

Abstract
We examined the changes that arise when neurotransmitter release is inhibited in a subpopulation of hippocampal neurons in coculture with normally active neighbors. Subsets of neurons were presynaptically silenced by chronic expression of tetanus toxin light chain tagged with cyan fluorescent protein (TNTCFP). Surprisingly, silenced neurons formed as many presynaptic terminals as their active neighbors when grown together on glial microislands. However, silenced neurons could not recruit the AMPA-type glutamate receptor subunit GluR1 as efficiently when competing with active neighbors. The immunofluorescence intensity ratio of GluR1 at synaptic puncta versus shaft was reduced by 22% opposite TNTCFP-expressing terminals compared with active neighbors. In contrast, this effect is abolished when vesicular release is blocked in all neurons. Local presynaptic inhibition by TNTCFP did not change the synaptic level of the AMPA receptor subunits GluR2 or GluR2/3 or of the PSD95 (postsynaptic density 95) family scaffolding proteins. Thus, neurotransmitter release selectively regulates the AMPA receptor population on a synapse-by-synapse basis but is not essential for an axon to efficiently compete for synaptic territory in a simple model system. These results demonstrate precise input specificity of postsynaptic receptor composition via differential activity among neighbor synapses.
Introduction
Activity is key in the development and refinement of the nervous system. In the mammalian visual system, spontaneous activity and sensory experience change the functional connectivity among neurons. The relative activity of competing inputs skews this connectivity; closure of one eye reduces the influence of that eye and increases the influence of the open eye in the organization of the cortex (Wiesel, 1982; Katz and Shatz, 1996). At the neuromuscular junction (NMJ), activity aids in the elimination of all but one axon innervating each postsynaptic endplate (Lichtman and Colman, 2000). In this way, competition between inputs shapes the organization of the nervous system. However, it is not known whether synaptic competition occurs among all neuron types nor whether common rules apply.
Activity also influences synaptic efficacy, partly through changes in postsynaptic protein complement. Most notably, the synaptic density of AMPA-type ionotropic glutamate receptors (GluRs) is regulated by activity. In cultured spinal neurons, chronic activity blockade increases miniature EPSC amplitude and synaptic levels of multiple AMPA receptor subunits (O'Brien et al., 1998). Such homeostatic effects are thought to maintain differences in strengths of individual synapses and be mediated by a global cell activity sensor (Turrigiano and Nelson, 2000). In contrast, long-term potentiation (LTP) and long-term depression (LTD) are changes in synaptic efficacy induced by local activity. LTP and LTD are accompanied and likely mediated by synaptic insertion or endocytosis of AMPA receptors (Bredt and Nicoll, 2003; Malenka, 2003). Subunit-specific rules may apply; for example, GluR1 is selectively required for receptor insertion during LTP (Zamanillo et al., 1999; Shi et al., 2001). Although numerous studies support input specificity of LTP and LTD, how locally this specificity is maintained is unclear. Several forms of LTP and LTD spread along individual dendritic arbors in cultured neurons and in hippocampal slices (for review, see Bi and Poo, 2001). Even for conventional LTP induced at the Schaffer collateral to CA1 synapse, input specificity breaks down at distances of less than ∼70 μm (Engert and Bonhoeffer, 1997). Whether transmitter release influences synaptic efficacy or postsynaptic receptor composition at individual synapses is unknown.
We examined local changes that arise when a subset of presynaptic sites are selectively inhibited in hippocampal cultures. To do this, we expressed the light chain of tetanus toxin tagged with cyan fluorescent protein (TNTCFP). Tetanus toxin cleaves vesicle-associated membrane protein 2 (VAMP2)/synaptobrevin, barring it from soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex formation, and preventing vesicular release of neurotransmitters (Sweeney et al., 1995; Schiavo et al., 2000). We find that synapses from neurons expressing TNTCFP are maintained in normal numbers even in the presence of active neighbors. However, the concentration of the GluR1 AMPA receptor subunit at inhibited synapses is reduced. This effect is seen only in the presence of neighboring synapses in which vesicular release is not compromised. Thus, the subunit composition of AMPA receptors is specifically controlled at each synapse based on its relative activity.
Materials and Methods
Cell cultures. For experiments in Figures 1 and 4, 5, 6, 7, Banker-style hippocampal sandwich cultures were used for optimal imaging (Goslin et al., 1998). Hippocampi were isolated from embryonic day 18 rat pups, trypsinized (0.25%), and mechanically dissociated. Neurons were plated at a density of 150,000 (see Figs. 4, 5, 6, 7) or 300,000 (see Fig. 1) cells per 60 mm dish onto glass coverslips coated with poly-l-lysine and dotted with paraffin wax feet. Neurons were allowed to adhere in MEM supplemented with 10% horse serum (plating media) and then placed cell-side down into dishes containing serum-free MEM with N2 supplements, 0.1% ovalbumin, and 1 mm pyruvate [N2.1 media; components from Invitrogen (Carlsbad, CA) and Sigma (St. Louis, MO)] and a feeder layer of astroglia prepared from postnatal day 0 (P0) rat pups. To prevent overgrowth of glia, neuron cultures were treated with cytosine arabinoside (5 μm; Calbiochem, La Jolla, CA) after 2 d in vitro (DIV). Cultures were grown up to 3 weeks, with one-third of the media replaced once per week. When applicable, treatment of cultures by chronic application of 1 nm tetanus toxin started the day after plating and was replenished after weekly feeding of the cultures and once more per week.
Microisland cultures used in Figures 2 and 3 were prepared as described previously (Bekkers and Stevens, 1991). Glass coverslips were coated with 0.15% agarose (Invitrogen) and then sprayed with small dots of a permissive substrate solution of collagen (0.2 mg/ml, Vitrogen 100; Cohesion, Palo Alto, CA) and poly-d-lysine (0.05 mg/ml; Sigma) in 17 mm acetic acid. Hippocampi from P0 mice were isolated and dissociated, and 200,000-250,000 cells were plated per 60 mm dish. After glia became confluent on islands, neurons were killed by exposure to 200 μm glutamate. To prevent glia overgrowth, cultures were treated with cytosine arabinoside (5 μm; Calbiochem). Neurons isolated from P0 mouse hippocampi using papain and mechanical dissociation were then plated onto the islands at 50,000-75,000 cells per dish. Twice a week, 3 ml of media was replaced with fresh media containing 4% fetal calf serum (HyClone, Logan, UT).
