

Abstract
Intact cholinergic innervation from the medial septum and noradrenergic innervation from the locus ceruleus are required for hippocampal-dependent learning and memory. However, much remains unclear about the precise roles of acetylcholine (ACh) and norepinephrine (NE) in hippocampal function, particularly in terms of how interactions between these two transmitter systems might play an important role in synaptic plasticity. Previously, we reported that activation of either muscarinic M1 or adrenergic α1 receptors induces activity- and NMDA receptor-dependent long-term depression (LTD) at CA3–CA1 synapses in acute hippocampal slices, referred to as muscarinic LTD (mLTD) and norepinephrine LTD (NE LTD), respectively. In this study, we tested the hypothesis that mLTD and NE LTD are independent forms of LTD, yet require activation of a common Gαq-coupled signaling pathway for their induction, and investigated the net effect of coactivation of M1 and α1 receptors on the magnitude of LTD induced. We find that neither mLTD nor NE LTD requires phospholipase C activation, but both plasticities are prevented by inhibiting the Src kinase family and extracellular signal-regulated protein kinase (ERK) activation. Interestingly, LTD can be induced when M1 and α1 agonists are coapplied at concentrations too low to induce LTD when applied separately, via a summed increase in ERK activation. Thus, because ACh and NE levels in vivo covary, especially during periods of memory encoding and consolidation, cooperative signaling through M1 and α1 receptors could function to induce long-term changes in synaptic function important for cognition.
Introduction
Cholinergic innervation from medial septum and adrenergic innervation from locus ceruleus are required for learning and memory processing in hippocampus (Blokland, 1995; Compton et al., 1995). Administration of cholinergic or adrenergic antagonists impairs memory in humans (Lasser et al., 1989; Preston et al., 1989; Little et al., 1998). Similarly, mice lacking either the gene encoding the M1 muscarinic acetylcholine (ACh) receptor (Hamilton et al., 2001; Anagnostaras et al., 2003) or the gene encoding the α1 adrenergic receptor (Knauber and Muller, 2000; Spreng et al., 2001; Tanoue et al., 2002), as well as mice unable to synthesize norepinephrine (NE) because of a disrupted dopamine β-hydroxylase gene (Thomas and Palmiter, 1997), exhibit impairments in spatial memory and consolidation. Interestingly, ACh and NE can modulate one another's release both in vivo (Nilsson et al., 1992; Vizi and Kiss, 1998; Kiss et al., 1999) and in vitro (Mantovani et al., 1983; Tanganelli et al., 1989), via activation of presynaptic cholinergic receptors located on noradrenergic terminals and noradrenergic receptors present on cholinergic terminals (Nilsson et al., 1992; Vizi and Kiss, 1998; Kiss et al., 1999). However, much remains unclear about the precise roles of ACh and NE in hippocampal function, particularly in terms of how interactions between these two transmitter systems might affect synaptic plasticity thought to underlie learning and memory at the cellular level (Malenka and Bear, 2004; Whitlock et al., 2006).
We reported previously that activation of muscarinic M1 or adrenergic αl receptors induces long-term depression (LTD) of synaptic transmission at excitatory CA3–CA1 synapses in acute slices of rat hippocampus; we refer to these forms of LTD as muscarinic LTD (mLTD) (Scheiderer et al., 2006) and norepinephrine LTD (NE LTD) (Scheiderer et al., 2004), respectively. Both mLTD and NE LTD require presynaptic activity and NMDA receptor (NMDAR) activation, indicating that they share mechanistic properties. In addition, M1 and α1 receptors couple to the same G-protein, Gαq (Porter et al., 2002; Hague et al., 2003).
The intracellular signaling molecules which mediate induction of mLTD and NE LTD in hippocampus have not been identified, but we hypothesize that these plasticities share a common Gαq-coupled signaling pathway. If so, then coactivation of M1 and α1 receptors should be able to elicit summed activation of signaling molecules required for LTD induction. Thus, simultaneous activation of both M1 and α1 receptors might result in LTD of greater magnitude than that induced by activation of either receptor type alone. Even more intriguing is the possibility that weak stimulation of M1 and α1 receptors, at a level subthreshold for inducing LTD via activation of either receptor alone, could combine to induce LTD by converging activation of a shared signaling cascade. Interestingly, hippocampal ACh and NE levels can vary in vivo, so that combined activation of Gαq-coupled signaling may be important physiologically to allow induction of plasticity even when ACh and/or NE are present at low levels. For example, ACh and NE levels fluctuate across the sleep–wake cycle, as hippocampal function alternates between memory encoding during wakefulness and consolidation during sleep (Hobson et al., 1975; Kafka et al., 1986; Aston-Jones et al., 1991; Hasselmo, 1999). Cholinergic and/or noradrenergic signaling can also be reduced in hippocampus during aging and neurodegenerative diseases (Carlsson et al., 1980; Tomlinson et al., 1981; Collerton, 1986; Springer et al., 1987; Bickford-Wimer et al., 1988; Chan-Palay and Asan, 1989; German et al., 1992; Kasa et al., 1997). Therefore, that ACh and NE can share signaling mechanisms depending on the receptors activated raises the possibility that increased activity of one neurotransmitter may be able to compensate for loss of the other.
In this study, we tested the hypothesis that mLTD and NE LTD are independent forms of LTD, yet they require a common signaling pathway for their induction. After identifying this signaling cascade, we investigated the net effect of coactivation of M1 and α1 receptors on the magnitude of synaptic depression induced.
Materials and Methods
All experiments were conducted with an approved protocol from the University of Alabama at Birmingham Institutional Animal Care and Use Committee. All procedures are in compliance with guidelines put forth by the National Institutes of Health.
Slice preparation and electrophysiology.
