

Abstract
Bitter, sweet, and umami tastants are detected by G-protein-coupled receptors that signal through a common second-messenger cascade involving gustducin, phospholipase C β2, and the transient receptor potential M5 (TRPM5) ion channel. The mechanism by which phosphoinositide signaling activates TRPM5 has been studied in heterologous cell types with contradictory results. To resolve this issue and understand the role of TRPM5 in taste signaling, we took advantage of mice in which the TRPM5 promoter drives expression of green fluorescent protein and mice that carry a targeted deletion of the TRPM5 gene to unequivocally identify TRPM5-dependent currents in taste receptor cells. Our results show that brief elevation of intracellular inositol trisphosphate or Ca2+ is sufficient to gate TRPM5-dependent currents in intact taste cells, but only intracellular Ca2+ is able to activate TRPM5-dependent currents in excised patches. Detailed study in excised patches showed that TRPM5 forms a nonselective cation channel that is half-activated by 8 μm Ca2+ and that desensitizes in response to prolonged exposure to intracellular Ca2+. In addition to channels encoded by the TRPM5 gene, we found that taste cells have a second type of Ca2+-activated nonselective cation channel that is less sensitive to intracellular Ca2+. These data constrain proposed models for taste transduction and suggest a link between receptor signaling and membrane potential in taste cells.
Introduction
Taste receptor cells are modified epithelial cells that transduce sensory input into changes in the release of neurotransmitter onto afferent nerve fibers. For bitter, sweet, and umami tastants, stimuli bind to seven transmembrane receptors on the apical surface of the cells, where they initiate a second-messenger signaling cascade, in which phosphoinositide signaling plays a critical role (Margolskee, 2002; Montmayeur and Matsunami, 2002; Medler and Kinnamon, 2004). Exposure of taste tissue to bitter and sweet tastants activates phospholipase C (PLC), leading to hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol trisphosphate (IP3) (Hwang et al., 1990; Bernhardt et al., 1996; Spielman et al., 1996; Rossler et al., 1998; Huang et al., 1999). More direct evidence for the importance of phosphoinositide signaling in taste transduction comes from the observation that mice with a targeted deletion of the PLCβ2 gene have diminished sensitivity to bitter, sweet, and umami tastants (Zhang et al., 2003; Dotson et al., 2005). The transient receptor potential M5 (TRPM5) ion channel, which is specifically expressed in taste cells (Perez et al., 2002), is critical for this signaling cascade, and mice with a targeted deletion of TRPM5 have little or no ability to detect physiologically relevant concentrations of bitter or sweet substances (Zhang et al., 2003; Damak et al., 2006). Thus, it can be hypothesized that taste transduction involves a PLC signaling cascade that leads to activation of the TRPM5 ion channel, a signaling pathway that is similar, in many respects, to phototransduction in the fly eye (Montell and Rubin, 1989; Ranganathan et al., 1995; Hardie and Raghu, 2001).
How does PLC signaling regulate the activity of TRPM5 ion channels? This question has been addressed by studying the properties of cloned TRPM5 channels expressed in heterologous cells type with contradictory results. In particular, although one study showed that activation of rat TRPM5 channels was independent of an elevation of intracellular IP3 or the ensuing rise in intracellular Ca2+ (Zhang et al., 2003), other studies showed that mouse TRPM5 channels could be directly activated by intracellular Ca2+ (Hofmann et al., 2003; Liu and Liman, 2003; Prawitt et al., 2003) or by Ca2+ store depletion (Perez et al., 2002). To add to the uncertainty concerning the mechanism by which TRPM5 channels are activated, it is likely that heterologously expressed channels do not fully recapitulate the properties of native channels, which may be composed of multiple subunits (Chen et al., 1993).
To directly determine the mechanism by which native TRPM5 channels are activated, we measured ionic currents in taste receptor cells from genetically modified mice, in which TRPM5-dependent currents could be unequivocally identified. Our results show that native TRPM5 channels are directly activated by intracellular Ca2+, downstream of IP3-mediated release of Ca2+ from intracellular stores.
Materials and Methods
Transgenic mice.
Transgenic mice in which 11 kb of mouse Trpm5 5′ flanking sequence drives expression of enhanced green fluorescent protein (eGFP) (TRPM5–GFP) and mice carrying a targeted deletion of the TRPM5 gene (TRPM5−/−) were described previously (Clapp et al., 2006; Damak et al., 2006). The two strains of mice were mated to generate animals that were positive for the GFP reporter and were +/+, +/−, or −/− for TRPM5 expression. Animals were genotyped as described previously (Clapp et al., 2006; Damak et al., 2006). Additional TRPM5+/+ animals were generated by crossing these animals with wild-type mice of the B6D2 strain (Harlan, Indianapolis, IN).
Antibody generation and immunostaining of taste tissue.
The terminal 70 amino acids of mouse TRPM5 (amino acids 1088–1158) were fused to glutathione S-transferase using the vector pGEX4T1 (Pfizer, New York, NY). Fusion protein was purified over glutathione Sepharose 4B, analyzed by PAGE, and used to inoculate two rabbits (Strategic Biosolutions, Newark, DE). Unpurified polyclonal antibody was used at a dilution of 1:500. Taste tissue was harvested according to approved methods by the University of Southern California Institutional Animal Care and Use Committee. Sectioning and staining of frozen tissue sections was as described previously (Liman et al., 1999). Images were acquired with Zeiss (Oberkochen, Germany) 510 Meta confocal microscope.
Acute dissociation of taste receptor cells.
