

Abstract
Growth arrest-specific protein 6 (gas6) activity is mediated through the receptor tyrosine kinase family members Axl, Rse, and Mer, all of which are expressed in human oligodendrocytes. In this study, we examined whether recombinant human (rh) gas6 protects oligodendrocytes from growth factor (insulin) withdrawal or tumor necrosis factor-α (TNFα) cytotoxicity. In addition, we examined whether the effect was caspase-dependent, which receptor mediated the protective effect, and whether survival required Akt1 activation. Oligodendrocyte viability was assessed by O4 staining and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. Addition of rhgas6 to insulin-depleted cultures resulted in a significant increase in oligodendrocyte viability. Rhgas6 and caspase inhibitors also reduced active caspase-3 immunoreactivity relative to TNFα-only-treated cultures. In cultures treated with TNFα (100 ng/ml), the oligodendrocyte survival rate was 18% compared with cultures treated with TNFα and rhgas6 (64%) or the caspase inhibitors IETD-fmk [z-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethyl ketone] (65%) and zVAD-fmk (N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone) (63%). Increased phosphoAkt (Ser473) immunoreactivity was detected 15 min after administration of gas6 and TNFα to oligodendrocyte cultures but not in TNFα-treated cultures. The gas6 protective effect was abrogated by the Axl decoy receptor Axl-Fc, by the phosphatidylinositol 3 (PI3) kinase inhibitor LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one], and in Akt1−/− oligodendrocytes. Oligodendrocyte cultures established from wild-type and Rse−/− mice, but not from Axl−/− mice, were also protected from TNFα-induced cell death when maintained in rhgas6. We conclude that gas6 signaling through the Axl receptor and the PI3 kinase/Akt1 survival pathway protects oligodendrocytes from growth factor withdrawal and TNFα-mediated cell death.
Introduction
Axl, Mer, and Rse comprise a receptor tyrosine kinase family that is activated by growth arrest-specific protein 6 (gas6). These cell adhesion molecule-related receptors contain two Ig-like repeats and two fibronectin repeats in the extracellular domain, a kinase domain, and a cytoplasmic domain that, after autophosphorylation, can recruit signaling molecules (Kazlauskas, 1994; Stitt et al., 1995; Varnum et al., 1995; Ling et al., 1996; Braunger et al., 1997; Hasanbasic et al., 2004). gas6 is secreted by neurons and endothelial cells and is widely expressed in the CNS, suggesting that the interaction between gas6 and its receptors has physiological relevance (Li et al., 1996; Prieto et al., 2000). The affinity of gas6 for the receptors is Axl > Rse > Mer with Axl activated by gas6 in the low nanomolar range (Nagata et al., 1996). In situ hybridization studies performed in rat CNS demonstrated that Axl, Mer, and Rse receptors are expressed in the white matter during myelination (Prieto et al., 2000). We determined by reverse transcription-PCR that oligodendrocytes isolated from human fetal spinal cord express all three receptors by 20 gestational weeks (gw). Furthermore, immunoblot analysis of brain and fetal spinal cord homogenates confirmed the presence of all three receptors and gas6, supporting a role in human and rodent development (Shankar et al., 2003).
Growth factor-mediated survival of mammalian cells often involves the activation of the phosphatidylinositol 3-kinase (PI3K) pathway (Jones et al., 1999) that can promote cell survival by activating the serine/threonine kinase Akt (Franke et al., 1997; Datta et al., 1999). Akt protects cells of the CNS against growth factor withdrawal and cytotoxic cytokines (Takano et al., 2000). Oligodendrocytes require the PI3K/Akt pathway for survival, and overexpression of constitutively active Akt, but not mitogen-activated protein kinase kinase, protected oligodendrocytes from various cellular stresses, including tumor necrosis factor-α (TNFα)-induced injury (Vemuri and McMorris, 1996; Takano et al., 2000). Previously, we demonstrated that gas6 protects human fetal oligodendrocytes from apoptotic cell death in the absence of trophic support and dramatically enhanced human oligodendrocyte survival in vitro. The gas6 prosurvival effect was blocked by inhibitors of the PI3 kinase pathway but not the extracellular signal-regulated kinase pathway (Shankar et al., 2003).
In this study, we examined the effect of insulin withdrawal or the addition of the cytokine TNFα as initiators of oligodendrocyte injury. We also tested the hypothesis that recombinant human (rh) gas6 engagement of the Axl receptor activates the PI3 kinase/Akt prosurvival pathway protecting oligodendrocytes from TNFα-induced apoptosis. TNFα was studied because it is one of the major secreted cytokines associated with white matter injury in multiple sclerosis and periventricular leukomalacia (Bitsch et al., 2000; Rezaie and Dean, 2002). Thus, our goal was to determine whether gas6 would protect oligodendrocytes from TNFα-induced damage and to identify the receptor (Axl, Rse, or Mer) and signaling pathways involved.
Materials and Methods
Human oligodendrocyte cultures.
Human fetal spinal cord tissue was obtained from the Human Fetal Tissue Repository as approved by the Institutional Review Board of Albert Einstein College of Medicine and state and federal laws. Human oligodendrocyte cultures were generated from 21–23 gw spinal cords as described previously (Shankar et al., 2003). Cells (5 × 104) were plated on poly-l-lysine (Sigma, St. Louis, MO)-coated 24-well tissue culture wells or on 12 mm glass coverslips (Fisher Scientific, Hampton, NH). Oligodendrocyte cultures were grown for 5 d in a chemically defined (CD) medium consisting of DMEM, N2 supplement (5000 ng/ml insulin, 10 μg/ml transferrin, 5 ng/ml selenite, 16 μg/ml putrescine, and 6 ng/ml progesterone; Invitrogen, Carlsbad, CA), penicillin/streptomycin at 100 U/ml and 0.1 mg/ml, respectively, 10 ng/ml platelet-derived growth factor (PDGF), and 5 ng/ml neurotrophin-3 (NT-3). CD medium was supplemented with 400 ng/ml (5.6 nm) rhgas6 (Amgen, Thousand Oaks, CA) and cultured at 37°C and 5% CO2. The purity of cultures was determined by immunostaining cells with an O4 monoclonal antibody (Sommer and Schachner, 1981; Gard and Pfeiffer, 1989) or rabbit antiserum against 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP) (Raible and McMorris, 1989), the astrocyte-specific glial fibrillary acidic protein (GFAP), neuron-specific nuclear protein (NeuN) antibody, CD45 and CD11b and were determined to be 71% O4+CNP+ and 10% GFAP+ astrocytes. Cells were negative for NeuN and type III tubulin (neurons), CD45 and CD11b (microglia), and vimentin (early astrocytes and endothelial cells). Thus, the remaining 19% of the cells that could not be accounted for with the standard oligodendrocyte-specific markers were likely human glial or neuronal progenitor cells.
Primary mouse cortical oligodendrocyte cultures.