Transfection of neurons. Neurons were transfected at plating for all experiments except those shown in Figure 2. Neuronal suspensions were transfected with Lipofectamine 2000 (Invitrogen) using a modified version of the protocol of the manufacturer. DNA and Lipofectamine were separately mixed with plating media and then combined and incubated for 20 min at room temperature. Media and neurons were added, incubated for 1 h at 37°C in 5% CO2, plated onto poly-l-lysine-coated coverslips, and incubated until cells adhered. Coverslips were then flipped, cells down, back into 60 mm dishes with a glial feeding layer (omitted with microisland cultures). Microisland cultures for the experiment in Figure 2 were transfected at 1-2 weeks of age to increase transfection efficiency. Experiments were performed at least 48 h after transfection. Lipofectamine 2000 was used with a modified version of the protocol of the manufacturer. Fresh media was prewarmed and equilibrated in the incubator. DNA and Lipofectamine were separately mixed with plating media, combined, and incubated at room temperature for 20 min. Coverslips with adhered neurons were transferred cell-side up into dishes with the prewarmed media, and the DNA/Lipofectamine mixture was added. Cultures were incubated at 37°C in 5% CO2 chamber for 3.5-5 h and then returned to their original dishes.
Immunocytochemistry. Neurons were fixed with warmed 4% paraformaldehyde plus 4% sucrose in PBS for 15 min at room temperature, permeabilized for 5-10 min with 0.25% Triton X-100 in PBS, and stained as below. Alternatively, for immunocytochemistry involving the NMDA receptor NR1, neurons were fixed with cold methanol at -20°C for 10 min, followed immediately by a quick rinse in ∼0.025% Triton X-100 in PBS. Nonspecific staining was blocked for 45-60 min in 10% bovine serum albumin (BSA) at 37°C. Neurons were incubated in 3% BSA in PBS with primary antibody overnight at room temperature and 1 h at 37°C with secondary antibody. If biotin was used, streptavidin was applied in 3% BSA in PBS for 20-30 min at 37°C. Coverslips were mounted on glass slides with elvanol (Tris HCl, glycerol, and polyvinyl alcohol with 2% 1,4 diazobicyclo[2.2.2] octane). For surface GluR1 staining, live neurons were removed from the incubator and washed once in extracellular solution (in mm: pH 7.4, 168 NaCl2, 2.4 KCl, 10 HEPES, 10 glucose, 2.0 CaCl2, and 2.0 MgCl2) at room temperature. Primary antibody was applied in extracellular solution plus 3% BSA for 25 min at room temperature. Neurons were then washed twice in extracellular solution, fixed with paraformaldehyde, and stained as above.
The following primary antibodies against the indicated protein were used in the course of this work. Rabbit polyclonal antibodies included the following: synapsin 1 (0.2 μg/ml; Chemicon, Temecula, CA), synaptic ras-GTPase-activating protein (SynGAP) (1.2 μg/ml; Affinity BioReagents, Golden, CO), GluR1 C-terminus (0.13 μg/ml; Upstate Biotechnology, Lake Placid, NY), GluR1 N-terminus (1:1000; R. A. McIlhinney, Oxford University, Oxford, UK), GluR2/3 (1 μg/ml; Chemicon), and microtubule-associated protein 2 (MAP2) (1:50,000; S. Halpain, Scripps Institute, La Jolla, CA). Mouse monoclonal antibodies included the following: VAMP2/synaptobrevin (0.2 μg/ml; Synaptic Systems, Goettingen, Germany), postsynaptic density 95 (PSD95) [although the term PSD95 is used here for simplicity, this antibody recognizes PSD95, PSD93, synapse-associated protein 102 (SAP102), and SAP97] (37 μg/ml; Affinity BioReagents), GluR2 (30 μg/ml; Chemicon), NR1 (0.5 μg/ml; PharMingen, San Diego, CA), and calcium/calmodulin-dependent kinase IIα (CaMKIIα) (1:1000; Affinity BioReagents). Guinea pig polyclonal antibodies included vesicular glutamate transporter 1 (VGlut1) (1:500; Chemicon). Secondary antibodies used include Alexa Fluor 568- and 647-conjugated subtype-specific secondary antibodies (2-4 μg/ml) from Molecular Probes (Eugene, OR) and biotin- and 7-amino-4-methylcoumarin-3-acetic acid (AMCA)-conjugated secondary antibodies (4-15 μg/ml) from Vector Laboratories(Burlingame, CA). Texas red-conjugated streptavidin (0.7 μg/ml; Vector Laboratories) was used to visualize biotin-conjugated secondary antibodies.
FM styryl dye staining. Cycling of synaptic vesicle was labeled by uptake of FM styryl dyes (Korkotian and Segal, 2001) at CFP- or TNTCFP-expressing synapses in transfected high-density 13 DIV Banker-style cultures. Coverslips were loaded into an imaging chamber in wash solution plus 2 mm CaCl2, containing the following (in mm): 119 NaCl, 2.5 KCl, 4 MgCl2, 25 HEPES, and 30 glucose, pH 7.4. Neurons were then exposed to FM4-64 (10 μm; Molecular Probes) for 45 s in high potassium stimulation buffer with the following (in mm): 90 KCl, 29 NaCl, 2 CaCl2, 2 MgCl2, 25 HEPES, and 30 glucose, pH 7.4. Stimulation solution was then exchanged for wash solution without calcium. Synaptophysin tagged with yellow fluorescent protein (SYNYFP), CFP, and FM images were acquired with a MicroMax cooled CCD camera (Roper Scientific, Tucson, AZ) on an inverted microscope (Eclipse 200; Nikon, Tokyo, Japan) equipped with 60× oil 1.4 numerical aperture (NA) objective using MetaMorph software (Universal Imaging, West Chester, PA). Images were prepared for printing with Photoshop (Adobe Systems, San Jose, CA).
Microscopy and analysis. Images for comparisons were acquired on the same rig with the same exposure settings. Image acquisition and/or analysis were performed blind to the experimental condition when possible. This includes all quantitative analysis presented in Figures 3 and 7.
