

Abstract
An intensely painful stimulus may lead to hyperalgesia, the enhanced sensation of subsequent painful stimuli. This is commonly believed to involve facilitated transmission of sensory signals in the spinal cord, possibly by a long-term potentiation-like mechanism. However, plasticity of identified synapses in intact hyperalgesic animals has not been reported. Here, we show, using neuronal tracing and postembedding immunogold labeling, that after acute noxious stimulation (hindpaw capsaicin injections), immunolabeling of Ca2+/calmodulin-dependent protein kinase II (CaMKII) and of CaMKII phosphorylated at Thr286/287 (pCaMKII) are upregulated postsynaptically at synapses established by peptidergic primary afferent fibers in the superficial dorsal horn of intact rats. In contrast, postsynaptic pCaMKII immunoreactivity was instead downregulated at synapses of nonpeptidergic primary afferent C-fibers; this loss of pCaMKII immunolabel occurred selectively at distances greater than ∼20 nm from the postsynaptic membrane and was accompanied by a smaller reduction in total CaMKII contents of these synapses. Both pCaMKII and CaMKII immunogold labeling were unaffected at synapses formed by presumed low-threshold mechanosensitive afferent fibers. Thus, distinct molecular modifications, likely indicative of plasticity of synaptic strength, are induced at different populations of presumed nociceptive primary afferent synapse by intense noxious stimulation, suggesting a complex modulation of parallel nociceptive pathways in inflammatory hyperalgesia. Furthermore, the activity-induced loss of certain postsynaptic pools of autophosphorylated CaMKII at previously unmanipulated synapses supports a role for the kinase in basal postsynaptic function.
Introduction
Postsynaptic autophosphorylation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) at Thr286/287, which renders the kinase autonomously active, is necessary and sufficient for the induction of NMDA receptor-dependent long-term potentiation (LTP) at many glutamatergic synapses (Lisman et al., 2002; Colbran and Brown, 2004). This autophosphorylation presumably accompanies a translocation of the enzyme to the postsynaptic density (PSD), where the active kinase may exert its effects on AMPA receptors, and on other downstream targets involved in synaptic potentiation. However, although natural stimulation may induce a reversible translocation of CaMKII to vertebrate synapses in vivo (Gleason et al., 2003), a corresponding activity-dependent increase in autophosphorylated CaMKII within the PSD has previously been observed only in unspecified synapses or after nonphysiological stimulation (Strack et al., 1997; Dosemeci et al., 2002; Rodrigues et al., 2004), and it is unknown whether such phosphorylation occurs also at topographically defined synapses in vivo. Dephosphorylation of CaMKII within the PSD is achieved by PSD-associated protein phosphatase 1 (PP1), which is likely involved in long-term depression and in depotentiation of synapses that have previously undergone LTP (Morishita et al., 2001; Colbran, 2004). Indeed, CaMKII and PP1 have been proposed to form within the PSD a bistable molecular switch that is responsible for the maintenance of LTP (Lisman and Zhabotinsky, 2001; Miller et al., 2005). Notably, however, negative regulation of postsynaptic CaMKII autophosphorylation by neuronal activity has not been demonstrated (Colbran, 2004).
CaMKII has been implicated in the induction of NMDA receptor-dependent sensitization of neurons in the spinal cord dorsal horn (Fang et al., 2002; Garry et al., 2003; Galan et al., 2004). This central sensitization is a likely mechanism underlying certain forms of hyperalgesia, the enhanced sensation of painful stimuli that may arise after intense noxious stimulation, and is commonly believed to involve LTP-like strengthening of primary afferent transmission that depends on activation of NMDA receptors and CaMKII (Kolaj et al., 1994; Sandkühler et al., 2000; Woolf and Salter, 2000; Willis, 2002; Ji et al., 2003; Yang et al., 2004; Pedersen et al., 2005). Thus, increased levels of CaMKII and CaMKII autophosphorylated at Thr286/287 (pCaMKII) would be expected at synapses from primary afferent fibers supplying the noxiously stimulated tissue. Primary afferent synapses in the dorsal horn constitutively contain pCaMKII within their PSD (Larsson and Broman, 2005), but it is unknown whether CaMKII may be modulated in these synapses in central sensitization. In fact, plastic changes at identified synapses after natural noxious stimulation in intact animals have not been observed, whether by molecular or electrophysiological methods. Therefore, we used capsaicin injection into rat hindpaw skin together with postembedding immunogold labeling and electron microscopy to investigate possible changes in the levels of pCaMKII and CaMKII at primary afferent synapses after acute noxious stimulation. In particular, the combination of transganglionic tracing of select thin-caliber primary afferent fibers with immunolabeling of substance P (SP) and calcitonin gene-related peptide (CGRP) allowed us to study molecular changes in synapses established by major identified and somatotopically defined populations of nociceptive and non-nociceptive primary afferent terminals.