All animal studies were approved by the Institutional Animal Care and Use Committee. Our Animal Welfare Assurance is on file with the Office of Laboratory Animal Welfare. Animals are maintained under barrier conditions in static microisolation cages. These colonies are tested quarterly and confirmed to be free of adventitious murine agents. Mice lacking the Axl (Axl−/−) or the Rse (Rse−/−) receptor were obtained from the Lemke laboratory (Lu et al., 1999) and backcrossed to C57BL/6J wild-type (WT) mice more than five generations. Akt+/− mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Axl+/−, Rse+/−, and Akt+/− heterozygotes were mated to obtain Axl−/−, Rse−/−, and Akt1−/− homozygotes and WT littermates. Oligodendrocyte cultures were established from postnatal day 1 WT, homozygous knock-out mice by a modification of previously established protocols (Knapp et al., 1987; Armstrong, 1998; Sperber and McMorris, 2001). Mice were decapitated, and the cerebral hemispheres were removed aseptically. The meninges were removed, and the cortices were dissected in a dish on ice containing Ca2+/Mg2+ free HBSS (Invitrogen). The minced tissue was incubated in prewarmed trypsin/EDTA solution (0.025% trypsin and 0.1 mm EDTA; Sigma) and shaken at 110 rpm on a Clay Adams nutator (Becton Dickinson, Parsippany, NJ) at 37°C for 20 min. Trypsinization was terminated by adding a 1:1 volume of 25 μg/ml soybean meal trypsin inhibitor (Sigma) for 5 min at room temperature (RT). The tissue was spun at 1000 rpm for 5 min at RT and incubated in 80 μg/ml DNase in HBSS containing 3.9% MgSO4 for 10 min at 37°C to digest extracellular DNA. The tissue solution was then mechanically triturated by pipetting up and down 15 times. The cell suspension was centrifuged at 1000 rpm for 5 min at RT, and the cell pellet was resuspended in 15 ml of DMEM containing 2% fetal bovine serum. The suspended cells were transferred to a T75 flask and incubated overnight in a 5% CO2, 37°C incubator to allow adherent cells including microglia and type 1 astrocytes to attach to the dish. Subsequently, nonadherent cells were collected and filtered twice through a cell strainer (40 μm pore size; Falcon, Franklin Lakes, NJ) and centrifuged at 1000 rpm for 5 min at RT. The pelleted oligodendrocytes were resuspended in a serum-free CD medium containing 10 ng/ml PDGF and 10 ng/ml basic fibroblast growth factor (bFGF). The 5 × 104 cells were plated on poly-l-lysine (Sigma)-coated 24-well tissue culture wells for the assessment of oligodendrocyte survival after treatments in the presence or absence of rhgas6 or on poly-l-lysine-coated coverslips (Fisher Scientific) for fluorescence microscopy. Cells were cultured at 37°C and 5% CO2. To allow for the differentiation of the oligodendrocyte progenitor cells, 3 d after plating, bFGF was excluded from the PDGF-supplemented CD medium and replaced with 5 ng/ml NT-3 for an additional period of 2 d before all experimentation. Immunocytochemistry performed on day 7 mouse oligodendrocyte cultures indicated that 77% of the cells were O4+CNP+ oligodendrocytes, 22% were GFAP+ astrocytes, and <1% were CD45 and CD11b+ microglia. No NeuN+ neurons were detected. All subsequent treatments were performed on differentiated murine oligodendrocytes that were O4+ and CNP+.
Immunocytochemistry.
Oligodendrocytes were plated in CD medium at 5 × 104 cells per well on poly-l-lysine-coated 24-well tissue culture plates or coverslips. To identify the expression of O4 and TNF receptor 1 (TNFR1) on human oligodendrocytes, immunocytochemistry was performed on day 7 murine oligodendrocytes. After treatments, oligodendrocyte cultures were fixed in prewarmed 4% paraformaldehyde for 30 min. Cultures were permeabilized with 0.25% Triton X-100 in 1× Tris-buffered saline [1× TBS (0.14 m NaCl, 0.001 m Tris, pH 7.4)] for 30 min, blocked in 10% goat serum in 5% nonfat dry milk or bovine serum albumin (BSA)/1× TBS for 1 h, and incubated in the primary antibodies overnight in a humid chamber at 4°C. The 0.25% Triton X-100 permeabilization step was excluded for the immunostaining of the cell surface markers O4 and TNFR1. The O4 antibody (Bansal et al., 1989) was routinely used at 1:25 and CNP antibody (Raible and McMorris, 1989) at 1:500. The TNFR1 antibody (Austral Biologicals, San Ramon, CA) was used at 1:100. Cleaved caspase-3 (Asp175) rabbit polyclonal antibody (Cell Signaling Technology, Beverly, MA), an antibody that detects endogenous levels of the large fragment (17 kDa) of activated caspase-3 resulting from cleavage adjacent to aspartic acid 175 was used at 1:300. After three 1× TBS washes, 5 min each, the cells were incubated for 1 h at RT in the corresponding secondary antibodies conjugated to Alexa dyes (rabbit anti-mouse IgM or goat anti-rabbit IgG; Invitrogen), diluted 1:500 in 5% nonfat dry milk/1× TBS. Coverslips were mounted on slides with Aquamount (Biomeda, Foster City, CA) and examined with a Leica AOBS laser scanning confocal microscope (Leica Microsystems, Bannockburn, IL).
TNFα treatment of oligodendrocyte cultures.
Oligodendrocyte cultures were treated with 100 ng/ml recombinant human or mouse TNFα (PeproTech, Rocky Hill, NJ) in the absence or presence of 400 ng/ml rhgas6. Soluble Axl-Fc (10 μg/ml) was used as a decoy to block the rhgas6 effect; TrkA-Fc (10 μg/ml) was used as a negative control (Stitt et al., 1995). The cell-permeable pan-caspase inhibitor zVAD-fmk (N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone) and the caspase-8 inhibitor IETD-fmk [z-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethyl ketone] (Calbiochem, La Jolla, CA) were used at 20 μm. The caspase inhibitors and TNFα were added simultaneously.
For the insulin withdrawal experiments, human or mouse oligodendrocytes were grown for 5 or 7 d in CD medium. On day 5 or 7, cells were washed twice in HBSS and incubated with CD medium containing varying concentrations of insulin (25–5000 ng/ml), 10 ng/ml PDGF, and 5 ng/ml NT-3 in the absence or presence of 400 ng/ml rhgas6; oligodendrocyte survival was determined 48 h later.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay and the assessment of oligodendrocyte cell survival.
To assess apoptotic cell death in the TNFα-treated oligodendrocytes incubated in the presence and absence of rhgas6 terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL), staining was performed using the Fluorescein In Situ Cell Death Detection kit (Roche Molecular Biochemicals, Indianapolis, IN). The TUNEL reaction preferentially labels cleaved genomic DNA generated during apoptosis by the addition of fluorescein dUTP at strand breaks. At 48 h after treatment, oligodendrocytes were fixed and permeabilized as described above for immunocytochemistry. O4 immunostaining was performed first, washed, and subsequently incubated in the TUNEL reaction mixture, prepared according to the recommendations manufacturer, for 1 h at 37°C. Omission of the terminal deoxynucleotidyl transferase in the label solution served as a negative control. Cells were examined with an Olympus Optical (Melville, NY) IX70 inverted microscope. For each treatment, 20 random, 200× magnification microscopic fields consisting of ∼400–450 cells in total were examined in duplicate wells of 24-well tissue culture plates. O4+TUNEL+ nuclei were expressed as a percentage of the total number of O4+ cells per individual field [% oligodendrocyte survival = 100 − %(O4+TUNEL+/total O4+)].
Western blot analysis.
Total protein homogenates were prepared from whole mouse brains at postnatal day 30 or for time course experiments at postnatal day 10–160, analyzed by SDS-PAGE on 10% gels, and electrophoretically transferred to nitrocellulose, blocked with 5% nonfat dry milk in 1× TBS (Shankar et al., 2003). Blots were incubated overnight at 4°C with affinity-purified rabbit polyclonal antibodies generated against Axl (Amgen) or Gas6 (Amgen). Visualization was by enhanced chemiluminescence (ECL) (Amersham Biosciences, Arlington Heights, IL).
O4/phosphoAkt (Ser473) double-label immunofluorescence microscopy of oligodendrocyte cultures and fluorescence intensity measurements.