Mouse taste cells were prepared using a modification of published methods (Spielman et al., 1989; Behe et al., 1990). In brief, we injected an enzyme cocktail (collagenase, 2 mg/ml; dispase, 5 mg/ml; Roche, Indianapolis, IN) between the epithelium and muscle layers of the tongue and allowed the tissue to incubate for 20 min in Tyrode's solution bubbled with 95% oxygen. After this treatment, the epithelium was peeled off and washed with Tyrode's solution, and a piece of tissue containing circumvallate papillae was isolated. This tissue was washed with dichlorofluorescein (DCF) (divalent-free solution) and incubated with DCF containing Pronase (1.5 mg/ml; Sigma, St. Louis, MO) and elastase (1 mg/ml; Worthington, Lakewood, NJ) for 20 min. The tissue was then washed with Tyrode's solution, triturated with a fire-polished Pasteur pipette, and plated onto a glass coverslip. GFP-positive (GFP+) taste receptor cells were identified under epifluorescence and were used within 5 h of plating. Tyrode's solution contained the following (in mm): 145 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 20 dextrose, 10 HEPES, and 1 pyruvate, pH 7.4. DCF contained the following (in mm): 65 NaCl, 26 NaHCO3, 20 KCl, 20 d-glucose, 2.5 NaH2PO4, 1 EDTA, and 1 pyruvate, pH 7.4.
Tissue culture and transfection of human embryonic kidney cells.
TRPM5 fused to eGFP (Liu and Liman, 2003) or enhanced cyan fluorescent protein (CFP) (Rizzo et al., 2004) was transiently transfected into cells human embryonic kidney 293 (HEK-293) by using Fugene (Roche Molecular Biochemicals, Indianapolis, IN) as suggested by the manufacturer. Transfected cells were identified under epifluorescence. Recordings were performed 24–72 h after transfection at room temperature.
Electrophysiology.
Patch pipettes were fabricated from borosilicate glass and fire polished to a resistance of 2–3 MΩ. Only recordings in which a gigaohm seal was achieved were used in the analysis. Excised patch recording was performed as described previously (Liu and Liman, 2003). Briefly, after formation of a gigaohm seal, the patch was excised into Ca2+-free solution and rapidly moved in front of a linear array of microcapillary tubes (Warner Instruments, Hamden, CT). For all experiments, the membrane potential was held at −80 mV. All recording were made with an Axopatch 200B amplifier, digitized with a Digidata 1322a, acquired with pClamp 8.2, and analyzed with Clampfit 8.2 (Molecular Devices, Palo Alto, CA). Records were sampled at 5 kHz and filtered at 1 kHz. Representative data shown in the figures was in some cases decimated 10- or 20-fold before exporting into the graphics programs Origin (Microcal, Northampton, MA) and Coreldraw (Corel, Eden Prairie, MN) to reduce the size of the files.
Solutions.
For whole-cell recording, the bath solution was as follows (in mm): 125 NaCl, 25 NaHCO3, 25 glucose, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, and 1 MgCl2 (bubbled with 95% O2, 5% CO2). Internal Cs+-based solution contained the following (in mm): 111 CsAsp, 22 CsCl, 11 HEPES, 2 MgATP, and 3.3 Na2ATP, pH 7.2. To this solution, we added the following: 2.5 mm 2,6-dimethyl-4-nitropyridine (DMNP)–EDTA, 0.75 mm CaCl2 (caged Ca2+) or 50 μm 1-(2-nitrophenyl)ethyl (NPE)-caged IP3 (caged IP3), 10 μm EGTA or 50 μm NPE-caged IP3, and 5 mm BAPTA. Internal K+-based solution contained the following (in mm): 120 KAsp, 20 KCl, 10 HEPES, 2 MgATP, 3 Na2ATP, 0.3 GTP, 2.5 DMNP–EDTA, and 0.75 CaCl2, pH 7.2. DMNP–EDTA, and NPE-caged IP3 were from Invitrogen (Carlsbad, CA).
For excised patch recording, the pipette solution contained the following (in mm): 150 NaCl, 10 HEPES, and 2 CaCl2, pH 7.4. Ca2+-free solution contained the following (in mm): 150 NaCl, 10 HEPES, and 2 HEDTA, pH 7.4. Cytosolic solutions with micromolar concentrations of free Ca2+ were obtained by adding Ca2+ (1.1, 1.5, or 1.9 mm) to obtain free Ca2+ concentrations of 5, 12, and 50 μm, respectively. Solutions containing 200 μm to 1 mm free Ca2+ were made by adding the appropriate amount of CaCl2 to a solution containing 150 mm NaCl and 10 mm HEPES, pH 7.4 with NaOH. For ATP blocking experiments, we added 100 μm Na2ATP to the solution containing 1 mm Ca2+; this is expected to yield a final concentration of 18 μm free ATP and 918 μm free Ca2+. All Ca2+ concentrations are reported as calculated with MaxChelator (www.stanford.edu/∼cpatton/maxc.html) and were confirmed by direct measurement with a Ca2+-sensitive electroprobe (Microelectrodes, Bedford, NH). For some experiments, we added the following biochemicals to the Ca2+-free solution: diC8-PIP2 (Echelon Bioscience, Salt Lake City, UT), 1,2-dioctanoyl-sn-glycerol (DOG), phosphatidic acid (PA) (Avanti Polar Lipids, Alabaster, AL), and IP3 (Invitrogen).
Flash photolysis of caged compounds.
All experiments were performed on an Olympus Optical (Tokyo, Japan) IX71 microscope. For most experiments, light from a xenon arc lamp was passed through a bandpass filter (380xv1; Chroma Technology, San Diego, CA) that was part of an LEP filter wheel/shutter (Ludl, Hawthorne, NY) operated under computer control (Simple PCI; Compix, Sewickley, PA). The shutter was opened for 2 s to uncage Ca2+ or 4 s to uncage IP3. For some experiments [e.g., uncaging Ca2+ using a K+-based intracellular solution (see Fig. 4C,D)], a uniblitz shutter (Vincent Associates, Rochester, NY) controlled light from a mercury arc lamp that was then passed through a 350/50× bandpass filter (Chroma Technology, San Diego, CA). The shutter was opened for 1 s to uncage Ca2+. To allow uniform dialysis of the cell with the caged compounds, we waited for 40 s after breaking into whole-cell recording mode before opening the shutter. Under these conditions, multiple responses could usually be obtained from the same cell. For analysis, only the first response from each cell was used, and the evoked current was measured as the difference between the peak current in response to the UV flash and the resting current before stimulation.
Ca2+ imaging.