For all experiments involving Banker-style cultures, images were taken on either an Axioskop or AxioPlan epifluorescent microscope equipped with a 63× oil 1.4 NA or 40× oil 1.3 NA objective (Zeiss, Thornwood, NY) using MetaMorph software. Images were acquired with a cooled CCD camera, either PhotoMetrics SenSys (Roper Scientific) or SPOT monochrome (Diagnostic Instruments, Sterling Heights, MI). Specific images for each of the fluorophores were obtained with the following filter sets from Chroma Technology (Brattleboro, VT): 31044v2 (CFP), 41028 (YFP), 41002c (Alexa 568), and 41008 (Alexa 647). Sometimes a fifth fluorophore (AMCA with Chroma Technology filter set 31000) was also used for qualitative assessment. Images for quantitative analysis were adjusted by subtracting matched dark-field images and then dividing by an image of a uniform fluorescent specimen to correct for any nonuniformity in illumination. Analysis was done using MetaMorph. Analyses were performed on excitatory synapses formed onto neurons likely to be pyramidal excitatory neurons by morphological criteria (Benson et al., 1994). For analysis of the synaptic enrichment of GluR1, GluR2, and GluR2/3, samples were colabeled with PSD95 (for GluR1 and GluR2/3) or SynGAP (for GluR2). A single antibody could not be used in all instances because of species limitations, but we found that these PSD95 and SynGAP antibodies give equally robust colocalized punctate immunoreactivity to mark glutamate postsynaptic sites. An unbiased mask of all glutamate postsynaptic areas was first generated by thresholding the corresponding PSD95 or SynGAP images. The region masks were then applied to the unthresholded GluR images, and mean synaptic intensity was determined. The mean intensity of nonsynaptic fluorescence along the associated dendrite shafts was also measured. The intensity of off-cell background fluorescence was subtracted from the synapse and shaft intensity values, and the synapse/shaft ratio was determined. To combine data from two or more independent experiments, synapse/shaft ratios in each condition of each culture were normalized to the median value of the nontransfected synapses of that condition.
All microisland cultures were imaged using a Fluoview500 confocal laser-scanning microscope equipped with a 60× oil PlanApo 1.4 NA objective (Olympus Optical, Melville, NY). Stacks were acquired in ∼0.5 μm Z steps by slow sequential scanning. Images were aligned when necessary using AutoDeblur/AutoVisualize 9.0 (Autoquant, Troy, NY). Maximum pixel projections were generated from stacks and analyzed using MetaMorph (Universal Imaging). For analysis of the number of synaptic puncta, a reasonable threshold was set by eye for each image, and resultant objects were measured by the program. Because puncta were often packed closely together, resulting in undercounting, dense fields were adjusted with manual counts.
All data processing was done using Microsoft (Seattle, WA) Excel. Student's t test, Mann-Whitney rank sum test, correlation coefficient, and Kruskal-Wallis one-way ANOVA on ranks analyses were performed with SigmaStat (Jandel Scientific, Corte Madera, CA) software. Images were prepared for printing with Photoshop, involving adjustments in brightness and contrast of images.
Electrophysiology. Paired whole-cell voltage-clamp recordings were done using 1- to 2-week-old microislands containing one transfected neuron and up to five nontransfected neurons. Microislands were perfused by gravity flow with extracellular solution (pH 7.4) containing the following: 168 mm NaCl2, 2.4 mm KCl, 10 mm HEPES, 10 mm glucose, 2.6 mm CaCl2, 1.3 mm MgCl2, and 100 μm APV. Recording electrodes were pulled from borosilicate glass (World Precision Instruments, Sarasota, FL) and had resistances of 1-3 MΩ. Electrodes were filled with a solution (pH 7.4) containing the following (in mm): 140 K-gluconate, 6.23 CaCl2, 8 NaCl, 2 MgCl2, 10 EGTA, 10 HEPES, 2 Na2ATP, and 0.1 Na2GTP. All recordings were done at room temperature. Recordings were done with Axopatch1D amplifiers and AxoGraph acquisition software (Axon Instruments, Union City, CA). Data were low-pass filtered at 5 kHz and acquired at 10 kHz. Neurons were held at -70 mV. EPSCs were evoked by brief (0.5-5 ms) depolarizations to 10-50 mV. Each neuron of the pair was stimulated for 20-100 trials. Neurons were stimulated at low frequency (0.1-0.2 Hz). After recording, neurons were fixed with paraformaldehyde, stained for MAP2, and imaged to confirm the number of neurons on each island. Analysis of responses of the unstimulated neuron in each pair was performed using AxoGraph. Only responses that were time locked to 5-10 ms after stimulus delivery were analyzed. The percentage of failures (number of stimulations that elicited no response/total number of stimulations administered × 100) observed for each recording was determined by manual inspection of the traces. All data processing was done using Microsoft Excel. Statistical analyses were performed with SigmaStat (Jandel Scientific) software.
Expression vectors. The enhanced YFP (EYFP) (Clontech, Palo Alto, CA) was fused in-frame to the C terminus of the rat cDNA encoding synaptophysin (GenBank accession number X063388; kindly provided by K. Buckley, Harvard University, Boston, MA) by PCR. The SYNYFP fusion protein was expressed from the EYFP-N1 plasmid (Clontech) under control of the cytomegalovirus promoter. The TNTCFP expression vector was a gift from Rob Burgess (The Jackson Laboratory, Bar Harbor, ME) and Josh Sanes (Harvard University, Boston, MA). It consists of enhanced cyan fluorescent protein (Clontech) fused to the C terminus of the synthetic tetanus toxin light chain (Eisel et al., 1993), also in the Clontech vector. Purified DNA was obtained using EndoFree plasmid kits from Qiagen (Valencia, CA).
Results
Tetanus toxin light chain-CFP expression inhibits synaptic vesicle fusion
We tested the ability of TNTCFP to block vesicular release of neurotransmitter. Tetanus toxin specifically cleaves VAMP2 and VAMP3/cellubrevin, preventing release of presynaptic vesicles containing neurotransmitter. In agreement with previous studies (Ahnert-Hilger et al., 1996; Martinez-Arca et al., 2001), long-term treatment of cultured neurons with bath tetanus toxin blocked evoked release and reduced spontaneous release by 90- fold but did not alter dendrite morphology or synapse density and yielded no detectable changes in synapse composition (Harms and Craig, 2005). Thus, TNTCFP expression is expected to selectively inhibit transmitter release but not neurite outgrowth or the basic mechanisms of synapse assembly. To identify the inhibited synapses formed by transfected neurons, we coexpressed SYNYFP. When expressed under our conditions, >75% of SYNYFP clusters chosen by a minimal size and intensity threshold colocalized with both synapsin and postsynaptic scaffolding protein (gephyrin plus PSD95 family) immunoreactive puncta, thus serving as a reliable marker for transfected synapses. In this way, we could examine the effect of inhibition at a definable set of synapses in an otherwise normally active culture. Although synaptophysin is not an essential protein, overexpression of SYNYFP may itself have an effect on synapses (Tarsa and Goda, 2002), so we compared the effects of expression of TNTCFP plus SYNYFP with expression of CFP plus SYNYFP

Selective losses of VAMP2 and FM dye uptake from TNTCFP-expressing synapses. a, Dissociated hippocampal cultures were transfected with SYNYFP and CFP (left column) or TNTCFP (right column) before plating, cultured for 9 d, and then fixed and stained for VAMP2 and synapsin. Active synapses formed by axons expressing SYNYFP and CFP show colocalization of synapsin and VAMP2 (arrows, left column). Inhibited synapses, expressing SYNYFP and TNTCFP, lack staining for VAMP2, whereas staining for synapsin was unchanged (arrows, right column). Note that VAMP2 and synapsin colocalize at synapses formed by nontransfected neurons(arrowheads). Scale bar, 10 μm.b, Transfected 9- to 13-d-old neurons were loaded into an imaging chamber and exposed to high potassium in the presence of FM4-64 for 45 s. After stimulation, neurons were washed and imaged. We observed little to no FM4-64 uptake at inhibited synapses expressing TNTCFP and SYNYFP (arrows, right column), whereas active synapses expressing CFP and SYNYFP showed robust vesicle cycling (arrows, left column). Note that synapses formed by nontransfected neurons also show robust FM4-64 uptake (arrowheads). Scale bar, 10 μm.