Human or mouse oligodendrocyte cultures were grown for 5 d on poly-l-lysine-coated coverslips as described above. By day 5 in culture, the oligodendrocytes exhibited morphological and antigenic differentiation (O4+CNP+). Cultures were washed and placed overnight in DMEM only without any trophic support. The following day, cultures were washed with HBSS and incubated in 100 ng/ml TNFα in DMEM (without insulin, PDGF, or NT-3) in the absence or presence of rhgas6 (400 ng/ml) for 15 min. Cells were fixed and blocked with 10% goat serum in 5% nonfat dry milk and incubated with the O4 monoclonal antibody (1:25) overnight at 4°C, washed three times in 1× TBS, and incubated with biotinylated anti-mouse IgM secondary antibody (1:200), followed by streptavidin Alexa 568. The secondary antibody and streptavidin Alexa 568 were diluted in 5% nonfat dry milk/1× TBS, and each incubation was performed for 1 h at RT. After O4 labeling, cells were permeabilized with 0.25% Triton X-100 in 1× TBS for 30 min at RT, blocked with 10% goat serum in 5% BSA, and incubated with anti-phosphoAkt1/PKBα (Ser473), clone 11E6 antibody (1:200 dilution in 5% BSA/1× TBS; Upstate Biotechnology, Lake Placid, NY) overnight at 4°C. Cells were washed three times and incubated with anti-mouse IgG1 Alexa 488 secondary antibody (1:500) for 1 h at RT. Coverslips were mounted with Aquamount (Biomeda). Images of random fields of O4 and phosphoAkt Ser473 immunostaining were collected using a Leica AOBS laser scanning confocal microscope at 600× magnification. Controls included complete labeling of either O4 or phosphoAkt antibody, followed by the secondary antibody of the second label and revealed no cross-reactivity. To ensure no crosstalk from one fluorescent channel to another, each channel was collected in the lambda scanning sequential mode with independent excitation and emission detection through its own narrow band filter.
To quantitate the phosphoAkt Ser473 immunofluorescence, low-magnification photomicrographs were obtained with an Olympus Optical 1X70 with a 20×, 1.0 numerical aperture planapo optics inverted microscope with a Photometrics (Roper Scientific, Tuscon, AZ) Censys cooled CCD camera; images were collected on a personal computer using IP Lab Spectrum software (Scanalytics, Fairfax, VA). Immunofluorescence measurements for phosphoAkt Ser473 and O4 were obtained from ∼80 cells in 15 random 200× magnification microscopic fields by NIH ImageJ software. phosphoAkt Ser473/O4 immunofluorescence quantification data were expressed as relative fluorescence intensity ± SD. Confirmation of data were obtained by repeating the experiment three times on independent material.
PI3 kinase/Akt inhibition using LY294002.
Human oligodendrocyte cultures grown for 5 d in CD medium in the presence of rhgas6 were incubated with 100 ng/ml TNFα for 48 h in the presence or absence of the PI3 kinase inhibitor LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one] (10 μm; Promega, Madison, WI). After 48 h, oligodendrocytes were fixed and immunofluorescence staining for O4 was performed followed by TUNEL staining. For each treatment, 20 random, 200× magnification fields consisting of ∼400 cells were examined in duplicate wells of 24-well tissue culture plates, and the percentage oligodendrocyte survival was determined.
Data analysis.
Data from each of the random microscopic fields, which were examined for O4+TUNEL+ oligodendrocytes, were analyzed as independent observations in an ANOVA model using Prism 2.01 software (GraphPad Software, San Diego, CA) to evaluate the effect of treatment. To validly combine data from multiple experiments in the analysis, the ANOVA model also included a factor to control for experiment to experiment variability, in addition to the main effect for treatment. Therefore, all p values corresponding to the effect of treatment are adjusted for between-experiment effects. Data are expressed as mean ± SD.
Results
rhgas6 is a survival factor for human oligodendrocytes after insulin withdrawal
The addition of rhgas6 to CD medium containing insulin at a final concentration of 5000 ng/ml significantly enhanced the survival of cultured human oligodendrocytes and reduced the number of TUNEL+ oligodendrocytes relative to CD medium lacking rhgas6 (Shankar et al., 2003). Here, we tested whether rhgas6 would maintain oligodendrocyte survival in CD medium with reduced insulin. Human (Fig. 1a) or mouse (Fig. 1b) oligodendrocytes were washed twice in HBSS and incubated with CD medium containing rhgas6 and varying concentrations of insulin (25–5000 ng/ml). Oligodendrocyte survival was determined 48 h later and compared with cultures similarly maintained in the absence of rhgas6. As demonstrated in Figure 1, a and b, the presence of rhgas6 in the medium (open circles) protected against insulin withdrawal with >60–75% oligodendrocyte survival observed in the cultures with lower insulin concentrations (25–100 ng/ml) for 48 h. When compared with the cultures grown in CD medium in the absence of rhgas6 (filled circles), rhgas6 significantly increased oligodendrocyte survival over the entire range of insulin concentrations examined. In the absence of rhgas6, oligodendrocyte survival was dramatically reduced in cultures with <100 ng/ml insulin in the medium. These data demonstrate that rhgas6 exerts its antiapoptotic effects independent of insulin-activated IGF-1 receptor (IGF1R) and that rhgas6 significantly enhances oligodendrocyte survival over that induced by PDGF, NT-3, and low insulin concentrations.

rhgas6 protects human and murine oligodendrocytes from insulin withdrawal induced-apoptosis. Human fetal oligodendrocytes (a) or murine oligodendrocytes (b) were grown in CD medium supplemented with insulin, PDGF, NT-3, with rhgas6 (a) or without rhgas6 (b) for 5 d on poly-l-lysine-coated plastic tissue culture wells in duplicate. On day 5 in culture, cells were washed twice and incubated in CD medium containing PDGF, NT-3, and the absence (Minus) or presence (Plus) of rhgas6 (400 ng/ml; 5.6 nm) with varying concentrations of insulin (25–5000 ng/ml) for 48 h. Apoptotic oligodendrocytes were detected by immunofluorescence staining with the oligodendrocyte-specific O4 antibody, followed by TUNEL staining. The number of O4+TUNEL+ double-labeled oligodendrocytes was counted from duplicate wells in 20 random, 200× microscopic fields and expressed as a percentage of the total O4+ cell numbers per field. Representative data are shown as percentage oligodendrocyte survival (mean ± SD) with ∼400 O4+ cells counted per treatment condition; three independent experiments were performed with similar results. ANOVA results indicated a significant effect of rhgas6 treatment (**p < 0.0001).
Variation in the concentration of rhgas6 and TNFα alters oligodendrocyte survival
We next examined whether rhgas6 would protect human oligodendrocytes against cell death induced by the cytotoxic cytokine TNFα. As illustrated in Figure 2, a and b, we observed numerous O4+TUNEL+ apoptotic oligodendrocytes 48 h after the addition of 100 ng/ml TNFα. In contrast, the coadministration of 100 ng/ml TNFα and 400 ng/ml rhgas6 significantly reduced the numbers of apoptotic oligodendrocytes (Fig. 2c). As shown in Figure 2d, addition of varying doses of TNFα (10–100 ng/ml) induced apoptosis in a concentration-dependent manner. In the absence of rhgas6 and in the presence of 100 ng/ml TNFα, 18.7 ± 4.4% oligodendrocytes survived, whereas the addition of 400 ng/ml rhgas6 led to a dramatic increase in cell survival to 64.3 ± 6.1%. Whereas the addition of varying concentrations of TNFα induced a dose-dependent decrease in oligodendrocyte survival, the coadministration of 10–100 ng/ml TNFα with rhgas6 significantly protected against oligodendrocyte apoptosis relative to TNFα alone.

