment in organotypic cocultures. A, Phase-contrast images of microglial-organotypic cocultures treated with NMDA. The middle panel shows strong proliferation of BV-2 cells transfected with c-fms [BV-2 (c-fms) + Hcs] compared with nontransfected cells [BV-2 (-) + Hcs] and BV-2 cells pretreated with LPS [BV-2 (LPS) + Hcs]. B, Quantitative analysis of total BV-2 cell numbers per well in cocultures containing BV-2 cells transfected with the control plasmid (pZeoSV), the c-fms plasmid, or pretreated with LPS, with (right) and without (left) the addition of NMDA, after 24 and 48 h. Hcs, Hippocampal cultures. Error bars represent SD.
Because LPS-pretreated BV-2 cells failed to protect neurons from excitotoxic injury in coculture, we examined the expression profile of these cells. Previously, we showed that LPS induces the expression of proinflammatory cytokines such as macrophage inflammatory protein-1α (MIP-1α) by BV-2 cells (Murphy et al., 1995). Transfection with the c-fms construct also induces MIP-1α production by BV-2 cells (Mitrasinovic et al., 2001). However, in the present study, LPS treatment of BV-2 cells resulted in no significant change in expression of M-CSF (p > 0.10) and actually downregulated the expression of M-CSFR by 51% (p < 0.05) in comparison with untreated cells. This is in contrast to c-fms transfection, which results in increased M-CSF and M-CSFR expression (Mitrasinovic et al., 2001).
To test whether neuroprotection conferred by c-fms-transfected microglia occurred only in the coculture system, we used biolistics to induce c-fms overexpression in microglia endogenous to the organotypic culture, as demonstrated previously (Mitrasinovic et al., 2004). Biolistic transfection with the CD11b/c-fms construct resulted in a significant average reduction of over 94% in neurotoxicity compared with control plasmid and CD11b/EGFP transfections (Fig. 4) (Scheffé-corrected p < 0.05). In fact, there was no significant difference between mean CD11b/c-fms-induced PI staining and that observed in untreated organotypic cultures.

Biolistic expression of the M-CSFR on microglia in organotypic hippocampal cultures protects neurons from NMDA. The y-axis shows PI signal expressed as a percentage of nontransfected hippocampal cultures (Hcs). Other conditions included biolistic transfection with the control vector (VCD11b), with the plasmid containing an EGFP cDNA (GFP), with the plasmid resulting in overexpression of M-CSFR on microglia in the slice culture (c-fms), or nontransfected slices not treated with NMDA [Hcs NMDA (-)]. Error bars represent SD.
We observed increased numbers of c-fms transfected BV-2 cells near organotypic slices in the coculture system after NMDA treatment (Fig. 5A, middle). In untreated cocultures, BV-2 cells transfected to overexpress c-fms showed significantly greater proliferation after 24 and 48 h than both control vector-transfected cells and LPS-treated cells (Fig. 5B, left) (Scheffé-corrected p < 0.05). A similar pattern was seen in coculture with organotypic slices treated with NMDA (Fig. 5B, right), although at both 24 and 48 h, c-fms-induced proliferation was significantly greater than in cocultures not treated with NMDA (t tests; p < 0.01 for both).
We tested whether neuroprotection was a function of cell density by coculturing wild-type BV-2 cells at a density equivalent to that observed with c-fms-transfected cells, which proliferate rapidly. NMDA-induced neurotoxicity in cocultures containing a higher density of wild-type BV-2 cells was 84.4% (SD, 35.0) of that in cocultures with the standard starting density. This difference was not statistically significant. To test whether c-fms-induced neuroprotection might be a result of nonspecific activation of BV-2 cells in combination with increased cell density, we compared cocultures containing LPS-treated BV-2 cells using the standard starting density to cocultures containing a density typically present in cocultures with c-fms-transfected BV-2 cells. NMDA-induced neurotoxicity in cocultures containing an increased density of LPS-treated BV-2 cells was 93.26% (SD, 20.5) of that in cocultures containing the standard BV-2 starting density. This difference was not statistically significant.