Hippocampal slices (400 μm) were prepared from 3- to 4-week-old Sprague Dawley rats as described previously (Scheiderer et al., 2004). Rats were deeply anesthetized using inhalation anesthesia (halothane or isoflurane) and rapidly decapitated. Brains were removed and placed in ice-cold artificial CSF (aCSF) containing “high sucrose” and low Na+ and Ca2+ (in mm): 85 NaCl, 2.5 KCl, 4 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 75 sucrose, 2 kynurenic acid, and 0.5 ascorbate. The use of this solution during the slicing procedure enhances neuronal survival by limiting excessive excitation. Coronal slices from the dorsal hippocampus were cut using a vibratome (Vibratome, St. Louis, MO). After completion of the slicing procedure, slices were incubated for an additional 30 min in the high-sucrose aCSF, and then transferred to a standard aCSF containing (in mm) 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26 NaHCO3, 10 glucose, and 2 mm kynurenic acid, saturated with 95% O2–5% CO2, pH 7.4, for an additional 30 min before beginning experiments. For recordings, slices were transferred to a submersion chamber and were continuously perfused at 3–4 ml/min with standard aCSF without kynurenic acid (28–30°C). Extracellular dendritic field potentials were recorded in stratum (s.) radiatum of area CA1 (Axoclamp 2B or Axopatch 1D; Molecular Devices, Sunnyvale, CA) using standard methods (Scheiderer et al., 2004). A stainless-steel or tungsten bipolar stimulating electrode (FHC, Bowdoinham, ME) was placed in s. radiatum to stimulate Schaffer collateral axons, and a glass microelectrode filled with either 2 m NaCl or aCSF was placed nearby in s. radiatum to record extracellular dendritic field potentials [field EPSPs (fEPSPs)]. Baseline transmission was acquired using 0.2 Hz (100 μs duration) stimulation frequency and the stimulus intensity was adjusted to generate a fEPSP of 0.8–1.0 mV in amplitude. After acquisition of a stable baseline of transmission (at least 20 min), agonists were bath applied to induce LTD. mLTD was induced via a 10 min application of carbachol (CCh) (50 μm) or the selective M1 agonist [4-(m-chlorophenylcarbamoyloxy)-2-butynyltrimethylammonium] (McN-A-343) (50 μm) (adapted from Kirkwood et al., 1999; see also Scheiderer et al., 2006) and NE LTD was induced by a 10 min bath application of the α1 agonist methoxamine (Methox; 40 μm) (Scheiderer et al., 2004, 2006). Receptor antagonists and inhibitors of the signaling molecules were bath applied at least 10 min before application of agonists to induce LTD.
Data analysis.
Data were filtered at 2 kHz and acquired using data acquisition software written in Labview (National Instruments, Austin, TX). The initial slope of the fEPSP was measured and plotted versus time, with each point representing the average of five consecutive data points. Statistical significance between groups was performed with Student's t test. Significant differences were determined at p < 0.05. All data are presented as mean ± SEM. Only experiments with <5% change in the original baseline were included in the analysis. The magnitude of the LTD induced was measured at 30 min after agonist removal.
Western blot.
Acute hippocampal slices and chemical solutions were prepared as for electrophysiological recordings. After a 1 h recovery period (as for electrophysiological recordings), the slices were placed in an oxygenated bath (at 28–30°C) that contained either aCSF alone, or agonists with or without antagonists, conditions that will either induce or prevent induction of mLTD or NE LTD. Induction of both mLTD and NE LTD requires presynaptic activity (i.e., activation of CA3 axons) (Scheiderer et al., 2004, 2006). However, when the CA3 region remains intact in the slice, LTD is induced in the absence of electrical stimulation, because agonist application causes oscillatory activity in CA3 pyramidal cells.
To measure changes in extracellular signal-regulated protein kinase (ERK) phosphorylation during induction of LTD (i.e., during agonist application), it was necessary to immediately dissect the CA1 region from the slice after the 10 min exposure to agonist. The tissue was then homogenized in lysis buffer containing 50 mm HEPES pH 7.3, 150 mm NaCl, 1.5 mm MgCl2, 1 mm EGTA, 10% glycerol, 0.2 mm NaVO4, 100 mm NaF, 50 mm β-glycerophosphate, 1 mm dithiothreitol, 1 mm benzamidine, 0.01 mg/ml leupeptin, 0.1 mg/ml aprotonin, 0.5 μg/ml pepstatin A, and 1% Triton X-100. Samples containing 15 μg of protein were resolved on 10% SDS-PAGE and transferred to polyvinylidene difluoride membrane, blocked, and incubated with the following antibodies according to the manufacturer's protocol: phospho-p44/42 (ERK1/2, Thr202/Tyr204) E10, 1:3000 dilution; total p44/42 (ERK1/2), 1:3000 (Cell Signaling Technology, Danvers, MA). Blots were then incubated in HRP-conjugated secondary antibody (1:3000; Bio-Rad, Hercules, CA). Bands were detected using enhanced chemiluminescence (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Densitometric quantification of immunopositive bands was performed with ImageJ software (NIH). ERK activation was measured as an increase in phosphorylation of ERK2 (Vanhoose et al., 2002) and was normalized to total ERK2; treatments caused no change in total ERK levels. Statistical analysis was performed using Student's t test with significance determined at p < 0.05.
Chemicals.
All drugs used in both electrophysiological and Western blot experiments were made as 1000× stock solutions in DMSO or water and diluted immediately before use. All drugs were obtained from Sigma (St. Louis, MO), except CCh, which was acquired from Calbiochem (La Jolla, CA) and 3-(4-chlorophenyl) 1-(1,1-dimethylethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (PP2), acquired from Tocris Bioscience (Ellisville, MO), and the muscarinic receptor toxin 7 (MTx-7), acquired from Peptides International (Louisville, KY).
Results
mLTD and NE LTD are independent plasticities
We reported previously and show here in Figure 1, A and B, that a 10 min application of either CCh (50 μm), a broad spectrum cholinergic agonist, or Methox, an α1 adrenergic agonist (40 μm), induces LTD at CA3–CA1 synapses recorded in acutely prepared hippocampal slices (CCh: 76 ± 2% of baseline fEPSP slope, n = 7, p < 0.05; Methox: 87 ± 5% of baseline fEPSP slope, n = 4, p < 0.05); we have termed these plasticities mLTD and NE LTD (Scheiderer et al., 2006). Additionally, we have shown that mLTD requires activation of M1 mAChRs; it can be induced using the selective M1 receptor agonist McN-A-343 (50 μm) and prevented by the selective M1 toxin MTx-7 (100 nm) (Scheiderer et al., 2006). Note the larger transient depression occurring during CCh application compared with during application of Methox. Because CCh is a nonselective mAChR agonist, it activates additional, non-M1 mAChRs located presynaptically that decrease glutamate release but are not required for induction of LTD (Scheiderer et al., 2006); Methox only activates postsynaptically located α1 receptors involved in LTD.