We first tested whether long-term expression of TNTCFP in cultured hippocampal neurons was effective in cleaving VAMP2. We transfected a subset of neurons at the time of plating with expression vectors for TNTCFP or CFP and SYNYFP and assayed after 1-2 weeks. Transfected cultures were fixed and labeled for VAMP2 and synapsin, a presynaptic protein that is insensitive to cleavage by tetanus toxin. Synapses formed by nontransfected or CFP plus SYNYFP-transfected axons onto a nontransfected dendrite were marked by VAMP2 and synapsin. In contrast, synapses that were positive for TNTCFP and SYNYFP showed a loss of VAMP2, whereas synapsin was unchanged (Fig. 1a). This loss of VAMP2 was also observed later in the lifetime of the culture, including after 3 weeks of culture, the age at which additional analyses were performed (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Thus, VAMP2 is depleted from the presynaptic structures formed by neurons that express TNTCFP.
We next examined whether exocytic and endocytic cycling of synaptic vesicles was compromised in neurons expressing TNTCFP by activity-dependent uptake of FM dye (Cochilla et al., 1999). Transfected neurons were loaded into an imaging chamber and exposed to high extracellular potassium in the presence of FM4-64 for 45 s. We observed little to no FM4-64 uptake at inhibited synapses expressing TNTCFP and SYNYFP (Fig. 1b). In contrast, untransfected synapses and synapses expressing CFP and SYNYFP showed robust vesicle cycling. Therefore, normal vesicle cycling is impaired with presynaptic TNTCFP expression.

Expression of TNTCFP inhibits evoked neurotransmission. a, Evoked release was determined from neuron pairs grown on microislands with one transfected neuron and up to five nontransfected neurons. Pairs of neurons were recorded in the whole-cell voltage-clamp configuration. Each neuron was stimulated by a brief depolarization, and the evoked postsynaptic current was recorded in the unstimulated neuron. b, Sample paired cell recordings: each trace represents an average of 20 trial recordings from different pairs. Stimulation of nontransfected (Nontrans.) or CFP-transfected neurons evoked an EPSC in a neighboring recorded cell, with a varying polysynaptic component. Nontransfected and TNTCFP-transfected cells could exhibit large postsynaptic currents during stimulation. However, stimulation of TNTCFP-expressing cell did not evoke these responses in nontransfected neighbors. c, The failure rate of each axon, or the percentage of stimuli that did not evoke a postsynaptic current in the unstimulated neuron, was assessed. Stimulation of nontransfected neurons (No expression) elicited postsynaptic responses in 18 of 23 cells. Likewise, stimulation of CFP-expressing neurons elicited postsynaptic responses in 10 of 10 cells. Stimulation of TNTCFP-expressing neurons elicited a postsynaptic response in only 2 of 15 cells, and these responses were similar in amplitude to spontaneous miniature events (∼50 pA). Data for nontransfected presynaptic neurons (No expression) includes pairs in which the postsynaptic neuron was untransfected, expressing CFP, or expressing TNTCFP; no differences were found among groups with respect to postsynaptic expression.
Finally, we examined evoked neurotransmitter release from neurons transfected with TNTCFP or CFP. For these experiments, we performed paired whole-cell voltage-clamp recordings from neurons grown on glial microislands. We used microislands with up to six neurons, in which one neuron was transfected. In these culture conditions, the chance of two neurons being synaptically connected is ∼75% (Fig. 2c, No expression). Each neuron of the pair was stimulated by a brief depolarization, and evoked postsynaptic currents were recorded in the unstimulated neuron- (Fig. 2a,b). The failure rate for each stimulated neuron (the percentage of trials that did not evoke a postsynaptic current in the unstimulated neuron) was assessed (Fig. 2c). Stimulation of non-transfected neurons elicited a postsynaptic current in 18 of 23 cells. Likewise, stimulation of CFP-expressing neurons elicited a postsynaptic current in 10 of 10 cells. However, stimulation of TNTCFP-expressing neurons elicited a postsynaptic current in only 2 of 15 cells. Moreover, a total of only four postsynaptic current events were evoked by over 800 stimuli applied to TNTCFP-expressing neurons, and these responses were similar in amplitude to spontaneous miniature events. TNTCFP-transfected neurons had functional synapses on them as evidenced by the evoked postsynaptic currents (Fig. 2b). Additionally, TNTCFP-transfected neurons were capable of firing action potentials (data not shown). These data indicate that evoked neurotransmitter release is almost completely abolished in neurons expressing TNTCFP. Together, these three experiments demonstrate that TNTCFP expression strongly inhibits vesicular release of neurotransmitter from TNTCFP-transfected synapses.

Inhibited neurons form an equal number of synapses as controls. a, Neurons were transfected with SYNYFP and TNTCFP or CFP and grown on microislands with up to six nontransfected neurons for 10-12 d. The number of neurons on a microisland was assessed by MAP2 staining and phase contrast. An island with two neurons is shown here. The number of SYNYFP puncta was counted as an estimate of the number of morphological synapses formed by the transfected neuron. Scale bar, 10 μm. b, Inhibited neurons formed as many presynaptic structures as their active neighbors (CFP, n = 20 cells; TNTCFP, n = 10 cells; p = 0.64, t test). c, The number of morphological synapses formed by a single axon did not vary with the number of neurons on an island. The synapse number did not vary with neuron number when considering all neurons together (correlation coefficient, 0.16; n = 30 cells; p > 0.1) or active (correlation coefficient, 0.06; n = 20 cells; p > 0.1) or inactive (correlation coefficient, 0.45; n = 10 cells; p > 0.1) neurons separately.