It has been proposed that increased microglial M-CSF and M-CSFR expression is a cause of CP-induced neurotoxicity (Hao et al., 2001, 2002). We directly tested the role of increased M-CSFR expression on CP-induced neuronal injury. Overexpression of c-fms rendered BV-2 cells neuroprotective in coculture with CP-injured slices. Neuronal injury in cocultures containing c-fms-transfected BV-2 cells was decreased by an average of 98% in comparison with cocultures containing wild-type or control plasmid-transfected BV-2 cells (Fig. 6A) (Scheffé-corrected p < 0.05) but was not significantly different from that observed in untreated slice cultures. We also noted increased proliferation of c-fms-transfected BV-2 cells when cultured with CP-injured slices (data not shown). Rather than promoting CP-induced neurotoxicity, microglial overexpression of the M-CSFR may be protective. These results also show that the neuroprotective effect of microglial M-CSFR overexpression is not limited to NMDA-induced injury.

A, Neuroprotection by c-fms-transfected exogenous microglia in the microglial-organotypic coculture system after treatment with the teratogen CP. Data points represent PI signal for CP-treated hippocampal cultures alone [Hcs CP (+)] and cocultures with nontransfected BV-2 cells (-), control vector transfected (pZeoSV), and c-fms transfected (c-fms). Also shown are results for untreated hippocampal cocultures containing nontransfected BV-2 cells (Hcs CP-). All data are expressed relative to PI signal in Hcs alone treated with CP. B, Loss of neuroprotection by BV-2 microglia overexpressing the M-CSFR in a two-level coculture system. Black data points represent PI signal in NMDA-treated cultures, whereas gray data points show signal for cocultures not treated with NMDA. All data are expressed as a percentage of PI signal in hippocampal cultures (Hcs) alone treated with NMDA. BV-2 (-), Nontransfected cells; c-fms, c-fms-transfected cells; LPS, LPS-pretreated BV-2 cells. Error bars represent SD.
To determine whether the neuroprotective effects of c-fms-transfected BV-2 cells were dependant on close proximity between exogenous BV-2 microglia and cells endogenous to the slice culture, we compared the direct coculture system (one-level) (Fig. 1A) to cocultures in which the BV-2 cells were grown on the bottom of the culture plate well and not in contact with the organotypic culture grown on the Millicell membrane above (two-level) (Fig. 1B). Results showed that c-fms-transfected BV-2 cells in the two-level system provided no neuroprotection in comparison with control or LPS-treated BV-2 cells (Fig. 6B, right plot) (ANOVA; p > 0.10). In parallel one-level cocultures with c-fms-transfected BV-2 cells, the characteristic decrease in NMDA neurotoxicity was observed as expected (Fig. 6B, left plot) (Scheffé-adjusted p < 0.05).
To characterize the transcriptional response of neurons subjected to excitotoxic injury in coculture with exogenous microglial cells, we used laser capture microdissection. We found a marked increase in expression of M-CSF and the M-CSFR in neurons captured from NMDA-treated hippocampal cultures grown with microglia overexpressing the M-CSFR compared with cultures grown with control or LPS-treated BV-2 cells (Fig. 7A,B) (Scheffé-adjusted p < 0.05). LPS-treated BV-2 cells resulted in a significant increase in neuronal BDNF (Fig. 7C) (Scheffé-adjusted p < 0.05), whereas the increase induced by microglial M-CSFR overexpression was not statistically significant. Pretreatment of BV-2 cells with LPS resulted in a significant increase in the neuronal expression of the NTRK2 full-length isoform, but coculture with cells overexpressing the M-CSFR caused a further significant increase in neuronal expression of this isoform (Fig. 7D) (Scheffé-adjusted p < 0.05). Significantly increased expression of the BDNF receptor isoform NTRK2 T1 was seen only in cultures containing BV-2 cells overexpressing the M-CSFR (Figs. 7E) (Scheffé-adjusted p < 0.05), whereas the LPS-induced change was not significant. The purity of the captured neuronal cells was verified with real-time RT-PCR. Whereas expression of NF-H was strong (CT value of 28 cycles), real-time RT-PCR signals for the glial markers GFAP, S100β, and CD11b were not above background levels.