mLTD does not require activation of adrenergic receptors, and NE LTD does not require activation of muscarinic receptors. A, Application of 50 μm CCh for 10 min results in LTD at hippocampal CA3–CA1 synapses measured in extracellular dendritic field potential recordings in acute slices prepared from 3- to 4-week-old rats (n = 7). B, Application of 40 μm Methox, a selective α1 agonist, for 10 min also induces LTD at these synapses (n = 4). C, mLTD induced with CCh (50 μm) is not altered by 10 μm prazosin, an α1 adrenergic receptor antagonist (n = 5). D, NE LTD induced with the Methox (40 μm) is not altered by 1 μm atropine, a broad spectrum muscarinic receptor antagonist (n = 5). Waveform traces are averages of 20 sweeps from 5 min before (thin line), during (dotted line), and 40 min after (thick line) agonist application. Error bars indicate SEM. Calibrations: 0.5 mV, 10 ms.
mLTD and NE LTD require presynaptic activity and activation of NMDARs for their induction (Scheiderer et al., 2004, 2006), M1 and α1 receptors couple to the same G-protein, Gαq (Porter et al., 2002; Hague et al., 2003) and ACh and NE modulate one another's release in vivo (Nilsson et al., 1992; Vizi and Kiss, 1998; Kiss et al., 1999). Therefore, given the strong potential for interaction, it is important to determine whether mLTD and NE LTD can be induced independently of one another. Perhaps only one form of LTD actually exists, and the other is induced indirectly as a consequence of either ACh or NE released in the slice after exposure to agonist. To confirm that mLTD and NE LTD are indeed separate plasticities, mLTD was induced (with CCh) in the presence of the α1 receptor antagonist prazosin (10 μm) and NE LTD was induced (using Methox) in the presence of the broad-spectrum muscarinic receptor antagonist atropine (1 μm). As shown, the magnitude of mLTD expressed is unaffected by blockade of α1 receptors with prazosin (Fig. 1C) (75 ± 1% of baseline fEPSP slope in the presence of prazosin, n = 6 vs 76 ± 2% of baseline fEPSP slope without prazosin, n = 7; p > 0.05). Likewise, NE LTD expression is not altered by blocking muscarinic receptors with the broad spectrum muscarinic receptor antagonist atropine (Fig. 1D) (78 ± 2% of baseline fEPSP slope in atropine, n = 5 vs 82 ± 3% of baseline fEPSP slope without atropine, n = 6; p > 0.05). These data clearly demonstrate that induction of mLTD occurs independently of α1 receptor activation and NE LTD induction occurs independently of M1 receptor activation.
mLTD and NE LTD induction require activation of ERK
Because M1 and α1 receptors are coupled to Gαq, it is likely that the signaling molecules required for induction of mLTD and NE LTD are the same. A signaling molecule downstream of Gαq-coupled receptors is the mitogen activated kinase, ERK. An increase in ERK phosphorylation is critical for induction of other forms of LTD at CA3–CA1 synapses, including metabotropic glutamate receptor (mGluR)-dependent LTD recorded in acute hippocampal slices (Gallagher et al., 2004) and low-frequency stimulation (LFS)-induced LTD recorded in vivo (Thiels et al., 2002). Therefore, we tested whether both mLTD and NE LTD require ERK activation. To prevent phosphorylation and activation of ERK, the mitogen-activated protein kinase kinase (MEK) inhibitor, 1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene (U0126) (20 μm), was bath applied during and after agonist application. Both mLTD and NE LTD were completely prevented (Fig. 2A,D) (mLTD: 101 ± 5% of baseline fEPSP slope in 20 μm U0126, n = 4 vs 74 ± 8% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 3; p < 0.05; NE LTD: 97 ± 4% of baseline fEPSP slope in 20 μm U0126, n = 7 vs 79 ± 6% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 6; p < 0.05). Furthermore, ERK activation is required during induction of mLTD or NE LTD because both plasticities were prevented when U0126 application was limited to 10 min before and during agonist application only (Fig. 2B,E) (mLTD: 112 ± 7% of baseline fEPSP slope in 20 μm U0126, n = 5 vs 81 ± 3% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 5; p < 0.01; NE LTD: 99 ± 1% of baseline fEPSP slope in 20 μm U0126, n = 4 vs 82 ± 4% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 5; p < 0.03). Interestingly, blocking mLTD induction with U0126 unmasked a significant potentiation of transmission (112 ± 7%; p < 0.01) that most likely is a consequence of activation of other mAChRs by the nonselective agonist CCh. Activation of M2 receptors have been shown to induce synaptic potentiation at CA3–CA1 synapses (Auerbach and Segal, 1996). A similar effect was not observed when NE LTD induction was prevented by U0126, probably because Methox activates α1 receptors, whose stimulation only induces LTD.

ERK activation is required for induction, but not expression, of mLTD and NE LTD. A, mLTD is prevented in the presence of the MEK inhibitor U0126 (20 μm; n = 4; p < 0.05). B, ERK activation is required during induction (red squares; n = 5; p < 0.01), but not expression of mLTD (n = 5; p > 0.05; black circles). C, CCh-induced increase in ERK2 phosphorylation (pERK2) is blocked by preincubation with the M1 antagonist, MTx-7 (100 nm; n = 3); *p < 0.02 vs control). D, NE LTD is prevented in the presence of U0126 (20 μm; n = 7; p < 0.05). E, ERK activation is required during NE LTD induction (n = 4; p < 0.03; red squares), but not during expression (n = 4; p > 0.05; black circles). F, Methox-induced increase in ERK2 phosphorylation is blocked by preincubation with the α1 antagonist prazosin (10 μm; n = 6; *p < 0.01 vs control). Phosphorylated ERK2 was normalized to total ERK2; treatments caused no change in total ERK2 levels. Error bars indicate SEM.