Materials and Methods
Animals and surgery.
Four adult male Sprague Dawley rats (∼300 g) were anesthetized with sodium pentobarbital (50 mg/kg, i.p.), after which 2% wheat germ agglutinin conjugated to horseradish peroxidase (WGA-HRP) was injected into digit 5 and the pad proximal to this digit on the ventral side of both hindpaws (0.5 μl per site). Three days later, the animals were again anesthetized with sodium pentobarbital (50 mg/kg, i.p.). In three of the rats, 10 or 15 μl of 3% capsaicin (dissolved in 15% ethanol and 7% Tween 80 in saline) was injected into the same skin sites as the tracer injections in one of the hindpaws (contralateral hindpaws and both hindpaws of one rat were left unstimulated). An intense inflammatory response was evident in the capsaicin-stimulated skin within a few minutes of capsaicin injection. Importantly, no vehicle was injected in the nonstimulated (control) hindpaws, because the trauma of the injection and the vehicle, in particular the ethanol that it contained, could by itself activate nociceptors (Baumann et al., 1991; Trevisani et al., 2002). All animals were kept anesthetized for 20 min after the capsaicin injection (or for an equivalent period of time in the nonstimulated rat), after which they were rapidly fixed by transcardial perfusion with PBS (300 mOsm; 20–30 s) and 1% paraformaldehyde and 1.25% glutaraldehyde in PBS (∼1 L; 30 min). Care was taken not to inflict any additional noxious stimulation of the hindlimbs before fixation. Experimental procedures were in accordance with institutional guidelines and were approved by the Animal Care and Use Committee of Malmö-Lund.
Tissue preparation.
The lumbar spinal cord was removed and parasagittal sections (150 μm) of the L4 and L5 segments were cut on a vibratome. Tracer substance was visualized by tetramethyl benzidine histochemistry performed at 4°C overnight. The reaction product was stabilized with phosphate buffer, pH 6.0, containing 3,3′-diaminobenzidine (0.5 mg/ml), CoCl2·6H2O (0.08%), and H2O2 (0.01%) for 10 min. The transganglionic labeling formed a patch centered in lamina IIi in the rostral one-half of medial sections through L5 (cf. Levinsson et al., 2002). Peroxidase-labeled sections of dorsal spinal cord were cryoprotected in glycerol overnight, plunged into liquid propane, and embedded by freeze substitution in Lowicryl HM20 (Electron Microscopy Sciences, Hatfield, PA), as described (Larsson et al., 2001). Serial ultrathin sections were cut and placed on single-slot Pioloform-coated nickel grids.
Postembedding immunogold labeling.
Postembedding immunogold labeling of CaMKII and pCaMKII was performed essentially as described (Larsson and Broman, 2005). Briefly, ultrathin dorsal horn sections were incubated in 50 mm glycine in Tris buffer containing 0.3% NaCl and 0.1% Triton X-100 (TBST), pH 7.4, for 10 min and in 2% human serum albumin in TBST (TBST-HSA) for 10 min. The sections were then placed in rabbit anti-CaMKII (4 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) or rabbit anti-pCaMKII (1:200; Promega, Madison, WI) in TBST-HSA for 2 h. After rinsing in TBST and TBST-HSA, the sections were incubated in goat anti-rabbit antibody conjugated to 10 nm colloidal gold (1:20; Amersham Biosciences, Uppsala, Sweden) in TBST-HSA for 1 h. In each experiment, sections from capsaicin-injected sides and controls were treated in parallel and incubated in the same drops of reagent.
Double immunogold labeling of SP and CGRP in adjacent sections was performed as above, except that the primary antibodies used were rat anti-SP (1:200; Chemicon, Temecula, CA) and rabbit anti-CGRP (1:400 or 1:800; Bachem AG, Bubendorf, Germany), and the secondary antibody solution contained goat anti-rat antibody conjugated to 5 nm gold (1:40; British Biocell, Cardiff, UK) and goat anti-rabbit antibody conjugated to 15 nm gold (1:20; Amersham Biosciences).
The anti-CaMKII antibody used is directed toward amino acids 1–300 of human CaMKII and recognizes all CaMKII isoforms. The anti-pCaMKII antibody is specific toward the α and β isoforms of CaMKII autophosphorylated at Thr286 (α) or Thr287 (β) and possibly also recognizes CaMKIIγ and/or CaMKIIδ phosphorylated at Thr287 (Lorenz et al., 2002). When antibodies were omitted from the primary antibody solution in the postembedding immunogold labeling procedure, we observed virtually no immunogold labeling, and specifically none associated with synapses. By immunocytochemistry, the pCaMKII antibody yields much reduced immunolabeling in the visual cortex of transgenic mice in which the Thr286 site of CaMKIIα had been mutated to an alanine (thus precluding phosphorylation of this site; residual labeling likely indicated other CaMKII isoforms phosphorylated at Thr287) (Taha et al., 2002), confirming the phosphospecificity of the antibody. Postsynaptic preembedding immunogold labeling using this antibody was shown to be increased in cultured hippocampal neurons after NMDA stimulation and phosphatase inhibition (Dosemeci et al., 2002). We also previously showed that rapid perfusion fixation does not affect the levels of immunolabeling with the antibody in the spinal cord (Larsson and Broman, 2005).