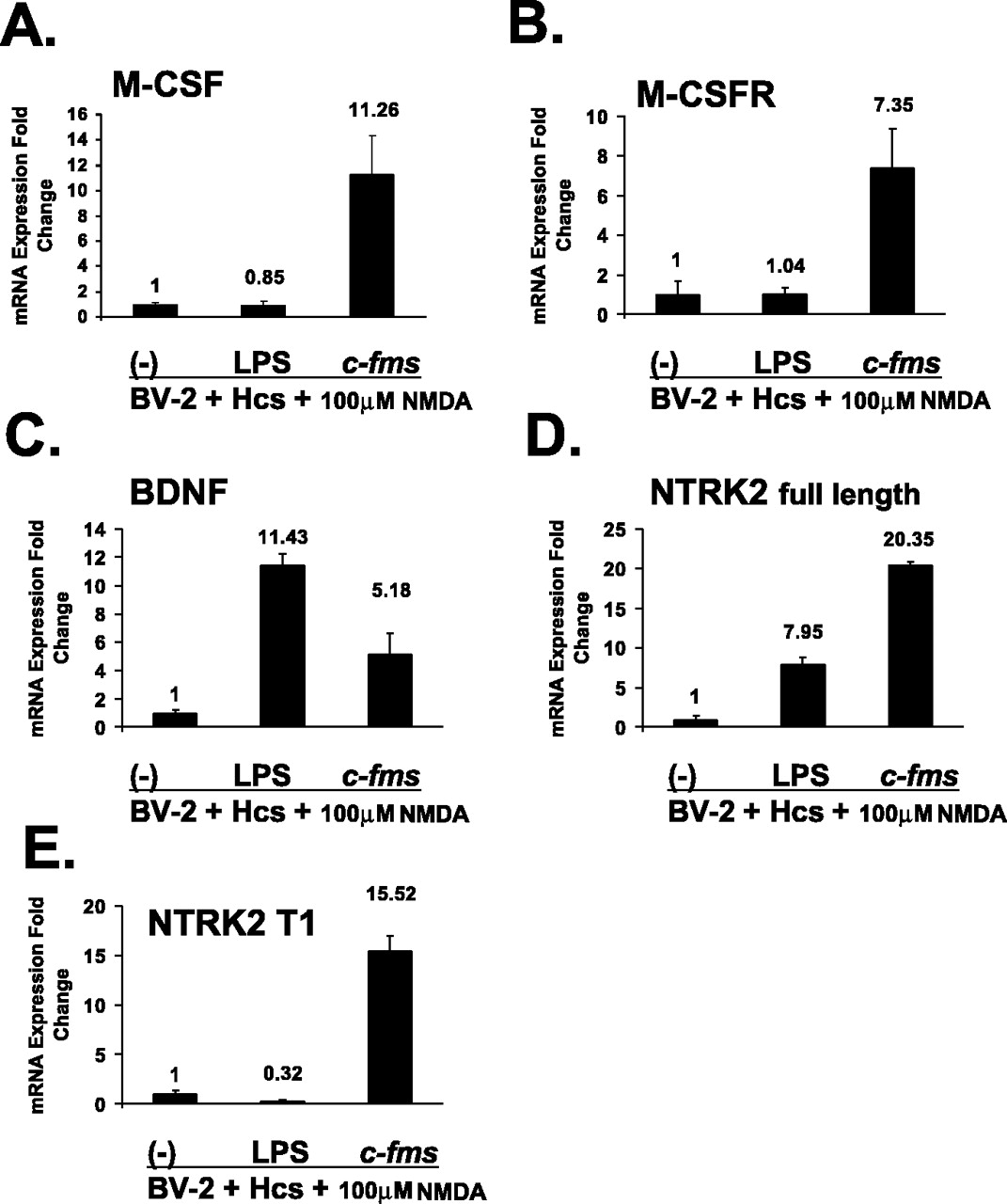
The effects of exogenous microglia on gene expression in neurons in microglia-organotypic cocultures treated with NMDA. A-E show results from neurons obtained with laser capture microdissection of cocultures. Data are presented for neuronal expression of M-CSF, M-CSFR, BDNF, NTRK2 full-length isoform, and NTRK2 T1 isoform. For each target, real-time RT-PCR results are shown for LPS-pretreated and c-fms-transfected BV-2 cells relative to expression levels in cocultures containing nontransfected BV-2 cells (-). Hcs, Hippocampal cultures. Error bars represent SD.