Next, we tested whether ERK activation is also required for expression of mLTD and NE LTD. The MEK inhibitor, U0126, was applied 15 min after agonist application, and the magnitude of neither mLTD nor NE LTD was affected (mLTD: 80 ± 3% of baseline fEPSP in 20 μm U0126, n = 5 vs 81 ± 3% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 5; p > 0.05; NE LTD: 84 ± 2% of baseline fEPSP slope in 20 μm U0126, n = 4 vs 82 ± 4% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 5; p > 0.05). Together, these data demonstrate that ERK activation is required for induction, but not expression, of both mLTD and NE LTD.
To confirm that ERK phosphorylation is elevated during agonist application to induce mLTD and NE LTD, Western blot analysis was performed on hippocampal CA1 regions after bath application of CCh (50 μm) or Methox (40 μm), respectively, using the same conditions as those to induce LTD in the electrophysiology experiments (see Materials and Methods). Note that in these studies, the primary band observed is ERK2 (p42), consistent with other reports (Thiels et al., 2002; Vanhoose et al., 2002; Topolnik et al., 2006) (therefore, only this band was analyzed). As shown in Figure 2C, ERK phosphorylation is significantly increased after treatment with CCh and this phosphorylation requires M1 receptor activation because it is blocked by preincubation with the M1 toxin MTx-7 (100 nm) (Fig. 2C) (ERK2 phosphorylation in CCh-treated slices, 250 ± 35% of control slices, n = 3, p < 0.02; CCh plus MTx-7-treated slices, 112 ± 21% of control slices, n = 3, p > 0.05). Activation of α1 receptors with Methox also significantly increases ERK phosphorylation, which is blocked by preincubation with the α1 antagonist prazosin (10 μm) (Fig. 2F) (ERK2 phosphorylation in Methox-treated slices, 279 ± 39% of control slices, n = 6, p < 0.01; Methox plus prazosin-treated slices, 145 ± 32% of control slices, n = 6, p > 0.05).
Neither mLTD nor NE LTD induction requires activation of the PLC signaling pathway
The canonical signaling pathway downstream of receptors coupled to Gαq is the phospholipase C (PLC) pathway, which increases IP3 and DAG, with subsequent release of Ca2+ from intracellular stores and activation of PKC, respectively. A further downstream consequence of this signaling pathway is activation of ERK. Therefore, induction of mLTD and NE LTD could require activation of the PLC pathway upstream of ERK activation.
Alternatively, previous studies demonstrate that M1 and α1 receptor activation, in addition to stimulating the PLC pathway, stimulates the Src family of tyrosine kinases (Lindquist et al., 2000; Rosenblum et al., 2000; Chan et al., 2005), which subsequently stimulates phosphorylation of ERK (Igishi and Gutkind, 1998; Berkeley et al., 2001; Vanhoose et al., 2002). Using specific inhibitors, we first tested whether mLTD and NE LTD require activation of the PLC pathway. The magnitude of neither mLTD nor NE LTD was affected by the PLC inhibitor 1-[6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,5-dione (U73122) (10 μm) (Fig. 3A,B) (mLTD: 70 ± 5% of baseline fEPSP slope in 10 μm U73122, n = 5 vs 71 ± 4% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 4; p > 0.05; NE LTD: 90 ± 4% of baseline fEPSP slope in 10 μm U73122, n = 5 vs 89 ± 3% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 3; p > 0.05). These findings imply that PLC, as well as signaling molecules downstream of PLC activation, are not required for mLTD or NE LTD induction. However, as an additional test of the PLC pathway, we investigated the impact of blocking PKC activation using the PKC inhibitor chelerythrine, and depleting intracellular calcium stores with thapsigargin (10 μm) and cyclopiazonic acid (CPA) (10 μm) on the magnitude of mLTD. Neither inhibiting PKC nor depleting intracellular Ca2+ stores had any effect on the magnitude of mLTD [72 ± 5% of baseline fEPSP slope in 10 μm chelerythrine, n = 5 vs 75 ± 6% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 5, p > 0.05; 79 ± 4% of baseline fEPSP slope in 10 μm thapsigargin, n = 5 vs 69 ± 1% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 3, p > 0.05; 73 ± 3% of baseline fEPSP slope in 10 μm CPA, n = 5 vs 71 ± 3% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 4, p > 0.05 (supplemental Fig. 1A–C, available at www.jneurosci.org as supplemental material)]. These data demonstrate that mLTD does not require activation of PLC, PKC, or release of Ca2+ from intracellular stores. Given these results and because NE LTD was also unaffected by blocking PLC, we did not pursue the roles of PKC or intracellular stores in induction of NE LTD.