Ca2+ imaging was performed on an Olympus Optical IX71 microscope equipped with an LEP filter wheel (Ludl Electronic Products) and ORCA ER camera (Hamamatsu Photonics, Shizuoka, Japan). Cells transfected with TRPM5 fused to CFP were loaded through the patch pipette with the low-affinity Ca2+ indicator Fluo-5F (20 μm). The fluorescence emission from Fluo-5F was detected using a yellow GFP filter set (Chroma Technology, Rockingham, VT). Images were captured and analyzed with simple PCI (Compix).
Results
TRPM5-dependent currents in taste receptor cells are activated by elevation of intracellular IP3
To understand the mechanism by which TRPM5 currents in taste receptors cells are activated, we used genetically modified mice in which these currents could be unequivocally identified. In one mouse strain, GFP expression was driven by the TRPM5 promoter (Clapp et al., 2006), allowing us to visually identify cells that have the capacity to transcribe the native TRPM5 gene. This strain was crossed to mice that carried a targeted deletion of the TRPM5 gene to generate mice that were +/+, +/−, or −/− for TRPM5 channel expression. To confirm fidelity of the GFP marker for TRPM5-expressing cells, we stained sections of the tongue containing circumvallate papillae with a polyclonal antibody directed against mTRPM5. In TRPM5+/+ and TRPM5+/− mice, all GFP-positive cells showed TRPM5 immunoreactivity (Fig. 1A,B), whereas no TRPM5 immunostaining was observed in sections from TRPM5−/− mice (Fig. 1C,D). Thus, GFP expression can be used to identify TRPM5-expressing cells or those that would have made TRPM5 had the gene not been deleted. Note that all mice studied were heterozygotic or homozygotic for expression of the GFP transgene, and no attempt was made to discriminate further between these two genotypes.

TRPM5 is expressed in GFP-positive taste cells in transgenic TRPM5–GFP mice. A, A section of circumvallate papillae from a transgenic mouse in which the TRPM5 promoter drives expression of GFP stained with an antibody against TRPM5 (red). Note that this mouse was TRPM5+/−. B, The same section showing complete coincidence between GFP fluorescence (green) and TRPM5 immunoreactivity (red). C, D, A section of circumvallate papillae from a TRPM5−/− mouse stained with an antibody against TRPM5 shows normal GFP expression (green) and complete absence of TRPM5 immunoreactivity (red), as expected. E, Isolated taste receptor cells from a mouse in which the TRPM5 promoter drives expression of GFP show intense green fluorescence. These cells are typical of those used for patch-clamp recording in this paper. Scale bars: (in D) A–D, 50 μm; E, 10 μm.
In acutely dissociated taste cells, bright GFP fluorescence was observed in ∼30% of morphologically identifiable taste receptor cells, consistent with the proportion of cells expected to express TRPM5 (Fig. 1E) (Perez et al., 2002). Whole-cell patch-clamp recording from these cells showed that they have voltage-gated sodium currents as expected (Akabas et al., 1988) (data not shown).
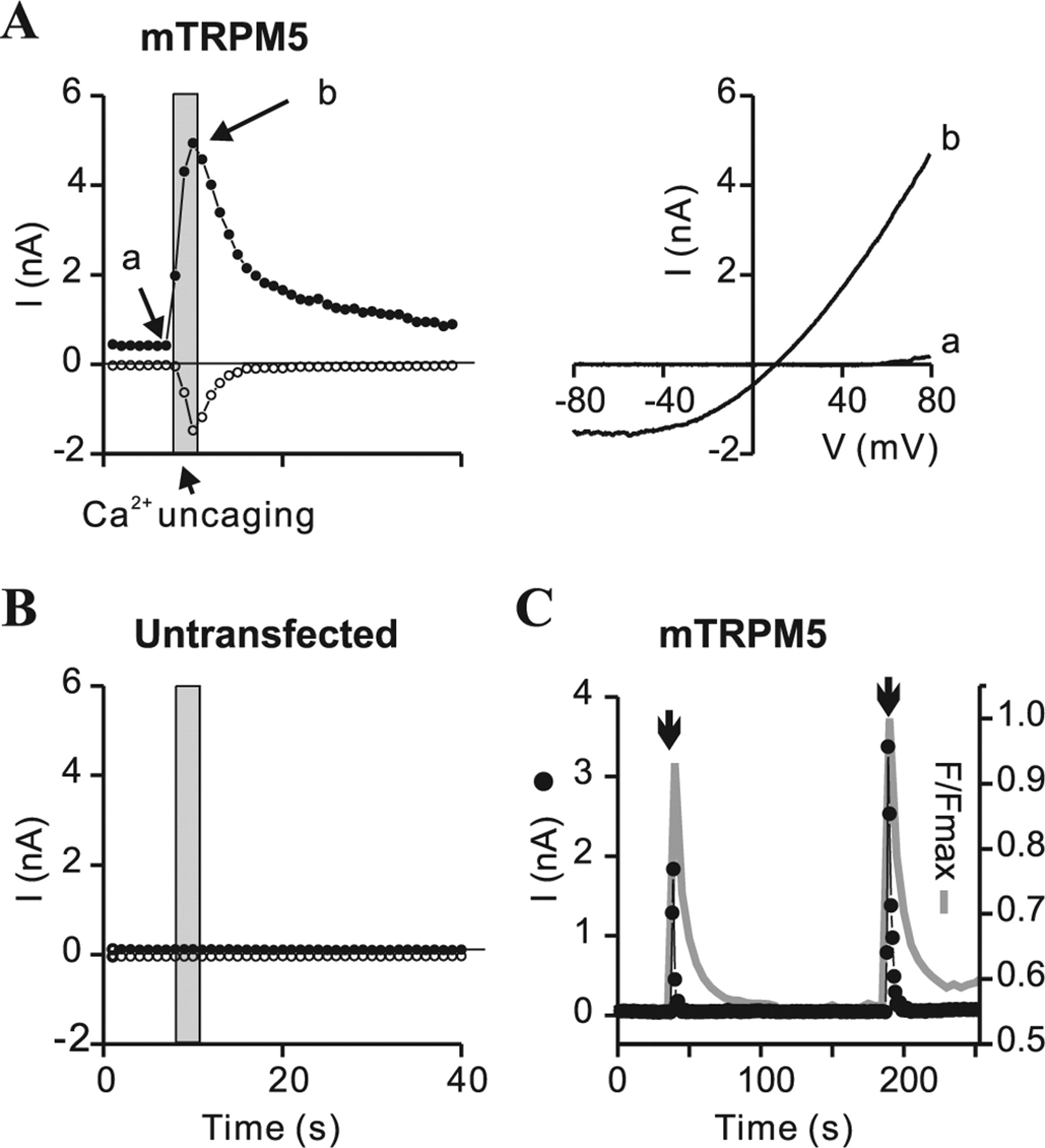
To determine whether native TRPM5 currents can be activated downstream of an elevation of IP3, we initially conducted experiments in which IP3, dissolved in a Cs+-based intracellular solution was dialyzed into taste cells. However, the presence of a relatively large outwardly rectifying current in the absence of IP3 in taste cells from both TRPM5+/+ and TRPM5−/− animals complicated analysis of the results (Fig. 2A,B and data not shown). We therefore, instead, perfused cells with an inactive form of IP3 (NPE-caged IP3) that could be activated by exposure to light of 380 nm. In response to release of caged IP3, a large current was evoked in taste cells from TRPM5+/+ animals (Fig. 2A,C). The evoked current had a reversal potential of 2.4 ± 2.9 mV (n = 7), indicating that it was nonselective among Na+ and Cs+, and it decayed with a time course that could be fit with a single exponential (τ of 2–7 s). No current was elicited by the same protocol in TRPM5−/− animals (Fig. 2B). A voltage-activated Na+ current was evident in most cells, and this served as a convenient indicator that the cells were healthy and that good electrical access in whole-cell recording mode had been achieved (Akabas et al., 1988). Thus, we can conclude that the current observed in TRPM5+/+ cells during uncaging IP3 depends on TRPM5 expression and most likely represents the native TRPM5 current. To confirm that, under the conditions of our experiments, flash photolyis of caged IP3 had the expected effect of releasing Ca2+ from intracellular stores and activating heterologously expressed TRPM5 channels, we recorded from HEK-293 cells transfected with mTRPM5. Flash photolysis of caged IP3 activated a large outwardly rectifying current in HEK-293 cells expressing TRPM5 (n = 5) but not in untransfected cells (n = 5) (Fig. 3A,B), and it caused a transient elevation of intracellular Ca2+, as determined by measuring fluorescent emission from the Ca2+ indicator Fluo-5F (n = 5) (Fig. 3C).