To test whether M-CSF expression by c-fms-transfected BV-2 microglia in the coculture system was essential for neuroprotection, we used a hammerhead ribozyme to decrease M-CSF production by the exogenous c-fms-transfected cells. Results showed an average 56% decrease in M-CSF mRNA by BV-2 cells treated with the M-CSF ribozyme, which was significantly less than that for control cells and significantly less than that for cells treated with a control ribozyme (Fig. 8A) (Scheffé-corrected p < 0.05 for both comparisons). In cocultures treated with NMDA, after transfection of BV-2 cells with the M-CSF hammerhead ribozyme, there was an average 53% decrease in c-fms-induced neuroprotection (Fig. 8B) (Scheffé-corrected p < 0.05). There was no significant loss of neuroprotection in cocultures with c-fms-transfected BV-2 cells treated with the control ribozyme. Because the M-CSF ribozyme inhibited microglial proliferation, we also tested cocultures that contained the same density of ribozyme-treated BV-2 cells as was found in a typical coculture with c-fms-transfected cells. Increased BV-2 cell density did not significantly change the inhibitory effects of the ribozyme on c-fms-induced microglial neuroprotection.

A, Decreased M-CSF expression by BV-2 microglia in monotypic cultures cotransfected with the c-fms plasmid and with the anti-M-CSF ribozyme (RzM-CSF). Data are also presented for a control ribozyme (RzCtrl). B, Attenuation of neuroprotective effect of BV-2 c-fms transfection in microglial-organotypic cocultures by cotransfection with the anti-M-CSF ribozyme. The y-axis represents PI signal relative to that in hippocampal cultures (Hcs) treated with NMDA alone (first data point). Also shown are results for coculture containing c-fms-transfected cells (second data point), c-fms-transfected cells treated with the anti-M-CSF ribozyme (third data point), c-fms-transfected BV-2 cells treated with the anti-M-CSF ribozyme and seeded at increased density (fourth data point), c-fms-transfected cells treated with the control ribozyme (fifth data point), and Hcs without cocultured BV-2 cells and without NMDA (sixth data point). The increased density data point represents cocultures containing numbers of ribozyme-treated cells equal to those typically found in a c-fms-transfected coculture not treated with the ribozyme. Error bars represent SD.
Discussion
Our results demonstrate that in a microglial-organotypic coculture system and after biolistic transfection of microglia in organotypic cultures, overexpression of the M-CSFR on microglia protects neurons in the slice cultures from excitotoxic and teratogen-induced injury. We showed previously that M-CSFR overexpression markedly alters microglial cytokine production, including upregulation of M-CSF, the ligand for the M-CSFR. Inhibition of M-CSF expression in exogenous microglia with a hammerhead ribozyme attenuated the neuroprotective effect of microglial M-CSFR overexpression in the coculture system, indicating that autocrine and/or paracrine effects of this cytokine may be important in neuroprotection. The lack of effect with a control ribozyme makes it unlikely that the ribozyme cotransfection procedure alone was responsible for the loss of neuroprotective properties induced by M-CSFR overexpression. Microglia pretreated with LPS, which were not neuroprotective, lacked increased M-CSF expression.
Although there is substantial evidence that activated microglia can have negative effects in neurologic disease (Spranger and Fontana, 1996; McGeer and McGeer, 1998; Glass and Wesselingh, 2001; Bamberger and Landreth, 2002; Garden, 2002), evidence exists that under certain circumstances, microglia can be neuroprotective. Several groups have found that microglia express the glutamate uptake transporter, which may help microglia protect nerve cells during excitotoxic injury (Kondo et al., 1995; Lopez-Redondo et al., 2000; Nakajima et al., 2001; Schwartz et al., 2003). Tong et al. (2000) demonstrated that in a coculture model, migration of macrophages toward neurons after administration of the human immunodeficiency virus-related toxin platelet-activating factor was neuroprotective. Watanabe et al. (2000) found that microglial supernatants protected dissociated neurons from excitotoxic injury. In another coculture model, microglial-neuronal contact changed the microglial phenotype from toxic to protective (Zietlow et al., 1999). Grafting of microglia into injured spinal cord results in neurite outgrowth and other regenerative neuronal responses (Rabchevsky and Streit, 1997). Microglial activation was recently associated with better neuronal survival in an optic nerve crush injury model (Shaked et al., 2004), possibly through recruitment of neuroprotective T-cells to the site of CNS injury (Schwartz and Kipnis, 2004). Non-neuronal cells, including microglia, appear to protect neurons in an animal model for amyotrophic lateral sclerosis (Clement et al., 2003). This and other evidence has led some authors to conclude that an entirely negative role for microglia is probably incorrect (Streit, 2002; Nakajima and Kohsaka, 2004).