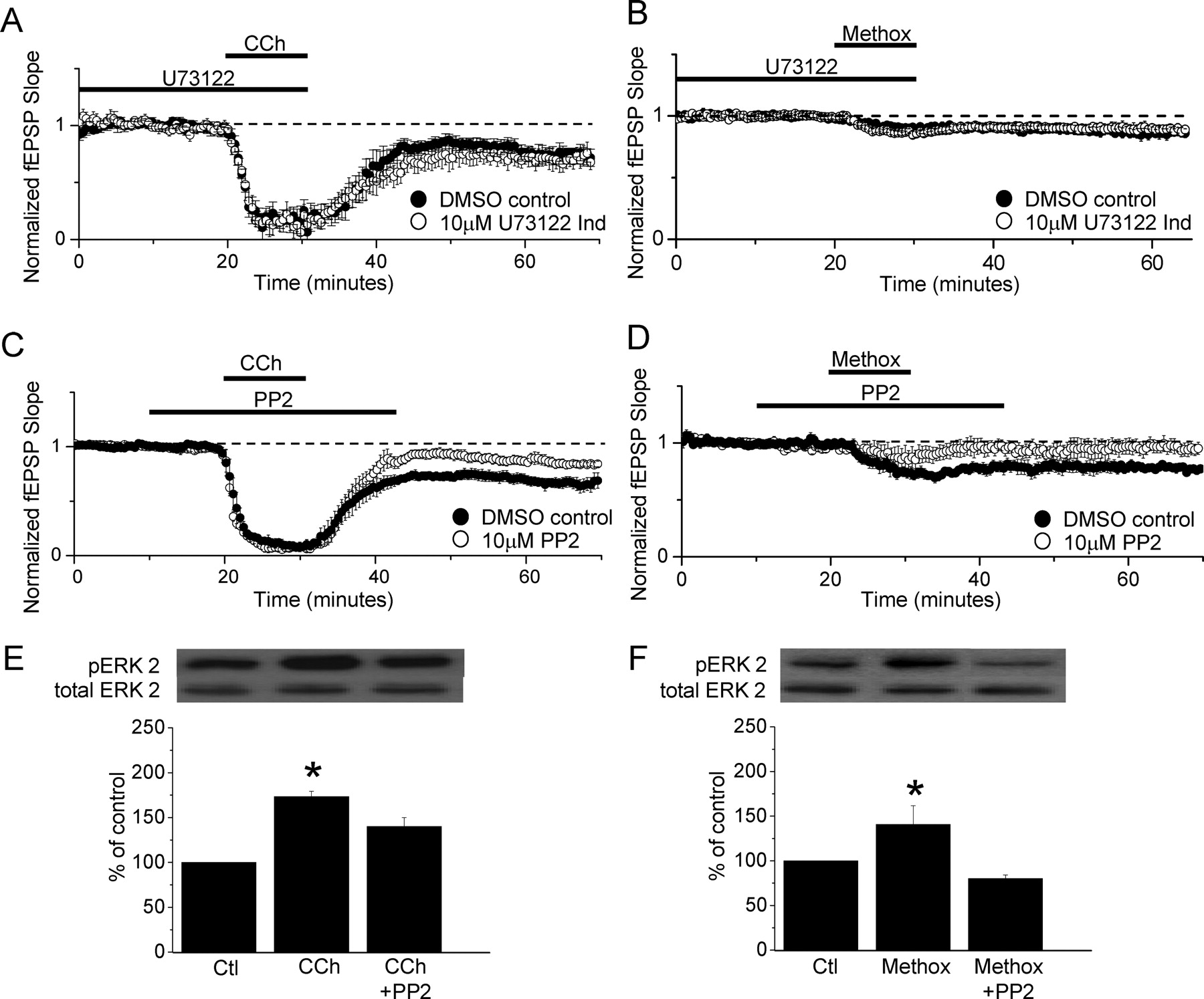
Activation of the Src kinase family, but not the PLC pathway, is required for induction of mLTD and NE LTD and increase in pERK. A, mLTD induction is unaffected by blockade of PLC activation using the PLC inhibitor U73122 (10 μm; n = 5 in U73122; n = 4 interleaved control slices; p > 0.05). B, NE LTD induction is unaffected by the PLC inhibitor U73122 (10 μm; n = 5 in U73122; n = 3 interleaved control slices; p > 0.05). C, mLTD is prevented by the Src kinase inhibitor PP2 (10 μm; n = 5; p < 0.01). D, NE LTD is also prevented by the Src kinase inhibitor PP2 (10 μm; n = 5; p < 0.02). All statistical comparisons are made to the amount of LTD induced in interleaved DMSO-treated control slices. E, CCh-induced increase in ERK2 phosphorylation (pERK2) is significantly decreased by the Src kinase inhibitor, PP2 (n = 4; *p < 0.001 vs control; CCh + PP2, p < 0.01 vs control; CCh vs CCh + PP2, p < 0.03). F, Methox-induced increase in ERK2 phosphorylation is significantly decreased by the Src kinase inhibitor PP2 (n = 4; *p < 0.05 vs control; Methox + PP2, p < 0.01 vs control; Methox vs Methox + PP2, p < 0.01). Error bars indicate SEM.
Both mLTD and NE LTD require activation of the Src family of tyrosine kinases
Because inhibiting the PLC signaling pathway had no effect on either mLTD or NE LTD, we next investigated the potential role of the Src family of kinases that are stimulated after activation of Gαq-coupled receptors and increase ERK phosphorylation. As shown in Figure 3C, the Src kinase inhibitor PP2 (10 μm) significantly inhibits mLTD (89 ± 4% of baseline fEPSP slope in PP2, n = 5 vs 70 ± 4% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 5; p < 0.01). We next tested whether the same is true for NE LTD. Indeed, NE LTD is prevented in the presence of PP2 (Fig. 3D) (100 ± 6% of baseline fEPSP slope in PP2, n = 5 vs 79 ± 3% of baseline fEPSP slope in interleaved DMSO-treated control slices, n = 5; p < 0.02).
Other investigators have shown that ERK phosphorylation occurs subsequent to Src activation (Peavy et al., 2001). To confirm that Src is upstream of ERK phosphorylation after M1 and α1 receptor activation, Western blot analysis of ERK phosphorylation was performed on hippocampal slices treated with either CCh (50 μm) or methoxamine (40 μm) in the presence or absence of the Src inhibitor PP2 (10 μm) (Fig. 3E,F, respectively). We show that both CCh and methoxamine induced ERK phosphorylation is significantly attenuate by the Src inhibitor PP2 (CCh: 173 ± 6% of control, p < 0.001 vs control; CCh plus PP2: 140 ± 10%, p < 0.01 vs control, p < 0.03 vs CCh; n = 4 for all groups; Methox: 140 ± 21%, p < 0.05 vs control; Methox plus PP2: 80 ± 4%, p < 0.01 vs control, p < 0.01 vs Methox; n = 4 for all groups). Together, these data demonstrate that the Src activation is upstream of ERK phosphorylation and that mLTD and NE LTD have similar requirements for the Src family of tyrosine kinases and ERK activation.