Synapse formation by inhibited neurons
Having shown that TNTCFP strongly attenuates vesicular release of neurotransmitter, we asked whether TNTCFP-transfected neurons were capable of forming as many synapses as cocultured control neurons. To address this question, we examined transfected and nontransfected neurons cocultured on microislands. Neurons were transfected with SYNYFP and either TNTCFP or CFP on day 0 and allowed to grow on microislands with as many as six control, untransfected neurons (Fig. 3a). In this culture system, synapses begin forming after 4-5 d in vitro. Microisland neurons exhibit spontaneous firing by 6 d in vitro and a gradual activity-dependent increase in evoked EPSC amplitude and decrease in release probability from 6 to 14 d (Chavis and Westbrook, 2001; Takada et al., 2005). We thus maintained the cultures in vitro for 10-12 d to allow for the formation of many synapses and for potential activity-dependent synaptic competition to occur. We then counted the number of SYNYFP puncta as an estimate of the number of morphological synapses that arose from the transfected neuron (Fig. 3b). Surprisingly, TNTCFP-transfected neurons exhibited as many morphological synapses as CFP-transfected neurons (TNTCFP, 829 ± 98 vs CFP, 762 ± 87 synapses per neuron; CFP, n = 20 cells; TNTCFP, n = 10 cells; p = 0.64, t test). Thus, neurotransmitter release is not essential for an axon to efficiently compete for synaptic territory in this system.
Interestingly, the number of SYNYFP puncta formed by a single axon did not vary with the total number of neurons on an island. This was true when considering all neurons together (correlation coefficient, 0.16; n = 30 cells; p > 0.1) or active (correlation coefficient, 0.06; n = 20 cells; p > 0.1) or inactive (correlation coefficient, 0.45; n = 10 cells; p > 0.1) neurons separately (Fig. 3c). Thus, the number of synapses formed by a single axon appears to be independent of the number of potential postsynaptic targets in this model system.
Postsynaptic composition of inhibited synapses
We addressed whether inhibiting a subset of synapses interferes with their ability to recruit synaptic proteins compared with other normally active synapses on the same dendrite. Before plating, <1% of the neurons in a culture were cotransfected with SYNYFP and either CFP or TNTCFP. These cultures were grown for 3 weeks, fixed, immunostained, and assayed for postsynaptic protein colocalization.
We first assessed the distributions of PSD95 family proteins (using an antibody that recognizes PSD95, PSD93, SAP102, and SAP97, referred to hereafter as PSD95 for simplicity) and SynGAP. PSD95 and SynGAP were localized postsynaptically to inhibited and neighbor noninhibited synapses, giving highly punctate synaptic distributions (Figs. 4, 5 and data not shown). For subsequent experiments, we used PSD95 or SynGAP to mark glutamate postsynaptic sites to assess levels of other potential activity-regulated proteins.
One key activity-regulated protein is CaMKIIα. In normal spontaneously active cultures, CaMKIIα is partially concentrated in postsynaptic spines as well as present diffusely throughout dendrite shafts. We found that CaMKIIα still localized to the postsynaptic sites of inhibited synapses and that there was no obvious difference in the level of this protein opposite silenced versus active presynaptic structures (Fig. 4). Thus, although CaMKIIα can be induced by NMDA receptor stimulation to translocate from the dendrite shaft to postsynaptic spines (Shen and Meyer, 1999), we find here that maintenance of the basal concentration of CaMKIIα in spines does not require local, synapse-specific activity.
Glutamate receptors themselves are redistributed by activity. NMDA receptors undergo long-term homeostatic regulation by activity. In response to chronic activity blockade, NMDA receptors redistribute to a more synaptic localization (Rao and Craig, 1997). However, in our studies, we saw no difference in the level of the essential NMDA receptor subunit NR1 at inhibited versus normally active synapses (Fig. 4). Thus, local changes in the activity of specific synapses do not mimic the culture-wide changes in NMDA receptor distribution resulting from global blockade of activity.
Effects of local inhibition on AMPA receptor subunits
Interestingly, immunostaining for the AMPA receptor subunit GluR1 showed a marked decrease specifically at presynaptically inhibited synapses (Fig. 5) but only when TNTCFP-transfected neurons were cocultured with untransfected neurons (see below). Qualitatively, this difference can be appreciated most readily by examining the overlay images of PSD95 and GluR1 (Fig. 5). Whereas dendrites innervated by neurons expressing CFP and SYNYFP show both GluR1 and PSD95 at most synapses (Fig. 5, left column, yellow), dendrites innervated by neurons expressing TNTCFP and SYNYFP have some synapses with an obvious paucity of GluR1 relative to PSD95 (Fig. 5 right column, red). These synapses are precisely those innervated by the inhibited presynaptic terminals (Fig. 5 right column, blue). Larger spines are reported to have a higher AMPA receptor content (Nusser et al., 1998; Takumi et al., 1999; Matsuzaki et al., 2001), and we observed this trend as well, but the difference in GluR1 content attributable to local inhibition is not related to synapse size. Qualitatively, when we compare PSD95 immunofluorescent puncta of similar size opposite inhibited versus active terminals, the associated GluR1 content is still markedly different (Fig. 5). Quantitatively, the effect of local inhibition is reflected in a difference in the synaptic enrichment of GluR1 but not of PSD95 (see below).

Locally inhibited synapses still recruit SynGAP, CaMKIIα, and NMDA receptors. Banker-style dissociated neurons were cotransfected before plating with SYNYFP and TNTCFP, grown for 3 weeks, fixed, and stained for SynGAP, CaMKIIα (left column), and the obligate NMDA receptor subunit NR1 (right column). In spontaneously active cultures, CaMKIIα is partially concentrated in postsynaptic spines (arrowheads, left column) and diffusely throughout the dendrite shaft. At inhibited synapses (arrows, left column), CaMKIIα still localized to postsynaptic sites, defined by SynGAP, as well as in the shaft. Likewise, NR1 was concentrated at both inhibited (arrows, right column) synapses and active (arrowheads, right column) synapses. The bottom row shows a triple color overlay. Scale bars, 10 μm.