Microglial cytokines may have neurotrophic effects under certain conditions. For example, it was shown recently that microglial tumor necrosis factor-α release induced by activation of the microglia P2X7 receptor is neuroprotective (Suzuki et al., 2004). We found previously that M-CSF applied directly to organotypic hippocampal cultures protects against excitotoxicity (Vincent et al., 2002a), and similar results have been obtained in vivo (Berezovskaya et al., 1996). Likewise, deletion of M-CSF expression renders neurons vulnerable to injury (Berezovskaya et al., 1995; Bruccoleri and Harry, 2000). The mechanism for M-CSF neuroprotection remains unclear. For microglia transfected to overexpress the M-CSFR cocultured with hippocampal slices, microglial M-CSF could be directly neuroprotective. Or, M-CSF could stimulate exogenous microglia to produce neuroprotectants by autocrine or paracrine mechanisms, or it could stimulate non-neuronal cells in the slice culture to express neuroprotectants or to decrease production of neurotoxins.
Microglial cytokines could also modify gene expression in neurons. Using pure samples of laser-captured slice culture hippocampal neurons, we found that only neuroprotective c-fms-transfected BV-2 cells induced increased expression of neuronal M-CSF and M-CSFR in NMDA-treated cultures. This may result in neuronal self-protection through autocrine and paracrine M-CSF signaling. Some, but not all, studies have reported neuronal expression of the M-CSFR that varies depending on age and anatomic region (Murase and Hayashi, 1998; Raivich et al., 1998). Injury induces neuronal M-CSFR expression locally (Wang et al., 1999). In our coculture system, excitotoxic injury strongly induced neuronal M-CSFR expression when activated microglia are present, which could augment the neurotrophic properties of microglial M-CSF.
Microglia may induce other potentially self-protective neuronal responses. Activation of NMDA receptors increases expression of BDNF and subsequent activation of NTRK2, resulting in neuroprotection in cerebellar granule cell cultures via an autocrine survival loop (Jiang et al., 2003). In our coculture system, only LPS-treated BV-2 cells significantly increased neuronal BDNF expression in coculture. Yet, neurons from cocultures containing c-fms-transfected microglia showed significantly greater expression of the NTRK2 full-length and T1 isoforms than did neurons from cultures containing LPS-pretreated cells. Whereas BDNF binding to full-length NTRK2 containing the tyrosine kinase domain is classically associated with neuroprotection (Rossler et al., 2004), the NTRK2 T1 truncated receptor is said to inhibit the effects of the full-length form (Haapasalo et al., 2002). However, NTRK2 T1 has been shown recently to promote dendritic growth (Yacoubian and Lo, 2000; Hartmann et al., 2004), indicating ligand-indepedent trophic effects of this isoform. Increased neuronal neurotrophin receptor expression induced by c-fms-transfected microglia could serve to protect nerve cells from excitotoxicity.
Conditioned medium from neurons injured by the teratogen CP was reported to increase microglial expression of M-CSF and the M-CSFR, leading to production of other inflammatory effectors by microglia (Hao et al., 2001, 2002). It was proposed that this microglial response augments CP-induced neurotoxicity. We directly tested whether microglia with increased expression of M-CSF and M-CSFR resulted in neuronal injury. In our system, increased expression of M-CSF and its receptor by microglia actually protected neurons from CP-induced injury. These results demonstrate that microglial activation need not result in neurotoxicity and that neuroprotection induced by microglial overexpression of the M-CSFR occurs with at least two distinct forms of neurotoxicity: NMDA and CP.