Quantitative analysis of immunogold labeling.
For both CaMKII and pCaMKII immunolabeling, in two experiments we analyzed sections from the dorsal horns ipsilateral and contralateral to the capsaicin injections, whereas in the third experiment, we used a dorsal horn section from the unstimulated animal as control.
Transganglionic tracing using WGA-HRP enabled the identification of terminations of primary afferent fibers projecting from capsaicin-stimulated skin sites and corresponding sites in unstimulated hindpaws. Synapses formed by peroxidase-labeled terminals were identified by screening an adjacent nonimmunolabeled section. Peroxidase-labeled terminals were found predominantly in inner lamina II (IIi), although a few such terminals were present in outer lamina II (IIo). Peroxidase-labeled terminals in lamina I were very rare. The majority of peroxidase-labeled terminals contained no or very few dense core vesicles. An important aim of the study was to investigate possible differences in CaMKII regulation between peptidergic and nonpeptidergic nociceptive fiber synapses; thus, to yield a more homogeneous sample of synapses, terminals containing more than three dense core vesicles, and therefore judged to be the central terminals of peptidergic type Ib glomeruli (Ribeiro-da-Silva et al., 1989; Ribeiro-da-Silva, 2004), were discarded from the analysis. The large majority of the remaining sampled terminals could be identified by their dark axoplasm and clear vesicles of heterogeneous size as dense sinusoid axon terminals [i.e., the central terminals of type Ia glomeruli, arising from isolectin B4-binding, nonpeptidergic C-fibers (Gerke and Plenderleith, 2004; Ribeiro-da-Silva, 2004)].
Synapses in lamina I formed by terminals of thin-caliber primary afferent fibers containing SP and CGRP were identified by screening the adjacent SP/CGRP double-immunolabeled section. Although SP+/CGRP+ terminals in lamina I were seldom peroxidase-labeled, we took advantage of the observation that primary afferent fibers show similar somatotopy throughout the dorsal horn (Willis and Coggeshall, 2004). Thus, SP+/CGRP+ terminals, which showed either a simple synaptic configuration or were the central terminal of type Ib glomeruli, were sampled within a rostrocaudal area in lamina I defined by the rostrocaudal extension of transganglionic labeling in lamina II (see Fig. 1). The border between lamina I and lamina IIo was not well defined and therefore lamina I was defined as the portion of the dorsal horn within 50 μm of the white matter border. Thus, some sampled SP+/CGRP+ terminals may have been located in lamina IIo.
Presumed low-threshold mechanosensitive primary afferent terminals were identified by morphological criteria in the nonimmunolabeled section by their glomerular arrangement and light axoplasm containing small, clear vesicles and numerous mitochondria (Maxwell and Réthelyi, 1987; Willis and Coggeshall, 2004; Larsson and Broman, 2005). Synapses formed by such terminals were sampled in laminas IIi–IV within the same rostrocaudal boundaries as the other synaptic populations (see Fig. 1).
After identification of a terminal, electron micrographs of its synapses were obtained at 80,000× in an adjacent CaMKII or pCaMKII immunogold-labeled section. For quantitative analysis of CaMKII and pCaMKII immunolabeling, the postsynaptic plasma membrane, or cleft face of the PSD, was outlined and locations were recorded of the center of gold particles that could be orthogonally projected onto this outline. Postsynaptic gold particles within 100 nm of the postsynaptic membrane were considered to be associated with the PSD. Indeed, nearly all such particles lay immediately over the PSD or were within ∼25 nm (the spatial resolution limit of the immunogold labeling) (Matsubara et al., 1996) of the cytosolic face of the PSD. Analysis of the subsynaptic distribution of immunogold labeling was performed essentially as described (Larsson and Broman, 2005).