To quantify the difference in GluR1, we analyzed four independent sets of cultures transfected with TNTCFP or CFP and SYNYFP at day 0 and then fixed and immunolabeled for GluR1 and PSD95 3 weeks later. We also compared control cultures with those chronically treated with tetanus toxin in the bath to determine whether any effects were specifically attributable to local blockade. Images of nontransfected pyramidal cell dendrites contacted by transfected axons were acquired and analyzed blind to the experimental treatment (TNTCFP vs CFP and control bath vs tetanus toxin bath). The area of PSD95 immunofluorescence was used to define the postsynaptic area. For each field, we measured the average immunofluorescence intensity of GluR1 and of PSD95 at transfected (Fig. 5, arrows) and nontransfected (Fig. 5, arrowheads) synapses along the same dendrites. We compared the average intensity of immunofluorescence at synaptic sites with that of the dendrite shaft to control for possible differences in GluR1 expression between cells. Generally, the concentration of synaptic PSD95 was ∼3.5 times greater than that in the shaft and ∼2 times greater for AMPA receptor subunits. We combined data from multiple experiments by normalizing all values in each condition of each culture to the median value of the nontransfected synapses of that condition. In this way, we examined the synaptic enrichment of GluR1 at CFP-expressing synapses relative to their nontransfected neighbors and at TNTCFP-expressing synapses relative to their nontransfected neighbors in both control and tetanus toxin-supplemented bath (see Fig. 7) (Table 1).
Concentration of proteins at active versus inactive synapses
We found that the concentration of GluR1 was reduced by 22% opposite TNTCFP-expressing terminals compared with active synapses [1.02 CFP normalized to nontransfected neighbor vs 0.78 TNTCFP normalized to nontransfected neighbor; n > 80 cells and n > 1900 synapses each; p < 0.01 Kruskal-Wallis one-way ANOVA on ranks (see Fig. 7b)]. PSD95 at the same synapses was unchanged [1.01 CFP normalized to nontransfected neighbor vs 1.03 TNTCFP normalized to nontransfected neighbor (see Fig. 7e)]. The local regulation of GluR1 content was abolished when neurons were grown with tetanus toxin in the bath [1.17 CFP normalized to nontransfected neighbor vs 1.03 TNTCFP normalized to nontransfected neighbor; n > 25 cells and n > 350 synapses each (see Fig. 7b)]. Thus, the relative decrease in GluR1 opposite inputs from TNTCFP-transfected neurons only occurs when some synapses are unaffected by tetanus toxin. Furthermore, non-normalized values of the GluR1 immunofluorescence ratio at synapses versus shaft were not different between control bath and tetanus toxin bath groups, as determined in independent experiments (GluR1 1.76 control bath vs 1.67 tetanus toxin bath; n = 32 cells each, n > 3500 synapses each; p > 0.1 ANOVA) (Harms and Craig, 2005). Thus, synaptic enrichment of GluR1 was only reduced by blocking neurotransmitter release at a subset of synapses and not by global blockade of activity via bath application of tetanus toxin.
To ascertain whether the change in synaptic receptor levels represented a decrease in receptors on the membrane surface, we stained live neurons with an antibody that recognizes the GluR1 N terminus. Surface staining also revealed a local decrease in GluR1 levels specifically opposite TNTCFP-positive terminals, similar to the results obtained from fixed neurons [1.00 nontransfected vs 0.83 TNTCFP transfected; n = 12 cells and n > 300 synapses each; p < 0.01 Mann-Whitney rank sum test (see Fig. 7a)]. Thus, surface GluR1-containing AMPA receptor levels are decreased at synapses from TNTCFP-transfected axons.
Hippocampal pyramidal neurons have AMPA receptors composed primarily of the GluR1 and GluR2 subunits, in addition to some GluR3 and perhaps low levels of GluR4, which is mainly expressed early in development (Craig et al., 1993; Wenthold et al., 1996). We asked whether the distribution of GluR2 was also regulated by local activity. Qualitatively, unlike GluR1, GluR2 did appear enriched at locally inhibited synapses. The synaptic enrichment of GluR2 was indistinguishable opposite TNTCFP expressing terminals compared with normally active untransfected neighbor terminals or sister CFP-expressing terminals (Fig. 6). This result was confirmed by measurements of GluR2 immunofluorescence performed as described above for GluR1 (except that SynGAP was used instead of PSD95 to mark glutamate postsynaptic sites). Local presynaptic inhibition by TNTCFP did not change the relative synaptic enrichment of the subunit GluR2 at individual synapses [1.09 CFP normalized to nontransfected neighbor vs 1.02 TNTCFP normalized to nontransfected neighbor; n ≥ 20 cells and n > 400 synapses each (Fig. 7c)]. Because we saw a decrease in GluR1 without a decrease in GluR2, we wondered whether GluR1/2 heteromeric receptors were being replaced by GluR2/3 heteromeric receptors. We used an antibody that recognizes both the GluR2 and GluR3 subunits. Similar to the results for GluR2, we saw no change in GluR2/3 levels at locally inhibited synapses [1.05 CFP normalized to nontransfected neighbor vs 0.98 TNTCFP normalized to nontransfected neighbor; n ≥ 16 cells and n > 300 synapses each (Fig. 7d)]. Thus, we show that GluR1 is decreased at synaptic sites with local blockade of neurotransmitter release, with no change in GluR2 or GluR2/3, indicating that relative activity levels direct the AMPA receptor subunit composition at individual synapses.