Strong coactivation of M1 and α1 receptors does not induce LTD of larger magnitude than that induced when each receptor is stimulated separately
Because the above findings indicate that mLTD and NE LTD require the same intracellular signaling cascade for their induction, we hypothesized that strong activation of one receptor type might maximally activate the mechanisms underlying the LTD during that round of agonist application, such that strong, simultaneous activation of both receptor types should induce LTD of no greater magnitude. This was tested by determining whether coapplication of CCh (50 μm) and Methox (40 μm) could induce LTD of greater magnitude than that elicited by either agonist applied alone. As anticipated, coapplication of 50 μm CCh and 40 μm Methox induce LTD of the same magnitude as when each agonist is applied individually to induce LTD (Fig. 4) (82 ± 3% of baseline fEPSP slope with CCh alone, n = 4; 85 ± 3% of baseline fEPSP slope with Methox alone, n = 3; 82 ± 4% of baseline fEPSP slope when agonists are coapplied, n = 6; p < 0.05, ANOVA). This confirms that strong stimulation of either M1 or α1 receptors induces a maximal amount of LTD that can be elicited, at least by a single agonist application, and that these concentrations (50 μm CCh and 40 μm Methox) maximally activate a shared mechanism underlying LTD. This is further supported by results from experiments in which even higher agonist concentrations were used (200 μm CCh and 200 μm Methox, each applied alone), and no greater LTD was induced than 50 and 40 μm, respectively (CCh: 72 ± 4% of baseline fEPSP slope with 200 μm CCh, n = 5; 73 ± 3% of baseline fEPSP slope with 50 μm CCh, n = 5; p > 0.05; Methox: 84 ± 4% of baseline fEPSP slope with 200 μm Methox, n = 4; 87 ± 3% of baseline fEPSP slope with 40 μm Methox, n = 4; p > 0.05) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The lack of an additive effect of receptor coactivation on the magnitude of LTD suggests the possibility that strong activation of a single receptor type saturates the available Src kinase–ERK signaling cascade, limiting the magnitude of LTD that can be induced by a single round of agonist application. Alternatively, a shared mechanism downstream of ERK activation could also limit the magnitude of LTD expressed after a single agonist application that strongly activates the receptor.

Strong coactivation of M1 and α1 receptors does not induce LTD of greater magnitude. A, Waveform traces are averages of 20 sweeps from 5 min before (thin line) and 40 min after (thick line) agonist application. B, CCh (50 μm) and Methox (40 μm) do not induce a greater magnitude of LTD when coapplied (n = 6) than when applied separately (CCh, n = 4; Methox, n = 3). Error bars indicate SEM. Calibration: 0.5 mV, 10 ms.
Coapplication of low concentrations of M1 and α1 receptor agonists induces LTD
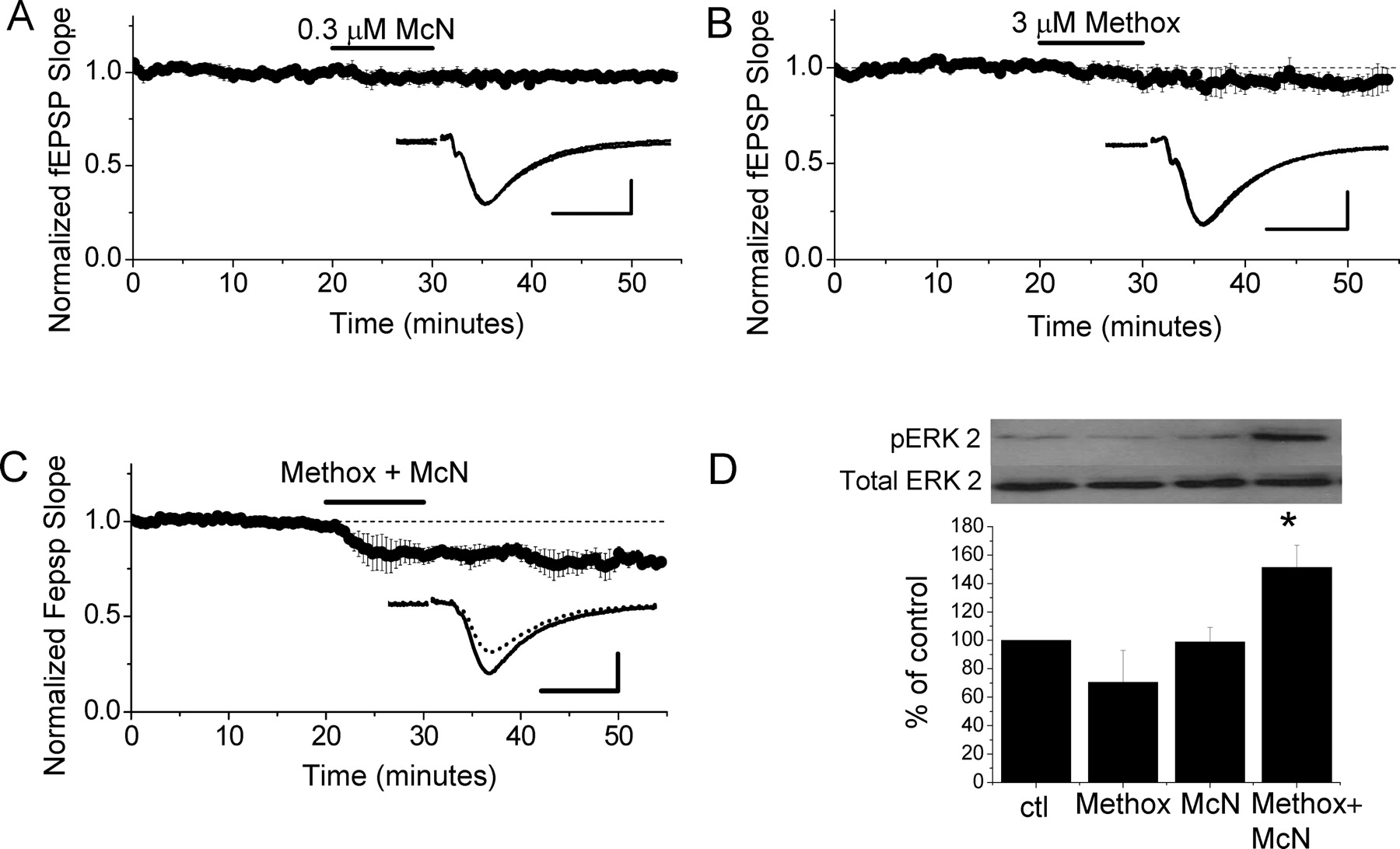
Because our data indicate that M1 and α1 receptors couple to the same signaling pathway to induce LTD, we hypothesized that simultaneous activation of weakly stimulated M1 and α1 receptors, using lower agonist concentrations that do not induce LTD when applied individually, might cause summed ERK activation and induce LTD. As shown in Figure 5A, selective activation of M1 receptors with 0.3 μm McN-A-343 does not induce LTD (98 ± 3% of baseline fEPSP slope; n = 4; p > 0.05). Similarly, application of 3 μm Methox to activate α1 receptors also does not induce a significant amount of depression (Fig. 5B) (94 ± 5% of baseline fEPSP slope; n = 5; p > 0.05). However, coapplication of the receptor selective agonists at these concentrations clearly induces LTD (Fig. 5C) (79 ± 3% of baseline fEPSP slope; n = 5; p > 0.05). Surprisingly, the magnitude of the LTD induced by M1 and α1 receptor coactivation is the same as that induced with either 50 μm CCh or 40 μm Methox when applied individually to induce LTD (p > 0.05) (Fig. 1). These findings suggest that the Src kinase–ERK signaling pathway downstream of M1 and α1 receptor activation must reach a threshold level of stimulation for LTD to be induced, and that this threshold can be reached either by summed activation of weakly stimulated M1 and α1 receptors or with strong stimulation of M1 or α1 receptors alone. Furthermore, once the threshold is reached, a maximum amount of LTD is induced for that round of agonist application.