Results
pCaMKII immunolabeling
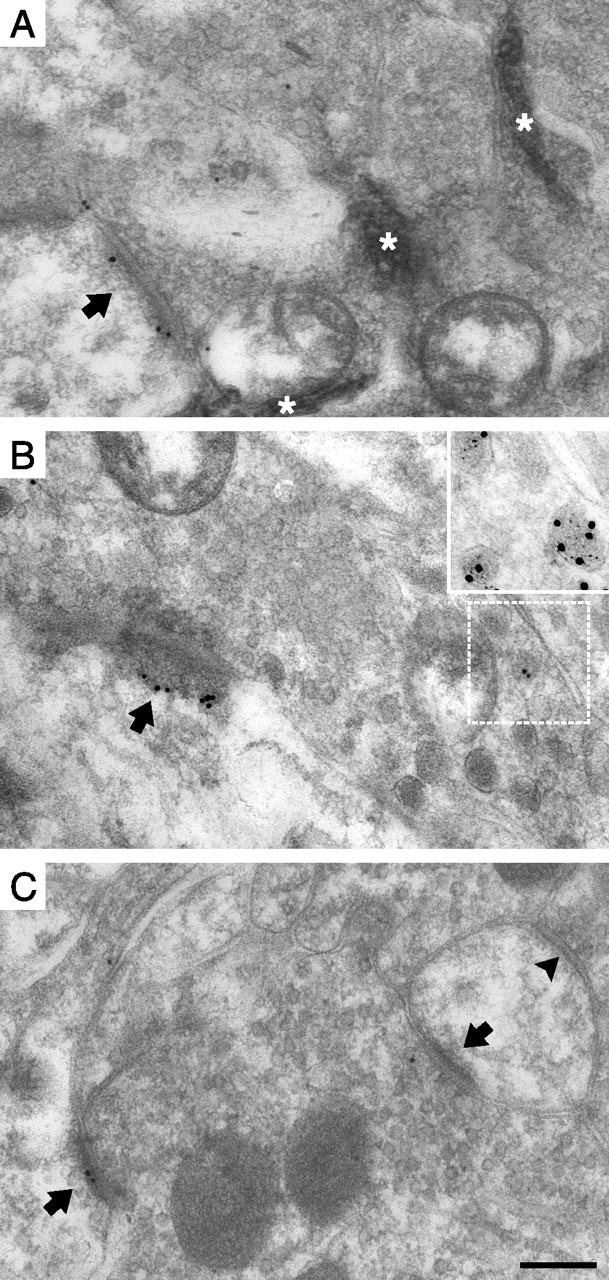
Transganglionic tracing using WGA-HRP allowed us to identify fine-diameter primary afferent terminations from the capsaicin-stimulated skin area (and the corresponding area of unstimulated hindpaws) in the superficial dorsal horn. To avoid confounding effects caused by inflammation induced by the injection trauma and the vehicle, control hindpaws did not receive vehicle injections. In the following, “capsaicin stimulation” thus refers to the combined peripheral stimulation conferred by injection of both capsaicin and vehicle. Peroxidase product was predominantly observed in nonpeptidergic presumed nociceptive C-fiber terminals in lamina IIi (Figs. 1, 2A,B). Surprisingly, synapses formed by peroxidase+ terminals in the dorsal horn ipsilateral to the capsaicin injections exhibited summed linear densities (number of PSD-associated gold particles per micrometer of postsynaptic membrane) of pCaMKII immunogold labeling on average 52% (range, 30–75%) of that of synapses in controls [contralateral dorsal horn (n = 2) or unstimulated animal (n = 1)] (Table 1, Figs. 2A,B, 3A). No difference was evident between those experiments using contralateral dorsal horn and that using a naive animal as controls, and therefore data from all experiments were pooled. The same pattern was evident for the absolute level of pCaMKII immunolabeling per synapse (Fig. 3B), showing that the reduction in linear density of synaptic immunolabeling was not attributable to rapid morphological changes such as synaptic enlargement.

Identification of topographically defined primary afferent synapses in the spinal cord dorsal horn. A, Sites of cutaneous injections of WGA-HRP and capsaicin in the glabrous hindpaw skin, indicated by ×. In contralateral hindpaws and both hindpaws of a control animal, only tracer injections were performed at these sites. B, Locations, in a parasagittal dorsal horn section, of synapses formed by primary afferent fibers supplying capsaicin-stimulated rat hindpaw skin, from pCaMKII immunolabeling experiment 3. Some marked locations represent more than one sampled synapse within a 16 × 16 μm field. Roman numerals (I–III) denote the different spinal laminas. The black line shows the border between white matter and lamina I. Peroxidase+, Synapses formed by nonpeptidergic nociceptive primary afferent fibers, as identified by transganglionic tracing with WGA-HRP. SP+/CGRP+, Lamina I synapses formed by primary afferent fibers containing SP and CGRP, as assessed in an adjacent section immunolabeled for these substances. LTM, Synapses formed by presumed low-threshold mechanosensitive afferent fibers. Note that SP+/CGRP+ and LTM synapses were not sampled beyond the rostrocaudal extent of the peroxidase+ synaptic sample. Scale bar, 50 μm.