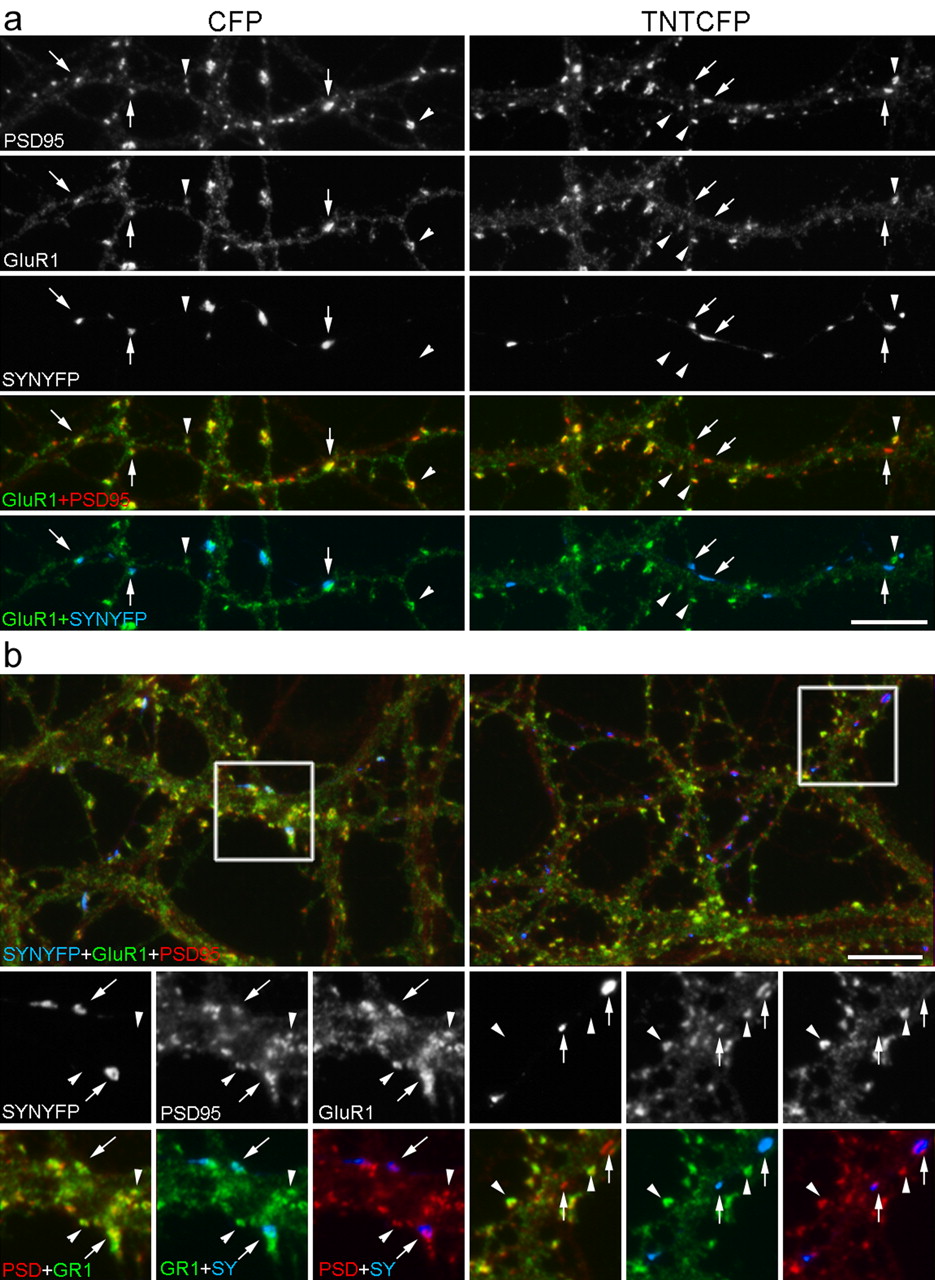
GluR1 is reduced at locally inhibited synapses. a, b, Banker-style dissociated neurons were cotransfected before plating with SYNYFP and either CFP (left column) or TNTCFP (right column), grown for 3 weeks, fixed, and stained for the AMPAR subunit GluR1 and the excitatory postsynaptic scaffolding proteins PSD95. GluR1 was found to colocalize at many active, CFP-expressing synapses (arrows, left column) and nontransfected synapses (arrowheads). In contrast, with TNTCFP expression, GluR1 was selectively reduced at inhibited synaptic sites (arrows, right column) compared with nontransfected synapses (arrowheads). Two examples are shown in a and b. Scale bars, 10 μm.
Discussion
We show here that expression of tetanus toxin light chain-CFP is a highly effective tool to locally inhibit transmitter release without grossly affecting general neuronal development or synapse assembly. Surprisingly, hippocampal neurons defective in transmitter release formed and maintained as many morphological synapses as active neurons when grown together in small groups on glial islands. Inhibited synapses were deficient in the amount of GluR1 that they were able to recruit but only in the presence of active neighbors. However, other synaptic proteins, including PSD95 family proteins, SynGAP, CaMKIIα, NMDA receptors, and AMPA receptor subunits GluR2 and GluR2/3, were present in normal amounts. Thus, differences in the ability to release neurotransmitter selectively regulate the AMPA receptor content of hippocampal pyramidal neurons on a synapse-by-synapse basis.
Local regulation of AMPA receptor subunit composition
Our results show that the level of GluR1 is controlled at individual synapses along a dendrite by neurotransmitter release. Chronically inhibited synapses have decreased levels of surface and total GluR1 without changes in PSD95. The dissociation of synaptic PSD95 and GluR1 levels is in contrast to previous studies proposing that the amount of PSD95 at the synapse regulates the concentration of synaptic AMPA receptors, particularly those containing GluR1 (El-Husseini et al., 2000; Beique and Andrade, 2003; Stein et al., 2003; Ehrlich and Malinow, 2004). We also see no changes in GluR2 or GluR2/3 staining at the synapse. Although we cannot rule out the possibility of accompanying changes in GluR4, or changes in GluR3 too subtle to be detected with the GluR2/3 antibody, the simplest interpretation of this data is local activity control of levels of homomeric GluR1 AMPA receptors. Although most AMPA receptors in these neurons are thought to exist as heteromers, some evidence indicates that there is a population of homomeric GluR1 receptors. Sequential immunoprecipitation shows that ∼8% of total AMPA receptor in adult CA1/CA2 neurons is homomeric GluR1, with the remainder being composed primarily of GluR1 with GluR2 and GluR2 with GluR3 (Wenthold et al., 1996). In addition, calcium-permeable AMPA/kainate receptors have been observed in hippocampal pyramidal neuron dendrites, likely in the form of homomeric GluR1 receptors (Ogoshi and Weiss, 2003).

GluR2 is unchanged at locally inhibited synapses. Banker-style dissociated neurons were cotransfected before plating with SYNYFP and either CFP (left column) or TNTCFP (right column), grown for 3 weeks, fixed, and stained for GluR2 and SynGAP. GluR2 was found to colocalize at both active, CFP-expressing synapses (arrows, left column) and inhibited, TNTCFP-expressing synapses (arrows, right column). The top row shows a triple color overlay. Scale bar, 10 μm.