rhgas6 prevents dose-dependent TNFα-induced oligodendrocyte apoptosis. Human fetal oligodendrocytes grown in CD medium plus rhgas6 for 5 d were washed twice and incubated in CD medium containing 5000 ng/ml insulin in the absence of rhgas6 (a, b) or the presence of rhgas6 (c) (400 ng/ml) and 100 ng/ml human recombinant TNFα for 48 h. Cells were fixed and immunostained with O4 (red), followed by TUNEL (green). A marked increase in O4+TUNEL+ oligodendrocytes was observed in cultures incubated with TNFα in the absence of rhgas6 (a). b shows a representative O4+TUNEL+ double-labeled oligodendrocyte at high magnification. Scale bars: a, c, 40 μm; b, 10 μm. d, TNFα (10–100 ng/ml) was administered in the absence or presence of rhgas6. The number of O4+TUNEL+ double-labeled oligodendrocytes was counted from duplicate wells in 20 random, 200× microscopic fields containing ∼350–400 O4+ oligodendrocytes in total. Data are shown as percentage oligodendrocyte survival (mean ± SD) of one of three independent experiments with similar results. ANOVA results indicated a significant effect on oligodendrocyte survival of TNFα alone or rhgas6 treatment alone or when rhgas6 was administered with TNFα (**p < 0.0001) when compared with minus gas6 or TNFα alone. e, Human oligodendrocytes were grown in CD medium plus 400 ng/ml rhgas6 for 5 d and washed with HBSS. Cultures were incubated for 48 h in CD medium containing 5000 ng/ml insulin with varying concentrations of rhgas6 (25–400 ng/ml; 0.36–5.6 nm) in the presence and absence of 100 ng/ml TNFα. Cells were fixed and stained with the O4 antibody, followed by TUNEL labeling. The numbers of O4+/TUNEL+ double-labeled oligodendrocytes were counted from 20 random, 200× microscopic fields from duplicate wells containing ∼500 O4+ oligodendrocytes in total. Data are shown as percentage oligodendrocyte survival (mean ± SD) obtained from one of two independent experiments with a second experiment yielding similar results. Significance relative to untreated or TNFα alone controls for all rhgas6 and rhgas6 plus TNFα treatments was **p < 0.0001 (ANOVA). f, Axl-Fc but not the TrkA-Fc decoy receptor blocks the rhgas6 protective effect. Human oligodendrocytes were grown as above and treated for 48 h with rhgas6 (400 ng/ml), Axl-Fc (10 μg/ml), or TrkA-Fc (10 μg/ml) in the absence and presence of TNFα (100 ng/ml). Data were obtained from 20 random, 200× microscopic fields from duplicate wells consisting of ∼500 O4+ oligodendrocytes in total. Data are expressed as percentage oligodendrocyte survival (mean ± SD) obtained from a single experiment (**p < 0.0001, unpaired Student’s t test; ns, not significant; p ≥ 0.05). g, Expression of TNFR1 in primary cultures of human oligodendrocytes. Top, A representative low-magnification field of O4+ (red) oligodendrocytes expressing TNFR1 (green). Nuclei were labeled with 4′,6′-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 10 μm. Bottom, O4+ oligodendrocyte with membrane expression of TNFR1 primarily at the cell surface with fainter staining in processes. Scale bar, 8 μm.
We examined whether varying concentrations of rhgas6 (25–400 ng/ml; 0.35–5.6 nm) would protect human oligodendrocytes from cell death 48 h after the administration of TNFα (100 ng/ml). As shown in Figure 2e (open bar), in the absence of rhgas6, 40.7 ± 6.0% of the oligodendrocytes survived. After administration of TNFα (black bar), there was an additional ∼20% reduction in oligodendrocyte survival (20.2 ± 5.3%) after 48 h. Addition of 25–400 ng/ml rhgas6 significantly increased oligodendrocyte survival at all doses in both the absence of TNFα (white bar with black dots) and the presence of TNFα (black bar with white dots). Approximately 35% of the oligodendrocytes survived TNFα cytotoxicity in rhgas6 concentrations of 25 and 50 ng/ml. When 100–400 ng/ml rhgas6 was added to the medium, 52–74% of the oligodendrocytes survived TNFα toxicity, demonstrating that rhgas6 protected human oligodendrocytes from TNFα-induced apoptosis in a concentration-dependent manner.
The specificity of the rhgas6 protective response from TNFα-induced apoptosis was examined by simultaneously administering TNFα and rhgas6 with the Axl decoy receptor Axl-Fc or a control decoy receptor TrkA-Fc. The Axl-Fc binds rhgas6 and, therefore, is a control to demonstrate that rhgas6 activity is a direct response. Figure 2f demonstrates that the addition of Axl-Fc eliminated the rhgas6-induced survival effect to oligodendrocytes in both the presence of rhgas6 alone and the presence of rhgas6 and TNFα. The percentage oligodendrocyte survival in the presence of the Axl-Fc was equivalent to the percentage survival observed after TNFα treatment in the absence of rhgas6. TrkA-Fc did not alter the rhags6 response, indicating that rhgas6 significantly protects oligodendrocytes from TNFα cytotoxicity.
Because TNFα-mediated apoptosis in oligodendrocytes is a result of TNFα binding to the TNFR1 (Hsu et al., 1996), we confirmed that the receptor was expressed on O4+ oligodendrocytes that were grown for 6 d in CD medium. As shown in Figure 2g, the O4+ oligodendrocytes (red) express the TNFR1 (green). TNFR1 membrane expression was primarily seen on the O4+ cell surface with fainter staining on the processes; robust O4 immunoreactivity was observed on both the oligodendrocyte cell body and the processes.