Postsynaptic pCaMKII immunolabeling at identified primary afferent synapses. A, B, Synapses formed by peroxidase+, nonpeptidergic C-fiber terminals in the dorsal horn of unstimulated controls (A) and after capsaicin stimulation (B). The arrows indicate PSDs. Note the nonimmunolabeled synapse in B. The asterisks indicate peroxidase reaction product. C, D, Synapses formed by SP+/CGRP+ terminals in control (C) and capsaicin-stimulated (D) dorsal horn. In D, SP/CGRP immunolabeling of a dense core vesicle in the presynaptic terminal in the adjacent section labeled for SP (5 nm gold) and CGRP (15 nm gold) is shown magnified in the inset, and the corresponding location in the pCaMKII immunolabeled section is indicated by the white frame. E, F, Immunolabeling of presumed LTM synapses in control dorsal horn (E) and in dorsal horn ipsilateral to capsaicin-stimulated skin (F). Scale bar: (in F) A–F, 200 nm; D, inset, 100 nm.
pCaMKII immunolabeling at primary afferent synapses in dorsal horn ipsilateral to capsaicin stimulation and in control dorsal horn

Postsynaptic pCaMKII immunolabeling is differentially affected at primary afferent synapses by cutaneous capsaicin injection. A, Summed linear densities of postsynaptic pCaMKII immunolabeling in the dorsal horn of controls and after capsaicin stimulation. Error bars indicate SD. ∗∗p < 0.01, ∗∗∗p < 0.001, ANOVA followed by Bonferroni’s post hoc test. n = 3 individual experiments. B, Number of gold particles per synapse. Data from all experiments were pooled. Error bars indicate SD. ∗p < 0.05, ∗∗p < 0.01, Kruskal–Wallis test followed by Dunn’s post hoc test.
Peptidergic nociceptive primary afferent fibers, which express SP and CGRP, constitute a major nociceptive input to intrinsic and rostrally projecting lamina I neurons expressing the SP-activated neurokinin-1 (NK1) receptor (McLeod et al., 1998; Todd et al., 2002). In contrast to peroxidase+ synapses, SP+/CGRP+ synapses in lamina I ipsilateral to capsaicin injections exhibited linear densities of pCaMKII immunolabeling on average 227% (range, 150–298%) of such synapses in controls (Table 1, Figs. 2C,D, 3A). There was a similar difference in the absolute number of gold particles per synapse (Fig. 3B).
Presumed low-threshold mechanosensitive primary afferent fiber (LTM) synapses from hindpaw skin were identified by morphological criteria in laminas IIi–IV. Such synapses did not show different pCaMKII levels between capsaicin-stimulated and unstimulated sides (Table 1, Figs. 2E,F, 3).
Subsynaptic distribution of pCaMKII immunolabeling
In some subdomains of a PSD, CaMKII holoenzymes may self-associate, thereby forming tower-like structures of stacked CaMKII molecules extending orthogonally from the cleft face of the PSD toward the cytosol, whereas other parts of the PSD may be devoid of CaMKII (Petersen et al., 2003). This suggests that there may exist within individual PSDs spatially segregated pools of CaMKII that could be independently regulated by Ca2+ transients during synaptic activity. Therefore, we investigated potential differences in the subsynaptic distribution of pCaMKII in capsaicin-stimulated versus control nociceptive synapses (Figs. 4, 5). In SP+/CGRP+ synapses, the postsynaptic increase in pCaMKII labeling appeared to occur up to 60 nm (although significant only at 0–20 nm) from the postsynaptic membrane, and was relatively even throughout the tangential extent of the PSD, at capsaicin-stimulated compared with control synapses (Figs. 4A, 5A), suggesting that pCaMKII was elevated throughout the PSD of such synapses by capsaicin stimulation. In synapses formed by peroxidase+ terminals, the decrease in pCaMKII immunoreactivity occurred preferentially within a domain ∼20–60 nm from the postsynaptic membrane, whereas the level of pCaMKII immunolabeling immediately subjacent to (within ∼20 nm of) the postsynaptic membrane did not differ substantially between capsaicin-stimulated and control sides (Fig. 4B). Moreover, the loss of pCaMKII appeared to be more pronounced in the peripheral part of the synapse than in the central part (Fig. 5B). Thus, spatially distinct CaMKII pools within the PSD of these synapses may differ in their response to capsaicin-induced activity. At LTM synapses, no differences in subsynaptic distribution between capsaicin-stimulated and control sides were detected (Figs. 4C, 5C).