GluR1 is reduced at locally inhibited synapses, where as GluR2, GluR2/3, and PSD95 are unchanged. The average intensity of surface GluR1 (a), total GluR1 (b), GluR2 (c), GluR2/3 (d), or PSD95 (e) was measured at transfected and nontransfected synapses along the same dendrites. Values were divided by the staining intensity of the dendrite shaft to control for differences in cellular expression. To account for experiment-to-experiment staining variation, we normalized all values in each condition of each culture to the median value of the nontransfected synapses of that condition. Thus, values reported here are normalized synapse/shaft ratios. We assessed these measures in both control (left column) and tetanus toxin-supplemented bath (right column). Local inhibition by TNTCFP did not change the level of the subunit GluR2 (c; n ≥ 20 cells and n > 400 synapses each) or GluR2/3 (d; n ≥ 16 cell and n > 300 synapses each) at individual synapses in either control bath or globally inhibited tetanus toxin bath conditions. The concentration of GluR1 was reduced by 22% opposite TNTCFP-expressing terminals compared with active neighbors (b; n > 80 cells and n > 1900 synapses each; p < 0.01, Kruskal-Wallis one-way ANOVA on ranks), although PSD95 at the same synapses was unchanged (e; n > 80 cells and n > 1900 synapses each). This occurred only in control bath and was abolished with global inhibition by bath application of tetanus toxin (b; n > 25 cells and n > 350 synapses each). This decrease with local inactivity was also reflected in surface GluR1 levels (a; n = 12 cells and n > 300 synapses each; p < 0.01, Mann-Whitney rank sum test), examined by staining live neurons with an antibody that recognized the extracellular N terminus of GluR1.
GluR1 confers unique properties to AMPA receptors that are not emulated by other AMPA receptor subunits. GluR1-containing AMPA receptors are selectively inserted during LTP (Shi et al., 2001). Furthermore, GluR1 knock-out mice lack adult CA1 LTP (Zamanillo et al., 1999) and show deficits in some forms of spatial memory (Reisel et al., 2002). Local alterations in subunit composition can change the kinetics of the AMPAR-mediated EPSC (Dingledine et al., 1999), stability or turnover of receptors (Lee et al., 2004), and protein-protein interactions with the variant intracellular domains of the different subunits (Malinow and Malenka, 2002; Song and Huganir, 2002). Such changes could in turn influence downstream signaling through the AMPA receptor complex after synaptic transmission. Local regulation of levels of GluR1 could also result in an alteration in local calcium signaling through GluR2-lacking AMPA receptors. Thus, local long-term inhibition of synaptic transmission may lead to local changes in responsiveness to induction of synaptic plasticity via alterations in GluR1 content. Although others have shown regulation of receptor composition in particular synapse populations (Rubio and Wenthold 1997; Toth and McBain 1998; Liu and Cull-Candy, 2000), this is the first study of which we are aware reporting localized synapse-by-synapse activity regulation of AMPA receptor subunit composition.
Perhaps the most striking finding here is the high spatial resolution of activity regulation of local AMPA receptor content. Inhibited synapses intermingled with neighbor active synapses, mere micrometers apart, exhibited precise synapse-specific regulation of AMPA receptor content. While our study was in progress, another group demonstrated synapse-by-synapse control of AMPA currents using focal glutamate uncaging (Matsuzaki et al., 2004). These functional changes might reflect local changes in AMPA receptor subunit levels, as we find with our longer-term local activity manipulation, although our effects appear to be independent of synapse size. These data show that hippocampal pyramidal neurons can exhibit precise input-specific activity regulation at the level of an individual synapse.
Synaptic competition
Competition between potential synaptic partners has been hypothesized to play an important role in the shaping of the organization of the nervous system (Wiesel, 1982; Katz and Shatz, 1996). Competition between individual axons has been studied at the neuromuscular junction, although different studies have yielded apparently contrasting results regarding the role of activity. Although several studies have reported that the more active axons take over more synaptic territory (Ridge and Betz, 1984; Balice-Gordon and Lichtman, 1994; Buffelli et al., 2003), others have reported the opposite, that inactive axons are favored (Callaway et al., 1987, 1989). It may be that more active axons tend to dominate at individual junctions, but this effect is balanced by limited resources of individual neurons and effects of scale and spacing (Barber and Lichtman, 1999). In the olfactory bulb, inhibition of only a subset of olfactory sensory neurons results in their aberrant targeting followed by apparent loss of the inhibited axons, suggesting that synapse stability and neuronal survival in this environment may be sensitive to relative levels of neurotransmitter release (Yu et al., 2004). However, our study did not show a change in the number of synapses formed by an inactivated axon. This suggests that hippocampal neurons, at least in our system, do not use an activity-dependent mechanism to determine the distribution of synaptic space among competing axons. Why do we not see a decrease in synapses formed by inhibited axons, even after 2 weeks? We cannot rule out the possibility that there may be a change in synapse number at other times in these cultures or that only some types of neurons have the requirement and machinery to exhibit synaptic competition. Perhaps resource and spacing issues are more important than relative activity in determining axonal synaptic territory in hippocampal microislands. In any case, these data imply that competition in the CNS may not be a simple scenario in which more activity leads to takeover of more synaptic space (Hata et al., 1999).
Others have found that decreasing neuronal excitability in a subset of cultured neurons before synapse formation caused the inhibited neurons to recruit fewer inputs (Burrone et al., 2002). This effect was blocked by growing the neurons in TTX, implying that it is dependent on different levels of postsynaptic activity between neurons. We found that blockade of presynaptic release does not lead to changes in the number of synapses formed by a given neuron; however, it does change the amount and/or composition of AMPA receptors at inhibited sites. Moreover, this local reduction in GluR1 content occurred only in the presence of neighbor active synapses and was abolished when all neurons were inhibited by bath application of tetanus toxin. These results suggest that synaptic competition between hippocampal neurons may occur with differential excitability of potential postsynaptic partners but not with differential release from potential presynaptic partners. This implies that regulation of synapse number is controlled by a mechanism intrinsic to the postsynaptic cell, which in some way senses its depolarization state. In contrast, synapse composition is regulated via a mechanism sensitive to relative levels of presynaptic release of neurotransmitter from neighboring inputs, implying a competitive mechanism of some sort. Whereas GluR1 is present in excess throughout the neuron, there could be a limited pool of GluR1 available for distribution to synapses, weighted according to relative activity. Alternately, there could be a more complicated mechanism underlying this competition, similar to the putative punishment and protection signals proposed to mediate competition at the NMJ (Lichtman and Colman, 2000). Additional experiments manipulating presynaptic release and postsynaptic excitability in other ways will be required to test the generality of these ideas.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grants NS34448 and NS33184. We thank Huaiyang Wu for excellent preparation of neuron cultures.
Correspondence should be addressed to Ann Marie Craig, Brain Research Centre, University of British Columbia, 2211 Wesbrook Mall, Vancouver, British Columbia, Canada V6T 2B5. E-mail: amcraig{at}interchange.ubc.ca.
K. J. Harms' present address: Department of Neuroscience, Brown University, Providence, RI 02912.
K. R. Tovar's present address: Vollum Institute, Oregon Health and Science University, Portland, OR 97239.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256379-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}