TNFα-induced caspase activation is inhibited by the addition of rhgas6
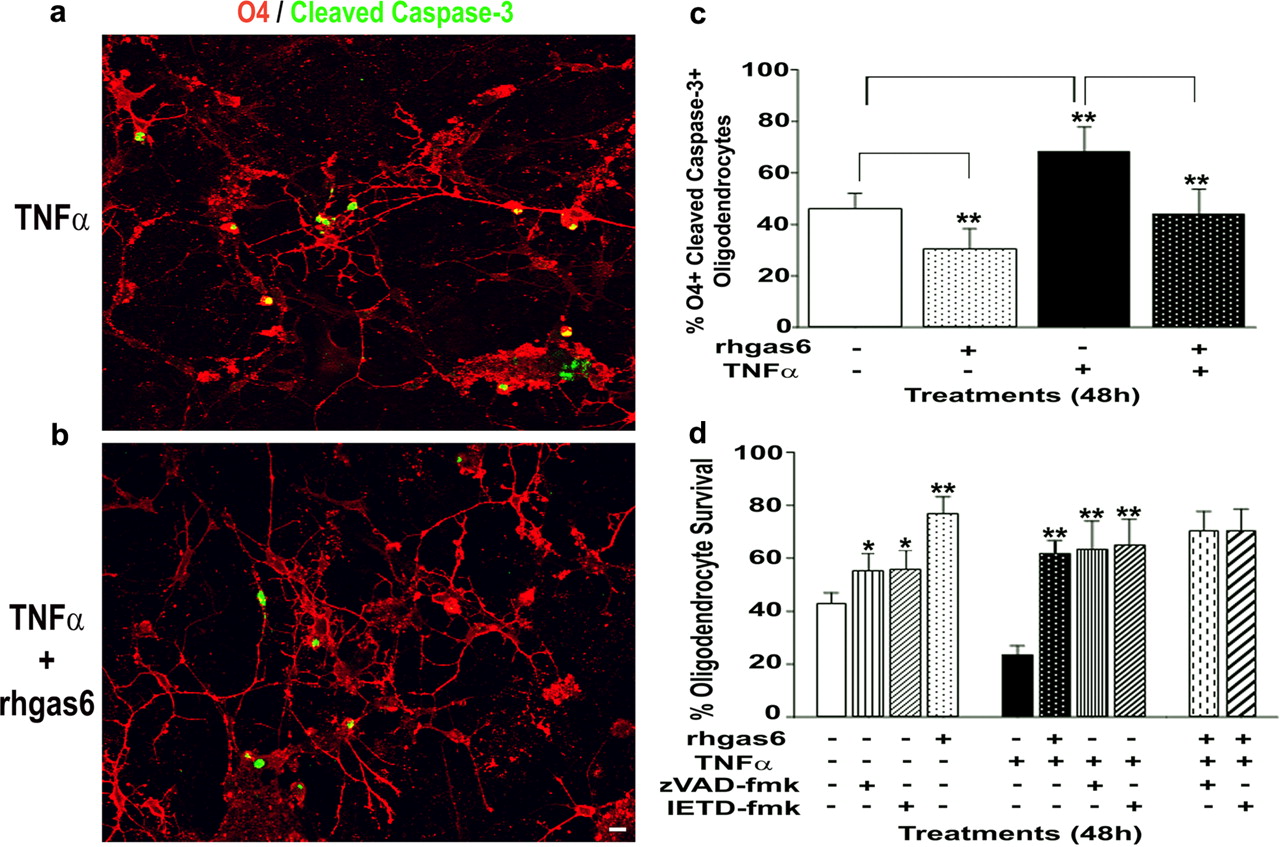
Figure 3a demonstrates that, 48 h after the addition of 100 ng/ml TNFα, there is an increase in active caspase-3 (green) detected within the O4+ (red) oligodendrocyte cultures. Quantification of the data showed that 68% of the oligodendrocytes were O4+/cleaved caspase-3+ (Fig. 3c). In contrast, cultures treated with rhgas6 and TNFα for the identical period of time had significantly fewer active caspase-3+ cells (Fig. 3b). Addition of rhgas6 and TNFα to the medium significantly decreased the numbers of O4+ oligodendrocytes with active caspase-3 to 43% (Fig. 3c). To determine whether the inhibition of caspases would enhance oligodendrocyte survival as effectively as rhgas6, day 5 human oligodendrocytes were washed twice and incubated in CD medium minus rhgas6 (Fig. 3d, open bar) plus 20 μm zVAD-fmk (stripes), IETD-fmk (diagonal), or rhgas6 (dots) and examined 48 h later. In the absence of TNFα, rhgas6 was most effective at supporting oligodendrocyte cell viability. In the presence of TNFα, rhgas6 was as effective as the caspase inhibitors in protecting the cultures from TNFα cytotoxicity. These results demonstrate that TNFα induces caspase-8 and downstream caspase activation in human fetal oligodendrocytes and that rhgas6 and caspase inhibitors can reduce TUNEL+ nuclei resulting from TNFα-induced cytotoxicity. These experiments not only show that caspase inhibitors can block TNFα-induced oligodendrocyte cytotoxicity but also stress the ability of rhgas6 to inhibit TNFα-induced caspase activation as effectively as the chemical caspase inhibitors.

rhgas6 is as effective as the pan-caspase inhibitor zVAD-fmk and the caspase-8 inhibitor IETD-fmk at protecting human oligodendrocytes against TNFα-induced apoptosis. a, TNFα treatment of human oligodendrocytes induces the activation of caspase-3. Human oligodendrocytes were grown in CD medium plus rhgas6 for 5 d, washed with HBSS, and incubated for 48 h in CD medium containing 5000 ng/ml insulin and TNFα (100 ng/ml) in the absence (a) and presence (b) of rhgas6 (400 ng/ml). Cells were fixed and immunostained with O4 antibody (red), followed by cleaved caspase-3 antibody (green). c, Quantitation of O4+/cleaved caspase-3+ oligodendrocytes after TNFα alone or with rhgas6 treatment. O4+/cleaved caspase-3+ double-labeled oligodendrocytes were counted from 20 random, 200× microscopic fields from duplicate wells containing ∼350 O4+ oligodendrocytes in total. Data are shown as percentage O4+/cleaved caspase-3+ oligodendrocytes (mean ± SD) obtained from one of two independent experiments with a second experiment yielding similar results (**p < 0.0001, ANOVA). d, Human oligodendrocyte cultures grown in rhgas6 for 5 d were washed with HBSS and incubated for 48 h in TNFα (100 ng/ml) in the presence or absence of rhgas6 (400 ng/ml), with and without the cell-permeable pan-caspase inhibitor zVAD-fmk or the caspase-8 inhibitor IETD-fmk (20 μm). Apoptotic oligodendrocytes were detected by immunofluorescence staining with the O4 antibody, followed by TUNEL labeling. O4+/TUNEL+ double-labeled oligodendrocytes were counted from 20 random, 200× microscopic fields from duplicate wells containing ∼400 O4+ oligodendrocytes in total. Data are shown as percentage oligodendrocyte survival (mean ± SD) obtained from one of two independent experiments (*p < 0.001, **p < 0.0001, ANOVA).
Rhgas6 protects human oligodendrocytes from cell death via the PI3 kinase/Akt pathway
To determine whether rhgas6 signaling protects against TNFα-induced toxicity via the PI3 kinase/Akt pathway, we examined whether the addition of TNFα alone or rhgas6 and TNFα would activate and hence phosphorylate the prosurvival kinase Akt. Human oligodendrocytes were placed overnight in DMEM, washed, treated with TNFα plus and minus rhgas6 for 15 min, and immediately fixed with 4% paraformaldehyde. Double-label immunofluorescence was performed with O4 and a phosphoAkt Ser473 antibody (Fig. 4a), and the relative phosphoAkt Ser473/O4 fluorescence intensity was compared with DMEM-only-treated cells arbitrarily set as 1.0 (Fig. 4b). TNFα alone did not significantly activate phosphoAkt. However, the presence of rhgas6 in the TNFα-treated cultures activated Akt relative to TNFα alone (Fig. 4b). When the PI3 kinase inhibitor LY294002 was simultaneously added to the medium with TNFα plus rhgas6, phosphoAkt immunoreactivity was reduced similar to the TNFα-only-treated cultures (Fig. 4b). To address whether rhgas6-induced Akt activation is enhanced in the presence of other survival-promoting factors, oligodendrocyte cultures were treated with insulin, IGF-1, PDGFα, or NT-3 in the presence and absence of rhgas6. rhgas6-induced Akt activation was neither additive nor synergistic in the presence of any of the survival-promoting factors (Fig. 4c).