Activation of a TRPM5-dependent current in taste receptors cells by elevation of intracellular IP3. A, Current activation in response to flash photolysis of caged IP3 in a TRPM5-expressing taste receptor cell (GFP+) from a TRPM5+/+ mouse. Time course of the current at +80 mV (filled circles) and −80 mV (open circles). The opening of the shutter on the UV lamp is indicated with the gray bar. Right shows the current–voltage relationship obtained from ramp depolarizations (1 V/s) before the flash (a) and at the peak of the response (b). B, No current was activated in a TRPM5−/− taste receptor cell in response to flash photolysis of caged IP3. C, Mean data from experiments such as those in A and B. Response amplitude was measured as the difference in the current after and before uncaging IP3. Note that inclusion of BAPTA (5 mm) severely attenuated the response to uncaging IP3 in TRPM5+/+ taste cells. Data are represented by the mean ± SEM. *p < 0.05, **p < 0.01, Student's t test.

Activation of heterologously expressed TRPM5 channels by elevation of intracellular IP3. A, B, Currents activated by flash photolysis of caged IP3 in HEK-293 cells transfected with mTRPM5 (A) or in untransfected HEK-293 cells (B). Filled and open circles represent the currents at +80 and −80 mV, respectively. Gray bar shows the time during which the shutter on the UV light source was open. Right panels show the current–voltage relationship obtained from ramp depolarizations (1 V/s) before the flash (a) and at the peak of the response (b). Similar results were obtained in five cells for each condition. C, Simultaneous measurement of the magnitude of the current (left axis; filled circles) and the change in intracellular Ca2+ (right axis; gray line) in response to uncaging IP3 in a HEK-293 cell transfected with mTRPM5. The Ca2+ indicator Fluo-5F (10 μm) was loaded through the patch pipette. Similar results were obtained in five cells. D, Mean magnitude of the response to uncaging IP3 in HEK-293 cells transfected with mTRPM5 in the presence and absence of 5 mm BAPTA (Vm of +80 mV). Data are represented by the mean ± SEM. *p < 0.05, Student's t test.
IP3 might act directly on TRPM5 channels or it could act indirectly by releasing intracellular Ca2+, which could be the stimulus that gates TRPM5 channels. This latter hypothesis predicts that the response to elevation of IP3 should be blocked by heavy buffering of intracellular Ca2+. In support of this possibility, we found that, in TRPM5-expressing taste cells (n = 6) (Fig. 2C) and in HEK-293 cells expressing TRPM5 (n = 5) (Fig. 3D), responses to uncaging IP3 in the presence of 5 mm BAPTA were severely attenuated compared with the responses measured when a low concentration of Ca2+ buffer was included in the pipette solution (10 μm EGTA).
Intracellular Ca2+ activates TRPM5-dependent currents in taste cells
To determine whether a rise in intracellular Ca2+ is sufficient to activate TRPM5-dependent currents in taste cells, we introduced DMNP–EDTA (caged Ca2+) in whole-cell patch-clamp recording mode and measured the electrical response when Ca2+ was released by flash photolysis. In TRPM5-expressing taste cells, flash photolysis of caged Ca2+ consistently activated a large current (Fig. 4A,C). With an intracellular solution that contained Cs+ and an external solution in which Na+ was the principle cation, this evoked current reversed at 5.5 ± 2.9 mV (n = 8) (Fig. 4A), consistent with nonselective permeability for Cs+ compared with Na+(see below). After activation, the current decayed with a variable time course (the time constant of decay ranged from <1 to 60 s). In contrast, in taste cells from TRPM5−/− animals, no current was evoked by uncaging Ca2+ (Fig. 4B,C). In HEK-293 cells transfected with mTRPM5, flash photolysis of caged Ca2+ led to a transient activation of a large current (Fig. 5A,C) and more prolonged elevation of intracellular Ca2+ (Fig. 5C). The current evoked in HEK-293 cells expressing mTRPM5 was somewhat more rectifying than that evoked in taste cells (I+80 mV/I−80mV was 3.9 ± 1.3 in HEK-293 cells, n = 5 and 1.9 ± 0.2 in taste cells, n = 8; p = 0.05, two-tailed t test). No current was elicited by uncaging Ca2+ in untransfected HEK-293 cells (Fig. 5B).