Differences in axodendritic distribution of pCaMKII immunolabeling between primary afferent synapses in control dorsal horn and in dorsal horn ipsilateral to capsaicin stimulation. Shown are the axodendritic distributions of pCaMKII immunogold labeling relative to the postsynaptic membrane (presynaptic gold particle distances negated), normalized by the number of synapses, for SP+/CGRP+ (A), peroxidase+ (B), and LTM (C) synapses. The dashed lines indicate the position of the outer surface of the postsynaptic membrane. Error bars indicate SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, two-way ANOVA followed by Bonferroni’s post hoc test. Pcaps, Statistical significance of capsaicin effect; Pdist, statistical significance of distance effect; Pi, statistical significance of interaction between capsaicin and distance effects.

Distribution of pCaMKII immunolabeling in the tangential axis of primary afferent synapses after capsaicin stimulation. A, Synapses formed by SP+/CGRP+ terminals. B, Peroxidase+ synapses. C, LTM synapses. central, Normalized number of gold particles located in the central one-half of the synapse. peripheral, Normalized number of gold particles found in the peripheral one-half of the synapse. Error bars indicate SD. ∗p < 0.05, ∗∗p < 0.01, two-way ANOVA followed by Bonferroni’s post hoc test. Pcaps, Statistical significance of capsaicin effect; Pdist, statistical significance of distance effect; Pi, statistical significance of interaction between capsaicin and distance effects.
CaMKII immunolabeling
Next, we sought to determine whether the observed pattern of pCaMKII at primary afferent synapses in capsaicin-stimulated and control sides was accompanied by similar differences in the postsynaptic levels of total CaMKII, and therefore performed immunogold labeling of sections from the same tissue blocks using a non-phosphospecific antibody. Because this antibody, like the anti-pCaMKII antibody, recognizes all CaMKII isoforms, it was possible to evaluate relative changes in the degree of CaMKII phosphorylation at the synapse. In control tissue sections, the overall CaMKII immunolabeling (assessed as particles/micrometer of postsynaptic membrane as well as particles/synapse) at PSDs of primary afferent synapses (Fig. 6, Table 2) resembled that of pCaMKII in control tissue (Larsson and Broman, 2005); notably, a significantly higher number of gold particles was observed at peroxidase+ synapses compared with LTM synapses (p < 0.05, repeated-measures ANOVA followed by Bonferroni’s post hoc test).
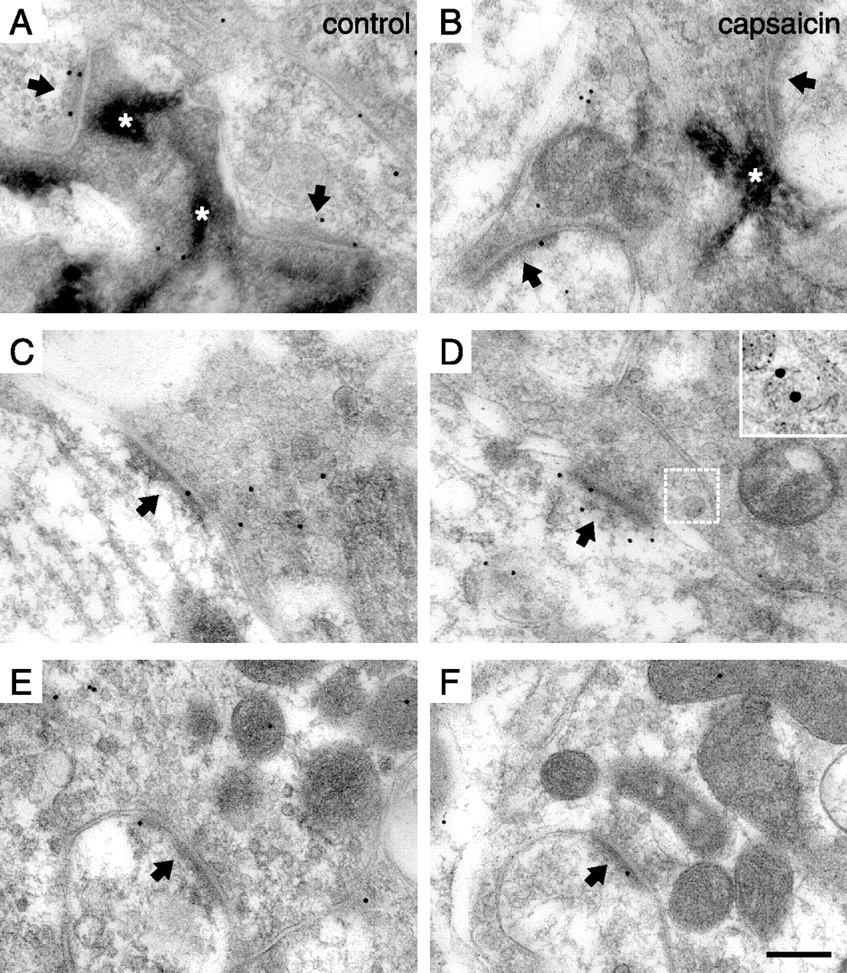
Postsynaptic non-phosphospecific immunolabeling of CaMKII at identified primary afferent synapses after cutaneous capsaicin injection. A, A synapse (arrow) formed by a peroxidase+, nonpeptidergic C-fiber terminal. The asterisks indicate peroxidase reaction product. B, A synapse (arrow) formed by an SP+/CGRP+ terminal. The inset shows SP/CGRP immunolabeling (SP, 5 nm gold; CGRP, 15 nm gold) of a presynaptic region containing dense core vesicles in the adjacent section labeled for these peptides; the corresponding location in the CaMKII-immunolabeled section is indicated by the white frame. C, A presumed LTM terminal forming one immunopositive and one immunonegative synapse (arrows). The arrowhead indicates a symmetric, presumed inhibitory synapse that lacks immunolabel. Scale bar: (in C) A–C, 200 nm; B, inset, 150 nm.