Coapplication of low concentrations of selective M1 and α1 receptor agonists induces LTD. A, Application of 0.3 μm McN-A-343 (McN), a selective M1 receptor agonist, does not induce mLTD (n = 4). B, Application of 3 μm Methox does not induce NE LTD (n = 5). C, Coapplication of 0.3 μm McN and 3 μm Methox induces LTD (n = 5). Waveform traces are averages of 20 sweeps from 5 min before (solid line) and 30 min after (dashed line) agonist application (A–C, insets). Calibrations: 0.5 mV, 10 ms. D, Application of either 0.3 μm McN or 3 μm Methox alone does not increase ERK2 phosphorylation (pERK2), but coapplication of the two agonists significantly increases ERK2 phosphorylation (n = 4; *p < 0.02 vs control). Phosphorylated ERK2 was normalized to total ERK2; treatments caused no change in total ERK2 levels.
To further test whether a threshold level of ERK phosphorylation is required to induce mLTD or NE LTD, Western blot analysis was used. Consistent with the electrophysiological data, treatment of hippocampal slices with either 0.3 μm McN-A-343 or 3 μm Methox alone fails to increase ERK phosphorylation; however, coapplication of the two agonists does significantly increase ERK phosphorylation (Fig. 5D) (ERK2 phosphorylation in Methox-treated slices, 70 ± 22% of interleaved control slices, n = 4, p > 0.05; McN-A-343-treated slices, 99 ± 10% of interleaved control slices, n = 4, p > 0.05; Methox plus McN-A-343-treated slices, 151 ± 16% of interleaved control slices, n = 4, p < 0.02). Interestingly, whereas the magnitude of LTD induced by combining weak stimulation of both M1 and α1 receptors is the same as that induced with either 50 μm CCh or 40 μm methoxamine alone (compare Figs. 1A,B, 5C), the magnitude of ERK phosphorylation, when weakly stimulating both receptors, appears to be lower than when strongly stimulating each receptor type individually (compare Figs. 2C–F, 5D). This suggests that the amount of ERK2 phosphorylation actually required for LTD induction is less than the amount phosphorylated by 50 μm CCh or 40 μm Methox, suggesting that the mechanism limiting the LTD magnitude (with strong receptor stimulation) is downstream of ERK activation.
The model shown in Figure 6 illustrates potential modulation of synaptic strength after activation of M1 and α1 receptors. Consistent with our data, weak M1 receptor (Fig 6A) or weak α1 receptor activation (B) is insufficient to stimulate Src kinase, phosphorylate ERK, and induce LTD. However, when combined, weak stimulation at both receptors sufficiently increases ERK phosphorylation, leading to induction of LTD. The LTD induced is of the same magnitude as that induced with either strong M1 or α1 receptor activation. This finding indicates that concurrent activation of two Gαq-coupled receptors can stimulate a threshold level of ERK activation to induce LTD, which may be physiologically important to allow induction of LTD in vivo, even when ACh and NE levels are low.

Proposed model of mLTD and NE LTD interaction. A, B, Our data demonstrates that a weak M1 receptor (A) or a weak α1 receptor (B) activation is insufficient to activate Src kinase, phosphorylate ERK, and induce LTD. C–E, However, when combined, weak stimulation at both receptors (C) sufficiently activates Src, which phosphorylates ERK and induces LTD at the same magnitude as that induced with strong M1 or α1 receptor stimulation alone (D, E). F, However, a combined strong activation of M1 and α1 receptor activation does not induce LTD of a greater magnitude than that observed with either agonist when applied alone, demonstrating that maximal activation of one receptor saturates the mechanisms required for LTD such that LTD induced by activation of the other receptor is occluded.
Discussion
The major finding of our study is that combined weak activation of M1 and α1 receptors can elicit summed activation of ERK to induce LTD. Although both the cholinergic and adrenergic systems are known to be critical for normal hippocampal learning and memory, this is the first demonstration that the two neurotransmitter systems can interact to induce hippocampal long-term synaptic plasticity.