We considered the possibility that neuroprotection induced by increased M-CSFR on microglia might occur only in the coculture system. Biolistic transfection was used to overexpress the M-CSFR on microglia endogenous to the slice culture using methods that we have described previously (Mitrasinovic et al., 2004). Results were similar to those obtained with the coculture system. Overexpression of EGFP by endogenous microglia did not result in neuroprotection, suggesting that ectopic expression alone is not sufficient to confer neuroprotective properties.
Coculture of microglia overexpressing the M-CSFR with NMDA-treated organotypic cultures resulted in greater proliferation of exogenous microglia than did coculture with untreated slices. A chemotactic response toward the injured slice was also observed. These responses were not observed when exogenous microglia were treated with LPS. We speculated that neuroprotection could be conferred by microglia overexpressing the M-CSFR merely as a result of increased cell numbers. However, when nontransfected BV-2 cells were seeded at an initial density equal to that of c-fms-transfected cells at the end of the experimental period, there was no neuroprotective effect. This suggests that the neuroprotective effects of microglia M-CSFR overexpression are not attributable primarily to increased cell numbers. Interestingly, LPS-treated BV-2 cells, even when seeded at increased density, showed neither a neurotoxic nor a neuroprotective effect in this system.
It is curious that LPS pretreatment of BV-2 microglia did not enhance NMDA neuronal toxicity, because LPS-activated microglia when cocultured with monotypic neurons can result in neurotoxicity (Xie et al., 2004). Aschner et al. (1999) suggested that neurons in dissociated cultures are particularly susceptible to negative effects of microglia as a result of the absence of normal neuronal-glial interactions. In fact, several reports suggest that astrocytic and neuronal factors are key in determining the toxic potential of microglia (Zietlow et al., 1999; Hailer et al., 2001; Smits et al., 2001).
We found that the neuroprotective effect of exogenous microglia overexpressing the M-CSFR occurred only when the microglia were in direct physical contact with the organotypic culture. In the one-level coculture system, some microglia come into direct contact with neurons (Mitrasinovic et al., 2001), although the majority remain on the surface of the slice culture. The local concentration of soluble factors such as M-CSF may be much higher when BV-2 cells are in direct contact with the slice culture than when diluted in the large volume of culture medium of the two-level system. Within tissue, cytokines can likely influence cells within a domain of up to 250 μm surrounding the effector cell (Francis and Palsson, 1997), which is approximately the thickness of a mature slice culture. Microglia on the surface of the slice are likely to have paracrine effects on cells located internally. Furthermore, in addition to a soluble form, M-CSF exists as a membrane-bound glycoprotein and extracellular matrix-bound proteoglycan that are biologically active (Fixe and Praloran, 1997). These M-CSF forms participate in cell adhesion and activation (Uemura et al., 1993; Yao et al., 2003) and could be important in neuroprotection. Membrane- or extracellular-bound M-CSF would not affect neurons or other cells in the slice culture in the two-level system, resulting in loss of neuroprotection.
In summary, we demonstrated that microglia overexpressing the M-CSFR protect neurons against excitotoxicity and teratogen-induced injury in a microglial-organotypic coculture system and after biolistic expression of the M-CSFR on microglia endogenous to the slice culture. It is unclear whether microglia showing increased expression of the M-CSFR could be neuroprotective in intact brain. However, because the M-CSFR expression is increased on microglia in AD, additional investigation of the effects of signaling through this receptor on microglial phenotype is warranted.
Footnotes
This work was supported by the National Institute of Mental Health (MH 57833) and by the Alzheimer's Association. O.M.M. is the recipient of an Alzheimer's Association New Investigator Award. We thank Jennifer Rhee, Grace Perez, Mark Rodgers, Angela Chen, and Feifei Zhao for assistance.
Correspondence should be addressed to Dr. Greer M. Murphy Jr, Associate Professor, Neuroscience Research Laboratories, Department of Psychiatry and Behavioral Sciences, Stanford University School of Medicine, Medical School Lab Surge Building, Room P-104, Stanford, CA 94305-5485. E-mail: gmurphy{at}leland.stanford.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254442-•$15.00/0
↵* A.G., C.C.R., and N.B.L. contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}