Activation of TRPM5-dependent currents in taste receptor cells by flash photolysis of caged Ca2+. A, B, Current activated in response to flash photolysis of caged Ca2+ in a taste receptor cell from a TRPM5+/+ mouse (A) and a TRPM5−/− mouse (B). The patch pipette contained a Cs+-based solution that was supplemented with DMNP–EDTA. Time course, shown on left, of the current at +80 mV (filled circles) and −80 mV (open circles). Gray bar shows the time during which the shutter on the UV lamp was open (2.4 s). The right panels show the current–voltage relationship before the flash (a) and at the peak of the response (b). C, Mean response to uncaging Ca2+ in taste cells from TRPM5+/+ and TRPM5−/− mice. The internal solution contained either Cs+ or K+, as indicated on the graph. Response amplitude was measured as the difference in the current after and before uncaging Ca2+. D, The current–voltage relationship of the current evoked in response to uncaging Ca2+ in a TRPM5+/+ taste cell dialyzed with an internal solution in which the principle cation was K+ (140 mm). The external solution was exchanged from one in which Na+ was the principal cation to one in which the principal cations were Ca2+ (50 mm) and NMDG (80 mm). The resting current before uncaging was subtracted. Data are represented by the mean ± SEM. *p < 0.05; **p < 0.01, Student's t test.

Activation of cloned TRPM5 channels by elevation of intracellular Ca2+. A, B, Current activated in response to flash photolysis of caged Ca2+ in HEK-293 cells transfected with mTRPM5 (A) or in untransfected HEK-293 cells (B). Filled and open circles represent the currents at +80 and −80 mV, respectively. Gray bar shows the time during which the shutter on the UV light source was open. Current–voltage relationship of the response in the transfected cell before the flash (a) and at the peak of the response (b) is shown on the right. C, Simultaneous measurement of the current at +80 mV (left axis; filled circles) and the change in intracellular Ca2+ (right axis) in HEK-293 cells transfected with mTRPM5–CFP in response to uncaging Ca2+ (indicated by the arrows). The Ca2+ indicator Fluo-5F (20 μm) was loaded through the pipette. Note that the current decayed faster than the Ca2+ signal and that multiple responses could be evoked in the same cell.
To determine whether TRPM5 currents could be activated under more physiological conditions, we dialyzed taste receptor cells with an internal solution in which K+ was the principal cation. Flash photolysis of caged Ca2+ elicited a current in cells from TRPM5+/+ animals (n = 6), but not in cells from TRPM5−/− animals (n = 5), indicating that the evoked current can be attributed entirely to the gating of TRPM5 channels (Fig. 4C). With Na+ as the principal cation in the external solution, the current reversed at 3.3 ± 4.8 mV (n = 6), indicating that the channels are equally permeable to Na+ and K+ (Fig. 4D). When the bath solution was exchanged for a solution containing 50 mm Ca2+ [80 mm N-methyl-d-glucamine (NMDG)], the reversal potential of the evoked current was shifted to −61.4 ± 3.8 (n = 6) mV, indicating that the channels have little or no permeability to Ca2+ (PCa2+/PK+ < 0.05) (Fig. 4D).
Native TRPM5 channels are Ca2+-activated, voltage-modulated cation channels
We recorded from inside-out membrane patches to determine whether Ca2+, IP3, or other potential second messengers could act directly at the plasma membrane to gate TRPM5 channels in taste receptor cells. In GFP-expressing cells from TRPM5+/+ animals, Ca2+ (50 or 100 μm) activated an inward current of >10 pA at −80 mV in 19 of 22 patches (Fig. 6). The magnitude of the inward currents was variable and, in one patch, was as large as 2 nA. Similarly, 15 of 16 patches from TRPM5+/− animals responded to Ca2+ (50 μm) (range up to 1.3 nA). In contrast, no response was elicited by exposure to other putative second messengers, including IP3, DOG (a synthetic analog of DAG), PA, or PIP2 (Fig. 6A,C). To confirm that this Ca2+-activated current represented the native TRPM5 conductance, we recorded from patches excised from GFP+ taste receptor cells from TRPM5−/− mice. An inward current in response to 50 μm Ca2+ was observed in only 5 of 25 patches (range up to 100 pA) (Fig. 6B,D), and these small responses were later found to represent a conductance that was clearly distinct from that of TRPM5 (see Figs. 8E, 9). The severe attenuation in the magnitude of the Ca2+-activated current in TRPM5−/− taste cells allows (Fig. 6D) us to conclude that the conductance activated by intracellular Ca2+ in TRPM5+/+ and TRPM5+/− taste cells primarily represents the native TRPM5 conductance.

TRPM5-dependent channels in excised patches from taste cells are activated by intracellular Ca2+. A, C, Current activation in response to intracellular Ca2+, but not other potential second messengers, in an excised patch from a TRPM5+/− taste receptor cell. Membrane potential was held at −80 mV. Chemicals tested were as follows: 50 μm Ca2+, 50 μm DOG, 10 μm IP3, 10 μm PA, and 10 μm PIP2. C, Average data from experiments as in A (n = 3). B, D, Currents evoked by 50 μm Ca2+ in excised patches from taste receptor cells from TRPM5+/+, TRPM5+/−, and TRPM5−/− mice. Membrane potential was held at −80 mV. D, Average data from experiments as in B. Data are represented by the mean ± SEM. **p < 0.01.