Immunolabeling of total CaMKII at primary afferent synapses in dorsal horn ipsilateral to capsaicin stimulation and in control dorsal horn
Linear density of PSD immunolabeling of total CaMKII in SP+/CGRP+ synapses ipsilateral to capsaicin stimulation was on average 167% (range, 138–197%) of such labeling at synapses in controls (Fig. 6, Table 2), thus exhibiting a pattern similar to that found for pCaMKII immunolabeling. This increase in postsynaptic CaMKII immunolabel occurred specifically 0–40 nm from the postsynaptic plasma membrane (Fig. 7A). At peroxidase+ synapses ipsilateral to capsaicin stimulation, postsynaptic CaMKII immunolabel was on average 81% (range, 74–95%) of that of peroxidase+ synapses in control sections (Fig. 6, Table 2). This overall difference between the sides was not statistically significant; however, analysis of the axodendritic distribution of CaMKII immunolabel revealed a statistically significant postsynaptic decrease specifically 10–30 nm from the postsynaptic membrane (Fig. 7B).

Axodendritic distribution of total CaMKII immunoreactivity at primary afferent synapses after capsaicin stimulation. The axodendritic distributions of CaMKII immunogold labeling relative to the postsynaptic membrane (presynaptic gold particle distances negated), normalized by the number of synapses are shown for SP+/CGRP+ (A), peroxidase+ (B), and LTM (C) synapses. The dashed lines indicate the position of the extracellular face of the postsynaptic membrane. Error bars indicate SEM. ∗p < 0.05, ∗∗p < 0.01, repeated-measures two-way ANOVA followed by Bonferroni’s post hoc test. Pcaps, Statistical significance of capsaicin effect; Pdist, statistical significance of distance effect; Pi, statistical significance of interaction between capsaicin and distance effects.
As in the case of pCaMKII immunolabel, no differences in CaMKII immunoreactivity were detected at LTM synapses between dorsal horn ipsilateral to capsaicin stimulation and control dorsal horn (Figs. 6, 7; Table 2). Furthermore, there were no differences between ipsilateral sides and controls in the tangential distribution of CaMKII immunolabel in any of the synaptic populations (data not shown).
Discussion
Here, we provide evidence of parallel changes in pCaMKII and total CaMKII at identified (with respect to both somatotopy and type) spinal nociceptive synapses after a noxious stimulus, thus confirming that postsynaptic translocation and phosphorylation of this kinase may occur concomitantly as a result of physiologically relevant afferent stimulation in vivo. Importantly, however, synapses of peptidergic and nonpeptidergic fine-diameter afferent fibers were shown to be differentially modulated with respect to both pCaMKII and CaMKII levels in a common model of central sensitization and inflammatory hyperalgesia (Willis and Coggeshall, 2004), suggesting distinct modes of plasticity of these parallel pathways in this form of hyperalgesia.
Both peptidergic and nonpeptidergic primary afferent neurons in the rat express the capsaicin receptor, TRPV1, although significant proportions of either population of nociceptor may lack the receptor (Tominaga et al., 1998; Guo et al., 1999; Michael and Priestley, 1999), and thus both types of nociceptor were likely directly activated by capsaicin in our study. However, because immunolabeling ipsilateral to the capsaicin-stimulated hindpaw was compared with that of unstimulated controls that had not received any vehicle injections, the observed effect likely depends partly on nociceptor activation induced by puncture and distention of the skin and by the constitutents of the vehicle. Notably, ethanol, which was included in the vehicle, may induce and potentiate TRPV1 receptor responses (Trevisani et al., 2002). Furthermore, capsaicin activation of peripheral TRPV1-positive nerve endings induces secondary release of inflammatory mediators and protons, which may activate also nociceptors that lack TRPV1 (Millan, 1999; Liu et al., 2004). Thus, the observed modifications of primary afferent synapses, rather than being a specific capsaicin-mediated effect, was presumably the result of a general acute inflammatory state and the activation of nociceptors associated with such a state. Nonetheless, capsaicin responses may be larger in nonpeptidergic rat primary afferent neurons (Liu et al., 2004), which could possibly underlie some of the observed difference in changes of pCaMKII and CaMKII levels between primary afferent synaptic populations; however, one would then expect a larger Ca2+ influx, and thus a larger increase of pCaMKII and CaMKII, at nonpeptidergic compared with peptidergic afferent synapses, rather than the observed, essentially opposite, difference. Similarly, activation of purinergic P2X3 receptors, selectively expressed by nonpeptidergic nociceptors (Vulchanova et al., 1998), would presumably enhance the postsynaptic Ca2+ at synapses established by these fibers. However, a possible explanation for the observed differences could be the activation of NK1 receptors preferentially expressed in dendrites postsynaptic to peptidergic nociceptors (McLeod et al., 1998; Todd et al., 2002). Indeed, concurrent activation of such receptors is essential for LTP in lamina I projection neurons (Ikeda et al., 2003).