The rhgas6 protective effect against TNFα is mediated by Akt activation and is inhibited by treatment with the PI3 kinase inhibitor LY294002. All human oligodendrocytes were grown for 5 d in CD medium plus rhgas6. a, Human oligodendrocytes were washed twice with HBSS, incubated in DMEM (without insulin, PDGF, or NT-3) and 100 ng/ml TNFα in the absence or presence of rhgas6 (400 ng/ml) for 15 min, and fixed with 4% paraformaldehyde. Oligodendrocytes were double-labeled sequentially with the O4 (red) and phosphoAkt Ser473 (green) antibodies. Nuclei were labeled with DAPI (blue). Confocal microscopy was performed, and images were collected in Z-series. Scale bar, 8 μm. Low-magnification images (red/green merge) were obtained with a 20× objective. Scale bar, 10 μm. b, phosphoAkt Ser473 and O4 immunofluorescence measurements were obtained from 80 cells in 15 random, 200× magnification microscopic fields by NIH ImageJ software. Data are expressed as relative fluorescence intensity (mean ± SD) from one of three independent experiments with similar results (**p < 0.0001, ANOVA). Data are shown relative to minus gas6 (set as 1.0); TNFα alone did not significantly (ns) activate phosphoAkt at 15 min. c, Human oligodendrocytes grown in rhgas6 for 5 d were placed overnight in DMEM, washed twice with HBSS, treated with minus or plus gas6 supplemented with 10 ng/ml IGF-1, 5 ng/ml NT-3, or 10 ng/ml PDGF for 15 min, and then stopped by the addition of 4% paraformaldehyde. PhosphoAkt Ser473 and O4 immunofluorescence measurements were obtained from 32 cells in 10 random, 200× magnification microscopic fields by NIH ImageJ software, and data are expressed as relative fluorescence intensity (mean ± SD) from one of two independent experiments with similar results. d, Human oligodendrocytes placed overnight in DMEM, washed twice with HBSS, and treated with minus or plus gas6 for 5 min, 15 min, 30 min, 1 h, and 24 h. PhosphoAkt Ser473 and O4 immunofluorescence measurements were obtained from 32 cells in 10 random, 200× magnification microscopic fields by NIH ImageJ software, and data are expressed as relative fluorescence intensity (mean ± SD) from one of two independent experiments with similar results (**p < 0.0001, ANOVA). e, Human oligodendrocytes grown as above were washed with HBSS and incubated for 48 h in TNFα (100 ng/ml) in the presence or absence of rhgas6 (400 ng/ml) with or without the PI3 kinase/Akt inhibitor LY294002 (10 μm). Data are shown as percentage oligodendrocyte survival (mean ± SD with ∼400 O4+ cells counted from 20 random, 200× microscopic fields per treatment condition from duplicate wells) obtained from one of two independent experiments (**p < 0.0001, ANOVA).
To determine whether Akt activation was transient or sustained after rhgas6 stimulation, oligodendrocytes were placed overnight in DMEM only without any trophic support, washed with HBSS, and treated with DMEM plus rhgas6 for 5 min, 15 min, 30 min, 1 h, and 24 h time points. Relative phosphoAkt Ser473/O4 fluorescence intensities were obtained and compared with DMEM-only-treated cells. As shown in Figure 4d, a significant increase in relative phosphoAkt Ser473/O4 fluorescence intensity was observed 5 min after rhgas6 treatment; the maximal increase in phosphoAkt Ser473/O4 was observed at 15 and 30 min after rhgas6 treatment. By 1 h, Akt activation was reduced, but elevated levels of phosphoAkt persisted at 24 h. Thus, rhgas6 treatment of oligodendrocytes results in sustained Akt activation. We examined whether inhibiting the rhgas6-induced Akt activation with LY294002 would influence oligodendrocyte survival 48 h after treatment with TNFα. Figure 4e demonstrates that oligodendrocyte survival was compromised in the rhgas6 plus TNFα-treated cultures after the addition of LY294002. Thus, the rhgas6-dependent increase in oligodendrocyte survival is dependent on the PI3 kinase/Akt pathway, and LY294002 eliminated the rhgas6-induced survival effect.
Rhgas6 protection against TNFα-induced apoptosis is mediated through the Axl receptor
Immunoblot analysis of various developmental time points determined that Axl and gas6 are expressed throughout rodent brain development. Both Axl and gas6 were expressed at high levels in whole mouse brain protein homogenates at postnatal days 10–13 (Fig. 5a), and Axl remained high through day 30. Although gas6 and Axl levels decreased by day 60, protein levels were detectable in the adult mouse brain at day 160.
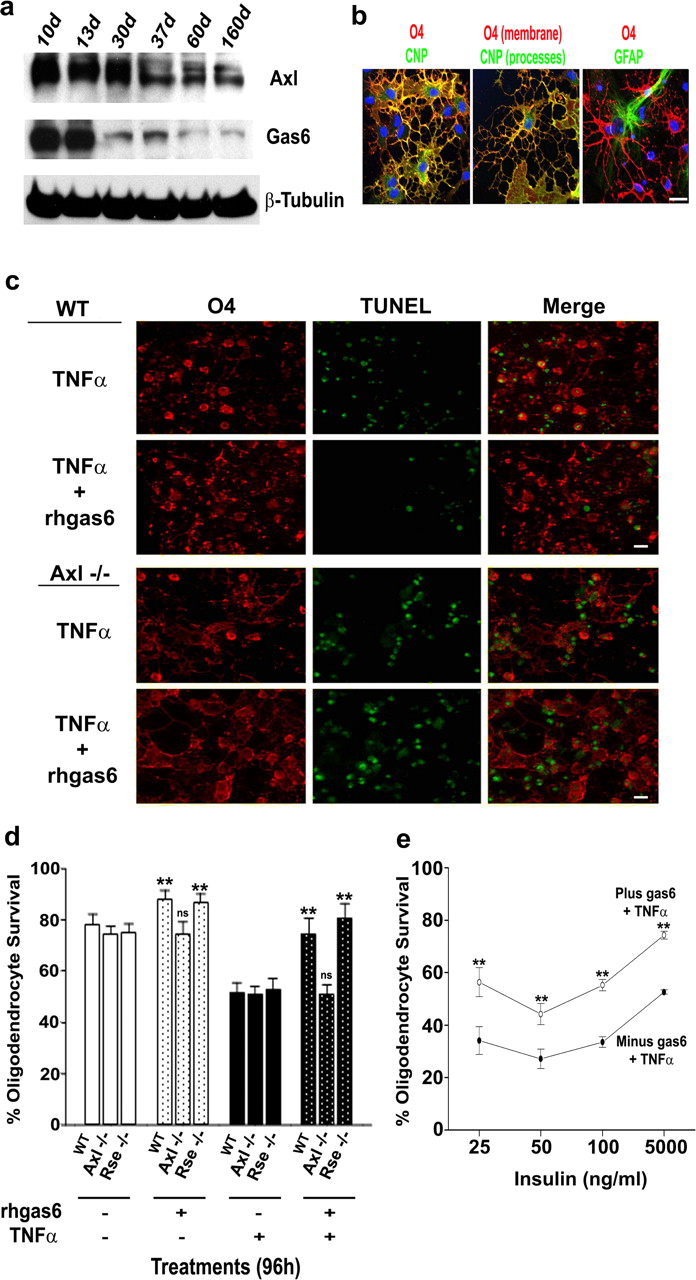
rhgas6 protects oligodendrocytes from WT and Rse−/− but not oligodendrocytes from Axl−/− mice against TNFα-induced apoptotic cell death. a, Developmental expression of Axl and Gas6 in mouse brain. A total of 50 μg of whole mouse brain protein homogenate was loaded per lane. Blots were incubated with affinity-purified antibodies Axl polyclonal antibodies (1:500; Amgen), Gas6 polyclonal antibodies (1:1000; Amgen), or β-tubulin (1:1000; Sigma). Axl migrates at 140 kDa, Gas6 at 86 kDa, and β-tubulin at 55 kDa. Visualization is by ECL. b, Mouse cortical oligodendrocytes grown for 7 d in culture were triple labeled with O4 (red), CNP (green), or GFAP (green) and DAPI (blue). Middle panel shows membrane extensions typical of mouse oligodendrocytes in vitro. Merged images show healthy oligodendrocytes with minimal astrocyte contamination. Scale bar, 16 μm. c, d, Rhgas6 protects oligodendrocytes from Axl+/+ and Rse−/− but not oligodendrocytes from Axl−/− mice against TNFα-induced apoptosis. Oligodendrocytes isolated from postnatal day 1 WT Axl−/−, and Rse−/− mice were grown for 5 d, washed with HBSS, and incubated for 96 h in DMEM (with 5000 ng/ml insulin) plus TNFα (100 ng/ml) in the presence or absence of rhgas6 (400 ng/ml). O4 and TUNEL labeling was performed sequentially, and low-magnification images (200×) were obtained on an Olympus Optical 1X70 microscope with a 20× objective (c). Scale bar, 10 μm. Results in d are based on duplicate wells of 20 random, 200× microscopic fields containing ∼600 oligodendrocytes in total. Data are shown as percentage oligodendrocyte survival (mean ± SD) of one of three independent experiments with similar results (**p < 0.0001, ANOVA; ns, not significant). e, Rhgas6 protects murine cortical oligodendrocytes from TNFα-induced cell death independent of insulin-activated IGFR. Oligodendrocytes were washed and placed in CD medium in the absence (minus) or presence (plus) of rhgas6 (400 ng/ml; 5.6 nm) with varying concentrations of insulin (25–5000 ng/ml) and in the presence and absence of 100 ng/ml TNFα for 48 h. Data are shown as percentage oligodendrocyte survival (mean ± SD) of one of three independent experiments with similar results (**p < 0.0001, ANOVA).