We measured the ionic selectivity of the native TRPM5 conductance in excised patches by changing the main intracellular cation while keeping the concentration of activating Ca2+ constant. Based on changes in the reversal potential of the current, we calculated the relative permeability of K+ to Na+ as PK+/PNa+ = 0.91 ± 0.13 (n = 3). The currents were relatively impermeable to the large monovalent cation NMDG+ (PNMDG+/PNa+ = 0.10 ± 0.04; n = 4) (Fig. 7A). Thus, based on these results, in combination with the data from whole-cell recording, we conclude that the native TRPM5 conductance is equally permeable to small monovalent cations and relatively impermeable to large monovalent cations or divalent cations (PCs+ = PK = PNa ≫ PNMDG or PCa2+). In a few patches in which small numbers of channels were present, opening of single channels could be resolved (Fig. 7B). These openings showed flickery gating, as reported previously for heterologously expressed TRPM5 (Liu and Liman, 2003). Amplitude histograms (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) gave a unitary current size of ∼1.4–1.7 pA, corresponding to a conductance of 17–21 pS. Because of the fact that the channels opened only briefly, this value probably slightly underestimates the true single-channel conductance.

Ion selectivity and conductance of TRPM5-dependent channels in taste cells. A, Ion selectivity of the Ca2+-activated current in an excised patch from a TRPM5+/− mouse. The cytoplasmic solution contained 100 μm Ca2+ to activate the current. Ion selectivity was assessed by measuring the current–voltage relationship after exchanging the major cation in the cytoplasmic solution. Na+ is 150 mm NaCl, K+ is 150 mm KCl, NMDG is 125 mm NMDG-Cl with 25 mm NaCl, and 0 Ca2+ is Ca2+-free 150 mm NaCl. The holding potential was −80 mV, and the voltage was ramped to +80 mV (1 V/s). Note the 45 mV change in the reversal potential when the solution was changed from Na+ to NMDG+ and absence of a shift when the solution was changed to K+. B, Single TRPM5 channels activated by intracellular Ca2+ in an excised patch from a TRPM5+/− animal (Vm of −80 mV). Note the flickery nature of the activity and small current amplitude of the open channels. C, Currents recorded in response to a voltage family in a patch from a TRPM5+/+ animal. Leak currents recorded before applying intracellular Ca2+ were subtracted. Right shows the steady-state current–voltage relationship, determined from experiments as in the left, normalized to the current at +100 mV. Filled symbols represent the mean, and error bars represent the SEM (n = 5). For B and C, the main cation in the cytoplasmic solution was Na+.
To determine whether native TRPM5 channels are voltage modulated, we recorded current responses to a family of voltage steps in the presence of a low concentration of intracellular Ca2+ (5 μm). Currents evoked in excised patches from TRPM5+/+ taste receptor cells showed a time-dependent activation after the voltage steps and a time-dependent relaxation during repolarization to −80 mV (Fig. 7C). Thus, endogenous TRPM5-dependent channels in taste cells are voltage modulated as are heterologously expressed TRPM5 channels (Hofmann et al., 2003; Liu and Liman, 2003).
Micromolar Ca2+ sensitivity of native TRPM5 channels
Ca2+ sensitivity of TRPM5-dependent channels in taste cells was assessed by sequentially exposing the intracellular surface of excised patches to increasing concentrations of Ca2+ (Fig. 8A–D). Dose–response curves obtained from pooled data showed that the currents in patches from TRPM5+/+ and TRPM5+/− mice were half-activated by 8 and 14 μm Ca2+, respectively (Fig. 8A–D). These values are somewhat lower than the half-activating concentration of Ca2+ reported for expressed TRPM5 channels under the same conditions (21 μm) (Liu and Liman, 2003). The currents were strongly outwardly rectifying when activated by a low concentration of Ca2+ (Fig. 8A,C), consistent with the observation that, when activated by 5 μm Ca2+, TRPM5-dependent channels are voltage modulated (Fig. 7C). At higher Ca2+ concentrations, the currents became more linear, indicating that there is an interaction between Ca2+ and voltage activation of the channels (Nilius et al., 2005).

Native TRPM5 channels are activated by micromolar concentrations of intracellular Ca2+. A–F, Currents evoked in response to increasing concentrations of intracellular Ca2+ in excised patches from taste receptor cells from TRPM5+/+ (A, B), TRPM5+/− (C, D), and TRPM5−/− (E, F) mice. Raw data shown in A, C, and E was obtained either immediately on patch excision (traces to the left) or after repeated exposure of the patch to intracellular Ca2+ (trace to the right). Note the pronounced decrease in the currents, indicating that the channels have desensitized. Insets show the I–V relationship at two calcium concentrations (indicated) for currents before desensitization. Note that the current in the cells from TRPM5+/+ and TRPM5+/− animals is strongly outwardly rectifying, whereas that in the cell from the TRPM5−/− animal is nearly linear. B, D, F, The Ca2+ dose–response relationship from experiments as in A, D, and G. Filled circles show the initial response to Ca2+, and open circles show the response after desensitization normalized to the peak response before desensitization. The sensitivity of the heterologously expressed channel before and after desensitization is shown by a dotted line (before desensitization, K1/2 of 21 μm, n = 2.4, Vmax of 105; after desensitization, K1/2 of 77 μm, n = 2.4, Vmax of 23) (Liu and Liman, 2003). Fits to data were as follows: +/+ initial (K1/2 of 8 μm; n = 3.2; Vmax of 100), +/+ desensitized (K1/2 of 48 μm; n = 1.4; Vmax of 11), +/− initial (K1/2 of 14 μm; n = 2.5; Vmax of 100), +/− desensitized (K1/2 of 133 μm; n = 1.2; Vmax of 10), −/− initial (K1/2 of 303 μm; n = 1.2; Vmax of 123), and −/− final (K1/2 of 283 μm; n = 2.2; Vmax of 76). Data are represented by the mean ± SEM.
Heterologously expressed TRPM5 channels desensitize in response to prolonged Ca2+ exposure (Liu and Liman, 2003). Similarly, we observed a decline in the maximal amplitude and a change in the Ca2+ sensitivity of native TRPM5 currents after prolonged exposure to intracellular Ca2+ (Fig. 8A–D).