It cannot be completely excluded that the observed changes in immunolabeling reflect changes in epitope accessibility (e.g., caused by stimulation-induced changes in CaMKII conformation or in its binding partners) rather than in epitope levels. However, the similar patterns of immunolabeling yielded by the phosphospecific and non-phosphospecific antibodies argue against this possibility. Furthermore, because of the high concentration of CaMKII within PSDs, it appears unlikely that modulation of CaMKII interactions with any one protein would have a substantial effect on the number of accessible epitopes.
Considering the roles of CaMKII and PP1 in synaptic plasticity (Morishita et al., 2001; Dosemeci et al., 2002; Lisman et al., 2002; Colbran, 2004; Colbran and Brown, 2004), these contrasting changes of pCaMKII and CaMKII levels at different nociceptive primary afferent synapses may correspond to differentially modulated efficacy of these synapses. The increase in pCaMKII and CaMKII levels at peptidergic nociceptive synapses thus suggest that these synapses are potentiated by CaMKII-dependent mechanisms, in keeping with the observation that LTP is inducible in vitro at SP-responsive lamina I projection neurons (Ikeda et al., 2003), which are extensively innervated by SP-containing afferents (McLeod et al., 1998; Todd et al., 2002) and may be necessary for hyperalgesia (Mantyh et al., 1997; Nichols et al., 1999; Vierck et al., 2003). In contrast, the observed molecular changes at nonpeptidergic C-fiber synapses in lamina IIi are difficult to interpret in a functional context, both because it is unknown what effects a decrease of postsynaptic pCaMKII may have on glutamatergic synaptic transmission, and because synaptic targets of nonpeptidergic C-fibers likely include both excitatory and inhibitory interneurons intrinsic to lamina II (Lu and Perl, 2003, 2005; Willis and Coggeshall, 2004).
A notable outcome of this study was the observation that synaptic activation may induce a loss of postsynaptic pCaMKII immunolabeling. This loss was not accompanied by a substantial general postsynaptic decrease in CaMKII levels, although a tendency toward such a decrease was noted and indeed was found significant within a subdomain of the PSD. Together, these results suggest that the degree of phosphorylation of individual CaMKII holoenzymes is to some extent decreased at nonpeptidergic C-fiber synapses after the low-frequency discharge induced in these afferents by the capsaicin injection. Interestingly, the loss of pCaMKII immunolabel, as well as the loss of total CaMKII immunolabel, occurred selectively at a distance >20 nm from the postsynaptic membrane, consistent with the notion of stacks of self-associated CaMKII molecules within the PSD (Petersen et al., 2003). Resistance to downregulation of phosphorylated CaMKII subunits subjacent to the postsynaptic membrane could conceivably be conferred by their binding partners in this subdomain of the PSD; for example, subunits activated by their binding to the NR2B subunit of the NMDA receptor could promote retained phosphorylation of neighboring kinase subunits (Bayer et al., 2001).
In conclusion, the present findings support a role for concurrent postsynaptic translocation and phosphorylation of CaMKII in glutamatergic synaptic plasticity in vivo. In addition, the demonstration that CaMKII may be differentially regulated at different primary afferent synaptic populations in vivo by capsaicin stimulation suggests significant differences in plasticity between two major nociceptive pathways in inflammatory pain.
Footnotes
-
This work was supported by the Swedish Research Council (Project 14276), the Crafoord Foundation, the Thorsten and Elsa Segerfalk Foundation, the Royal Physiographic Society, and the Magn. Bergvall Foundation. We thank Agneta Persson for expert technical assistance.
- Correspondence should be addressed to Max Larsson, Department of Experimental Medical Science, Division of Neuroscience, Lund University, BMC F10, SE-221 84 Lund, Sweden. Email: max.larsson{at}med.lu.se





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}