To determine whether the loss of the Axl or the Rse receptor would compromise murine oligodendrocyte survival, O4+CNP+ oligodendrocyte cultures (Fig. 5b) prepared from postnatal day 1 mouse brains were challenged with TNFα and rhgas6. In the absence of rhgas6, the percentage oligodendrocyte survival in the WT, Axl−/−, and Rse−/− cultures was >74% (Fig. 5d, white bars). When rhgas6 was administered to each of the cultures (stippled bars), there was a statistically significant increase in the percentage oligodendrocyte survival in the WT and Rse−/− mice but not in the Axl−/− mice, suggesting that, in the presence of exogenous rhgas6, oligodendrocyte survival can be enhanced only when the Axl receptor is present. Furthermore, when 100 ng/ml TNFα was administered to the cultures in the absence of rhgas6 (black bars), there was a dramatic decrease in oligodendrocyte viability that could be recovered when rhgas6 was administered to the WT and Rse−/− cultures but not the Axl−/− cultures (Fig. 5c,d, black stippled bars). These results suggest that the presence of the Axl receptor is crucial for rhgas6-induced oligodendrocyte survival after TNFα administration.
To address whether rhgas6 protected murine oligodendrocytes from TNFα-induced apoptosis independent of the survival induced by high insulin concentrations activating IGF1R, oligodendrocyte cultures were washed thrice to remove trophic factors, and the medium was replaced with CD medium containing 100 ng/ml TNFα plus and minus 400 ng/ml rhgas6 and a range of insulin concentrations (25–5000 ng/ml). After 48 h, the cultures were fixed and stained with O4 and TUNEL, and the percentage oligodendrocyte survival was quantified. When compared with TNFα alone, rhgas6 significantly increased oligodendrocyte survival at both high and low insulin concentrations (Fig. 5e). This demonstrated that rhgas6 could protect murine oligodendrocytes from TNFα toxicity at reduced insulin concentrations (25–100 ng/ml).
Because gas6 signaling to Axl protects oligodendrocytes from TNFα-induced cell death, we examined whether rhgas6 would activate Akt in the Axl−/− mice and whether rhgas6-treated oligodendrocytes from Akt1−/− mice would be protected from TNFα-induced cell death. Murine oligodendrocytes were placed overnight in DMEM, washed, and simultaneously treated for 15 min in growth factor-free DMEM with TNFα plus or minus rhgas6. Cultures were fixed, double-label immunofluorescence was performed with O4 and the phosphoAkt serine 473 antibodies, and the relative phosphoAkt and O4 fluorescence intensities were quantified. Figure 6a shows that, in the WT cultures, rhgas6 alone (white stippled bars) or rhgas6 plus TNFα (black stippled bars) induced an approximate threefold increase in phosphoAkt Ser473 relative to untreated cultures or cultures treated with TNFα alone. However, in the Axl−/− cultures, no increase in phosphoAkt was observed. To determine whether the Axl−/− oligodendrocytes were capable of Akt phosphorylation in response to other survival-promoting factors, we treated Axl−/− cultures for 15 min in growth factor-free DMEM or medium containing rhgas6, IGF-1, NT-3, or PDGF. Whereas rhgas6 lacked the ability to induce Akt phosphorylation in Axl−/− oligodendrocytes (Fig. 6b), a robust Akt activation was observed in cultures treated with IGF-1 (4.3-fold), NT-3 (4.2-fold), and PDGF (3.6-fold).

rhgas6 activates Akt in WT but not in Axl−/− murine oligodendrocytes; the survival effect requires activation of Akt1. a, Murine oligodendrocyte cultures (day 7) were placed overnight in DMEM, washed with HBSS, and incubated in 100 ng/ml TNFα in DMEM (without insulin, PDGF, or NT-3), in the absence or presence of rhgas6 (400 ng/ml) for 15 min. Oligodendrocytes were triple labeled with the O4, phosphoAkt Ser473, and DAPI. phosphoAkt Ser473 and O4 immunofluorescence measurements were obtained from 35 cells in 10 random, 200× magnification microscopic fields by NIH ImageJ software. Data are expressed as relative fluorescence intensity (mean ± SD) from one of three independent experiments with similar results. Data are shown relative to minus gas6 (set as 1.0). rhgas6 treatment significantly increased phosphoAkt Ser473 expression in TNFα-treated wild-type Axl+/+ oligodendrocytes (**p < 0.0001, ANOVA; ns, not significant). b, IGF-1, NT-3, and PDGF but not rhgas6 induce Akt phosphorylation in Axl−/− murine oligodendrocytes. Day 5 murine Axl−/− oligodendrocytes were placed overnight in DMEM, washed twice with HBSS, and treated with DMEM only or DMEM with either 10 ng/ml IGF-1, 5 ng/ml NT-3, 10 ng/ml PDGF, or 400 ng/ml rhgas6 for 15 min. phosphoAkt Ser473 and O4 immunofluorescence measurements were obtained from 35 cells in 10 random, 200× magnification microscopic fields by NIH ImageJ software. Data are expressed as relative fluorescence intensity (mean ± SD) from one of two independent experiments with similar results (**p < 0.0001, ANOVA). c, rhgas6 does not protect against TNFα-induced cell death in Akt1−/− oligodendrocytes. Oligodendrocytes isolated from postnatal day 1 Akt1−/− mouse brains were grown for 5 d, washed with HBSS, and incubated for 48 h in CD medium containing 100 ng/ml insulin plus TNFα (100 ng/ml) in the presence or absence of rhgas6 (400 ng/ml). O4 and TUNEL labeling was performed sequentially, and low-magnification images (200×) obtained on an Olympus Optical 1X70 microscope with a 20× objective. Results are based on duplicate wells of 20 random, 200× microscopic fields containing ∼300 oligodendrocytes in total. Data are shown as percentage oligodendrocyte survival (mean ± SD) of one of two independent experiments with similar results (ns, not significant).
Akt1 is the predominant Akt isoform in oligodendrocytes. Therefore, we treated oligodendrocyte cultures prepared from Akt1−/− mouse brains with TNFα plus and minus rhgas6 and examined whether rhgas6 protected against TNFα-induced cell death. Treatments were performed with 100 ng/ml insulin in the medium to determine whether the effect of rhgas6 on Akt1−/− oligodendrocyte survival was independent of insulin-activated IGF1R. As shown in Figure 6c, treatment of the Akt1−/− cultures with rhgas6 (white stippled bars) or rhgas6 plus TNFα (black stippled bars) did not alter oligodendrocyte survival relative to the untreated cultures or cultures treated with only TNFα. These results demonstrate that rhgas6/Axl signaling protects oligodendrocytes from TNFα-induced apoptosis by activating the PI3 kinase/Akt1 signaling pathway.