A TRPM5-independent Ca2+-activated nonselective cation channel in mouse taste receptor cells
Surprisingly, at very high intracellular Ca2+ concentrations (1 mm), a nonselective cation current was evoked in more than half of the patches excised from TRPM5−/− animals. When measured immediately after patch excision, this conductance was half-activated by 300 μm intracellular Ca2+, and it desensitized only slightly after prolonged exposure to intracellular Ca2+ (Fig. 8E,F). Under conditions in which Na+ was the main cation on both sides of the membrane, the current reversed at ∼0 mV (Fig. 8E). Replacement of intracellular Na+ with NMDG+ essentially abolished the outward current (n = 8) (Fig. 9A), indicating that the current was carried by cations. This was further confirmed by experiments in which we found that substituting intracellular Cl− with gluconate or methane sulfonate (n = 2; data not shown) had no effect on the magnitude or reversal potential of the currents. To further characterize the current in TRPM5−/− taste cells, we determined whether it was sensitive to block by intracellular ATP, which at micromolar concentrations blocks many Ca2+-activated nonselective cation channels (Thorn and Petersen, 1992; Liman, 2003) and blocks heterologously expressed TRPM4 channels (Nilius et al., 2004). ATP (18 μm free) blocked the Ca2+-activated currents in patches from TRPM5−/− taste cells (Fig. 9C), although the extent of block was highly variable (Fig. 9D). Under the same conditions, incomplete and variable block of heterologously expressed TRPM4 channels was also observed (data not shown). The Ca2+-activated channels in TRPM5−/− cells were further characterized by “box-like openings” and slow gating compared with the fast and flickery openings of TRPM5 channels (Fig. 9B) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). A conductance of 30 pS was calculated based on the current amplitude at −80 mV of −2.5 ± 0.1 pA (n = 6) (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material). Because we could not detect activation of this current by uncaging Ca2+ or IP3 in whole-cell recording mode, it is presently unclear what role it plays in taste cell signaling.

Properties of a TRPM5-independent Ca2+-activated channels in TRPM5−/− taste cells. A, Ca2+-activated current in an excised patch from a TRPM5−/− taste cell. Note that the outward current was eliminated when Na+ (150 mm) on the cytoplasmic side of the patch was replaced by NMDG (150 mm), indicting that the channels were permeable to Na+. Intracellular Ca2+ was 200 μm. B, Ca2+-activated channels in excised patches from taste cells from TRPM5+/− and TRPM5−/− animals (Vm of −80 mV). Note the slow gating and box-like openings of the channels in the recording from the TRPM5−/− cell. C, Block by intracellular ATP of the Ca2+-activated current in a patch from a TRPM5−/− taste cell. Intracellular Ca2+ was 1 mm, and free ATP was calculated to be 18 μm. D, Aggregate data from seven independent patches tested for sensitivity to ATP block. IATP is the current remaining after exchange to the ATP-containing solution and Imax in the current after ATP exposure. The fraction of current that was insensitive to block by ATP (IATP/Imax) was highly variable. Box plot shows the mean, SEM, and 90% confidence interval.
Discussion
Bitter, sweet, and umami tasting compounds are detected by taste receptor cells that share a common transduction cascade for which the ion channel TRPM5 is a critical element (Perez et al., 2002; Zhang et al., 2003; Damak et al., 2006). Despite its significance for taste, the properties of TRPM5 channels in taste cells have not been described previously, and any information about how the channels are regulated has been obtained solely from the study of heterologously expressed channels (Perez et al., 2002; Hofmann et al., 2003; Liu and Liman, 2003; Prawitt et al., 2003; Zhang et al., 2003; Liu et al., 2005; Talavera et al., 2005). In this report, we characterized native TRPM5 channels using transgenic animals that allowed us to identify those taste cells that normally express TRPM5 and to compare currents in these cells in the presence and absence of an intact TRPM5 gene. Our results show that elevation of intracellular IP3 activates TRPM5 currents in taste cells and that this response can be entirely attributed to an elevation of intracellular Ca2+. Detailed study in excised patches has allowed us to define the properties of native TRPM5 channels and to compare these properties with those of heterologously expressed channels. Our results show that the native channels, like expressed channels, are activated by micromolar concentrations of intracellular Ca2+ and are selectively permeable to monovalent cations. These studies have also allowed us to identify a second, distinct, Ca2+-activated cation current in taste cells.
In general, the properties of endogenous TRPM5 channels are very similar to those of channels expressed in heterologous cell types transfected with mouse TRPM5. Both native and expressed channels are activated in intact cells by an elevation of Ca2+ or IP3 and in excised patches only by elevation of cytoplasmic Ca2+ (Hofmann et al., 2003; Liu and Liman, 2003; Prawitt et al., 2003; Ullrich et al., 2005). Both native and expressed TRPM5 channels are nonselective among monovalent cations and relatively impermeable to divalent cations (Hofmann et al., 2003; Liu and Liman, 2003; Prawitt et al., 2003; Ullrich et al., 2005), and both native and expressed TRPM5 channels are, under the appropriate conditions, voltage modulated (Hofmann et al., 2003; Liu and Liman, 2003; Talavera et al., 2005). Native channels differ from heterologously expressed channels in that they are slightly more sensitive to activation by Ca2+ (K1/2 of 8 μm compared with 21 μm) and they show somewhat less outward rectification. Both of these properties endow the native channels with a greater capacity to convert small changes in intracellular Ca2+ to large changes in membrane potential. Differences in properties of native and expressed TRPM5 channels may reflect increased levels in taste cells of PIP2, which has been shown to increase Ca2+ sensitivity and decrease voltage-dependent gating of TRPM5 and TRPM4 channels (Liu and Liman, 2003; Zhang et al., 2005; Nilius et al., 2006).