Discussion
Our studies demonstrate that rhgas6 protects oligodendrocytes against growth factor withdrawal and TNFα-induced cytotoxicity by ligand activation of the receptor tyrosine kinase Axl. Human oligodendrocytes are more dependent on rhgas6 for their survival than murine oligodendrocytes, but, when oligodendrocytes are challenged with TNFα, rhgas6 is highly effective in maintaining survival in both species. The inability of rhgas6 to protect Axl−/− oligodendrocytes from TNFα-induced toxicity further supports our conclusion that Axl is the predominant receptor through which rhgas6 signals in oligodendrocytes. Axl receptor activation was shown to be more responsive to low doses of rhgas6 (Nagata et al., 1996), and our observation that as little as 0.35 nm rhgas6 protected oligodendrocytes from TNFα-induced cytotoxicity supports that study.
In a previous study, we showed that two independent inhibitors of PI3 kinase blocked the protective effect of rhgas6 and speculated that rhgas6 was signaling to Akt (Shankar et al., 2003). In this study, we demonstrate that rhgas6 signaling through the Axl receptor results in increased Akt phosphorylation at serine 473 in both human and murine oligodendrocytes and that the Akt activation was maintained when oligodendrocyte cultures were cotreated with TNFα and rhgas6. In addition, the rhgas6 protective effect was abrogated in oligodendrocytes from both Axl−/− and Akt1−/− mice, further demonstrating the importance of the Axl receptor and Akt1 in protection against TNFα. Several growth factors, including PDGF, high doses of insulin, and NT-3, increase cell survival by activating Akt. Doses of insulin above 250 ng/ml activate the IGF1R and lead to sustained activation of Akt (Ness and Wood, 2002). In our studies, the absence of insulin, or insulin concentrations below 100 ng/ml, dramatically decreased oligodendrocyte survival in both human and murine oligodendrocyte cultures. In contrast, oligodendrocytes maintained in rhgas6 with low concentrations (25–100 ng/ml) of insulin in the CD medium significantly reduced the numbers of TUNEL+ apoptotic cells. This demonstrates that rhgas6 potently increases oligodendrocyte survival independent of the insulin/IGF1R signaling.
Oligodendrocytes are sensitive to TNFα-mediated cytotoxicity in vitro and in vivo. In vitro administration of TNFα induced apoptotic cell death of primary oligodendrocytes (Selmaj and Raine, 1988; D’Souza et al., 1996; Scurlock and Dawson, 1999; Hisahara et al., 2000; Takano et al., 2000). In vivo, elevated levels of TNFα are found in multiple sclerosis plaques and in experimental autoimmune encephalomyelitis lesions (Powell et al., 1990; Genain et al., 1995; Brosnan and Raine, 1996). Transgenic mice expressing TNFα in the CNS showed increased oligodendrocyte apoptosis and primary demyelination (Akassoglou et al., 1997). TNFα-mediated apoptosis occurs as a result of TNFα binding to the TNFR1 receptor, which recruits FAS-associated death domain FADD, receptor-interacting protein RIP1, and TNF receptor-associated factor TRAF2/5 via the adaptor protein TNFR-associated death domain-containing protein TRADD (Hsu et al., 1996). FADD recruits and activates caspase-8, resulting in the cleavage and activation of caspase-3 and caspase-7 and subsequent apoptosis. Our studies indicate that the addition of TNFα to human and murine oligodendrocytes induces apoptosis. In our cultures, TNFα activated caspase-8 and caspase-3 and increased the number of active cleaved caspase-3+TUNEL+ oligodendrocytes. The effect was blocked with either the caspase-8 inhibitor IETD-fmk or the pan-caspase inhibitor zVAD-fmk. The presence of rhgas6 in TNFα-treated oligodendrocyte cultures significantly protected against caspase activation and cell death. There was an additional reduction in TUNEL+ oligodendrocytes when the caspase inhibitors were added to the rhgas6, TNFα-containing medium. However, rhgas6 was more effective at maintaining oligodendrocyte survival than the addition of the caspase inhibitors alone. This suggests that rhgas6 does not act to directly inhibit caspase activation but works upstream to activate signaling pathways such as PI3K/Akt. This was supported by our observation that increased phosphoAkt Ser473 was observed in both human and murine oligodendrocytes 15 min after the addition of TNFα and rhgas6 but not TNFα alone. Furthermore, the addition of the PI3 kinase inhibitor LY294002 blocked the protective effect of rhgas6 and the observed increase in phosphoAkt. Our observation of Akt activation in the rhgas6- and TNFα-treated human and murine oligodendrocyte cultures is supported by an in vitro study that demonstrated that, after gas6 stimulation, the Axl receptor undergoes autophosphorylation, resulting in tyrosine phosphorylation at three sites (Braunger et al., 1997). Two of the tyrosines phosphorylated form part of a consensus sequence pYXXM that may recruit PI3 kinase by direct binding of the Src homology 2 domain of the p85 subunit of PI3 kinase to Axl.
Axl, Rse, and Mer single, double, and triple knock-out mice have been generated and are viable but have varying degrees of impairments in cell viability and homeostasis (Lu et al., 1999; D’Cruz et al., 2000). The triple knock-out mice have neurologic abnormalities, major organ defects, physiological deficits, and autoimmune defects. Altered histology, increased apoptosis, and cellular degeneration are apparent in the CNS, and male mice produce no sperm (Lu et al., 1999; Lu and Lemke, 2001). To gain an understanding of the contribution of Axl or Rse receptors to oligodendrocyte survival, we generated oligodendrocyte cultures from Axl and Rse single knock-outs. Oligodendrocyte cultures from the Axl−/− or Rse−/− mouse brains appeared morphologically similar to the WT and exhibited normal survival and differentiation in culture medium supplemented with insulin, PDGF, and NT-3. However, when oligodendrocytes were challenged with TNFα, the absence of signaling through the Axl receptor prevented rhgas6 from exerting its survival effect, supporting an important role for Axl in cell survival.
The data obtained from the TNFα-treated differentiated oligodendrocytes from the Axl null mice strongly support our hypothesis that Axl is the primary receptor activated in response to low doses of rhgas6. Although we cannot rule out a role for the Rse or Mer receptors, we did not observe a significant change in cell survival in the Rse null oligodendrocytes expressing functional Axl and Mer receptors. Furthermore, examination of total brain homogenates from the Axl+/+ and Axl−/− mice did not indicate the upregulation of either the Rse or Mer receptor in the null animals (data not shown).
Western blot analysis of mouse brain homogenates prepared at several development ages determined that gas6 expression paralleled Axl expression, peaking early in development and leveling off in the mature animal. Several studies have demonstrated that gas6 is expressed and secreted by both neurons and endothelial cells and that gas6 is abundantly present in the CNS (Crosier and Crosier, 1997; Prieto et al., 2000). The expression of gas6 and Axl in adult animals suggests that the gas6/Axl signaling pathway may be able to protect cells from injury. Furthermore, the downstream Akt signaling pathway can be activated to protect against stress induced by insufficient trophic support or the release of cytotoxic cytokines. In summary, our results demonstrate that rhgas6 potently inhibits TNFα-induced oligodendrocyte apoptosis and identifies the gas6/Axl/PI3 kinase/Akt1 signaling pathway as an important mediator of oligodendrocyte survival after cellular stress and cytokine challenge.
Footnotes
-
This work was supported by National Multiple Sclerosis Society Grant RG3020 (B.S.-Z) and National Institutes of Health Grant NS11920 (C.F.B). We thank Dr. Brad Poulos, Einstein Human Fetal Tissue Repository, for material.
- Correspondence should be addressed to Dr. Bridget Shafit-Zagardo, Department of Pathology, Forcheimmer 516, 1300 Morris Park Avenue, Albert Einstein College of Medicine, Bronx, NY 10461. Email: zagardo{at}aecom.yu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}