We also identified a Ca2+-activated cation conductance in taste cells that is distinct from the TRPM5-dependent conductance. This conductance, which was detected in excised patch recording mode from TRPM5−/− cells, was relatively small and insensitive to activation by Ca2+. This may explain why we did not detect a response to uncaging Ca2+ or IP3 in TRPM5−/− cells. It is possible that this current is generated by channels that are encoded by the targeted TRPM5 gene, which is missing the first three exons, but we could detect no expression of the TRPM5 protein using a C-terminal-directed antibody in TRPM5−/− animals. It is more likely that this current is generated by channels encoded by a separate gene, possibly TRPM4, which is known to encode a Ca2+-activated nonselective cation channel. Heterologously expressed TRPM4 channels are relatively insensitive to intracellular Ca2+ (half activation of ∼100–300 μm in excised patches) (Nilius et al., 2004; Zhang et al., 2005), much like the channels observed in TRPM5−/− cells. Heterologously expressed TRPM4 channels are half-blocked by 2 μm free ATP, and this block is incomplete even at high ATP concentrations (Nilius et al., 2004); similarly, we found that the Ca2+-activated channels in TRPM5−/− cells could be blocked by 18 μm free ATP, although with variable efficacy. Finally, the conductance (∼30 pS) and gating kinetics of expressed TRPM4 channels (Launay et al., 2002; Zhang et al., 2005) and of the Ca2+-activated channels in TRPM5−/− taste cells are similar. Interestingly, the Ca2+-activated current in TRPM5−/− taste cells did not show any evidence of voltage dependence, in contrast to what has been reported for heterologously expressed TRPM4 and TRPM5 currents (Hofmann et al., 2003; Nilius et al., 2003). However, recent studies of native TRPM4 currents in mast cells has revealed that, in this context, the channels are not voltage dependent (Vennekens et al., 2007).
Our results predict that responses of taste cells to bitter, sweet, and umami tastants should be accompanied by the activation of a nonselective cation conductance. Despite decades of research, it is therefore curious that, in most studies, activation of a TRPM5-like current to tastants has not been observed. For example, taste cells from several species have been shown to depolarize in response to sweet compounds, but this had been attributed to block of voltage-activated K+ currents (Tonosaki and Funakoshi, 1988; Behe et al., 1990; Cummings et al., 1996; Gilbertson et al., 2001). Similarly, bitter compounds have been shown to block K+ channels in taste cells from frog, mudpuppy, and rat (Avenet and Lindemann, 1987; Cummings and Kinnamon, 1992; Ogura et al., 1997; Gilbertson et al., 2001). There are, as far as we are aware, just a few examples in the literature in which nonselective cationic currents were observed in response to taste stimuli. In one study, glutamate, presumably acting on umami receptors, was shown to cause a transient depolarizing response and to open cation channels in a subset of rat taste cells (Bigiani et al., 1997). In another series of studies, intracellular dialysis of IP3 or Ca2+ into frog taste cells was found to activate a nonselective cation conductance, and a similar conductance was evoked by sweet stimuli (Tsunenari et al., 1996; Okada et al., 1998, 2001); these currents are likely mediated by TRPM5. In studies that failed to observe activation of a cationic conductance, it is possible that dialysis of high concentrations of Ca2+ buffers into the cells interfered with Ca2+-dependent activation of TRPM5 channels.
Our work, in combination with that of others, suggests that IP3 and Ca2+ are second messengers for taste transduction, converting the biochemical signal generated by the activation of taste receptors into a change in ionic permeability of the plasma membrane. This hypothesis has its antecedents in other sensory systems. For example, early functional studies of vertebrate vision suggested that intracellular Ca2+ was the second messenger for phototransduction (Pugh, 1987). However, it was subsequently determined by patch-clamp recording that vertebrate phototransduction depends on changes in a cyclic-nucleotide gated (CNG) conductance (Fesenko et al., 1985; Haynes and Yau, 1985). CNG-regulated ion channels have been noted in taste cells (Kolesnikov and Margolskee, 1995; Misaka et al., 1997), but the following lines of evidence establish Ca2+ along with IP3 as key second messengers in taste transduction: (1) bitter, sweet, and umami tastes elicit an elevation of intracellular Ca2+ (Akabas et al., 1988; Bernhardt et al., 1996; Ogura et al., 1997; Caicedo et al., 2002); (2) PLCβ2, which generates IP3, is essential for normal bitter, sweet, and umami taste (Zhang et al., 2003; Dotson et al., 2005); and (3) TRPM5, which forms a Ca2+-activated nonselective conductance in heterologous cells (Hofmann et al., 2003; Liu and Liman, 2003; Prawitt et al., 2003) and in taste cells (this report), is essential for normal bitter, sweet, and umami taste (Zhang et al., 2003; Damak et al., 2006). Thus, according to our data and that of others, it can be hypothesized that, during taste transduction, receptors activate a PLC signaling cascade that culminates in an elevation of intracellular Ca2+, opening of TRPM5 ion channels, and membrane depolarization.
Future research will be needed to precisely define the molecular mechanisms acting downstream of TRPM5 to modulate neurotransmitter release in taste cells. Bitter-, sweet-, and umami-sensing taste cells do not express voltage-gated calcium channels or other components of Ca2+-evoked secretion and the mechanism by which the neurotransmitter ATP is released has been enigmatic (Medler et al., 2003; Finger et al., 2005; Clapp et al., 2006; DeFazio et al., 2006). In two recent papers, it has now been proposed that ATP is secreted by a nonvesicular pathway that involves the voltage-dependent opening of connexin or pannexin hemichannels (Huang et al., 2007; Romanov et al., 2007). Consistent with this possibility, release of ATP can be evoked in isolated taste cells by membrane depolarization without elevation of intracellular Ca2+ (Romanov et al., 2007). Thus, in taste cells, intracellular Ca2+ does not directly regulate transmitter release but instead may do so indirectly through its effects on the gating of TRPM5 ion channels.
Footnotes
-
This work was supported by National Institute on Deafness and Other Communication Disorders Grants 004564 (E.L.) and 003155 (R.M.). We thank S. Kinnamon, N. Chaudhari, D. Liu, and Y. Wang for help with taste cell isolation; D. Arnold, D. McKemy, and R. Chow for helpful discussions; and H. Waters for expert technical support.
- Correspondence should be addressed to Emily R. Liman, Program in Neurosciences, University of Southern California, 3641 Watt Way, Los Angeles, CA 90089. liman{at}usc.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}