Src kinase and ERK signaling during mLTD and NE LTD
Our finding that mLTD and NE LTD require ERK activation is consistent with previous data demonstrating that activation of M1 receptors (Berkeley, 2001; Berkeley et al., 2001; Chan et al., 2005) and α1 receptors (Vanhoose et al., 2002) can induce an increase in ERK phosphorylation in hippocampal pyramidal neurons. Several studies show that Gαq-coupled receptors can increase ERK phosphorylation after activation of the Src kinase family (Igishi and Gutkind, 1998; Peavy et al., 2001). Accordingly, activation of both M1 and α1 receptors stimulates the Src family of kinases, which subsequently leads to an increase in ERK phosphorylation (Lindquist et al., 2000; Rosenblum et al., 2000; Chan et al., 2005). Activation of Src kinases downstream of ERK phosphorylation has not been reported.
Signaling through ERK now appears to be a common mechanism underlying the induction of several forms of hippocampal synaptic plasticity, including NMDAR-dependent LFS-LTD (Thiels et al., 2002), mGluR LTD (Gallagher et al., 2004), high-frequency stimulation-induced LTP (HFS-LTP) (English and Sweatt, 1997), and theta burst stimulation-induced LTP (Winder et al., 1999). Although Src has been implicated in fewer hippocampal plasticities, its activation is required for HFS-LTP (Lu et al., 1998; Salter, 1998). Interestingly, agonist induced mGluR LTD, which can be blocked by simultaneously inhibiting mGluR1 and mGluR5 receptors (Volk et al., 2006), requires ERK activation, which does not require the PLC pathway (Fitzjohn et al., 2001; Schnabel et al., 2001; Gallagher et al., 2004), and it was reported previously that neuronal mGluR5 activation of ERK requires Src (Mao et al., 2005). Thus, activation of Src kinase and ERK appears to be a mechanism shared by several different Gαq-coupled receptors to induce multiple forms of long-term synaptic plasticity.
Cooperative action of muscarinic and noradrenergic agonists to induce LTD
The agonist concentrations used in our induction protocols (50 μm CCh and 40 μm Methox) appear to induce a maximum amount of LTD that can be elicited by a single application of agonist. This interpretation is supported by our data where the magnitudes of mLTD and NE LTD induced by a single agonist application are not different from one another; neither are their magnitudes increased by application of even higher agonist concentrations (200 μm) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), nor by simultaneous application of both agonists. In addition, coapplication of both agonists at concentrations too low to induce LTD when applied separately induces LTD, and this LTD is equal in magnitude to that induced by either agonist applied alone (at higher concentrations). These data indicate that there is a threshold level of ERK activation required for LTD induction. Furthermore, increasing phosphorylation of ERK past this threshold does not necessarily increase the magnitude of LTD, because weak stimulation of both receptors does not increase ERK phosphorylation as robustly as stronger stimulation of either receptor alone, although all three of these protocols induce equal amounts of LTD.
This report is the first demonstration that combined subthreshold stimulation of M1 and α1 receptors can significantly increase phosphorylation of ERK, leading to LTD at hippocampal synapses. In other studies, it has been reported that coapplication of a low concentration (200 nm) of CCh and the β adrenergic receptor agonist isoproterenol enhances induction of hippocampal LTP in response to brief 5 Hz electrical stimulation in an ERK-dependent manner, although application of either agonist alone does not (Watabe et al., 2000). Similarly, ACh and NE can act synergistically to increase the probability of electrically inducing LTP in visual cortex (Brocher et al., 1992). Furthermore, in the prefrontal cortex, simultaneous activation of group I and II mGluRs and dopamine receptors induces LTD by additively increasing ERK phosphorylation, whereas stimulation of either receptor alone does not (Otani et al., 1999). Together with the present report, these results suggest a role for ERK as a coincidence detector such that simultaneous low-level stimulation by multiple neurotransmitters acting at several similarly coupled G-protein-coupled receptors, as is likely to occur endogenously, can converge to induce synaptic plasticity.
The convergence of multiple receptor systems involved in hippocampal plasticity on common signaling mechanisms may also play a compensatory role in aging and neurodegenerative diseases by providing more than one way to induce plasticity. Interestingly, it has been demonstrated that cholinesterase inhibitors used to treat Alzheimer's disease increase release of hippocampal NE in vivo (Hatip-Al-Khatib et al., 2005; Shearman et al., 2006) and, conversely, inhibition of NE uptake increases hippocampal ACh release in vivo, and improves performance on memory tasks (Tzavara et al., 2006). Although further investigation is necessary to determine whether mLTD and NE LTD play a role in these effects, the similarity of these two forms of plasticity suggests that a decrease of either ACh or NE (or their receptors) could be compensated to some extent by the other system.
These data demonstrate that the cholinergic and adrenergic systems can act in concert to produce synaptic plasticity, and highlight the fact that it is difficult to examine long-term synaptic plasticity induced by one neurotransmitter without consideration of other transmitters that may be released concomitantly. This seems especially important considering the demonstration that even low agonist concentrations can exert a functional effect when presented simultaneously. Interestingly, ACh and NE levels are both high during active wakefulness, when new memories are encoded, and low during slow-wave sleep (Hobson et al., 1975; Kafka et al., 1986; Aston-Jones et al., 1991; Hasselmo, 1999). It is thought that during slow-wave sleep, lowered hippocampal ACh optimizes memory consolidation by releasing suppression of excitatory feedback connections (Kafka et al., 1986; Hasselmo, 1999). However, during REM sleep, when ACh is elevated in hippocampus, but both ACh and NE levels are low in cortex, activity is allowed to spread in cortex without hippocampal interference (Kafka et al., 1986; Dodt et al., 1991; Jones, 1991; Hasselmo et al., 1997; Hasselmo, 1999). Thus, hippocampal mLTD and NE LTD could potentially contribute to memory encoding, to memory consolidation during sleep, or both. The ability of ACh and NE to activate the same signaling pathway to induce LTD, either individually when present in relatively high concentrations or by acting synergistically at lower concentrations, may provide a way to effect synaptic depression in hippocampus even as ACh and NE levels fluctuate across the sleep–wake cycle.
Footnotes
-
This work was supported by National Institutes of Health (NIH)–National Institute on Aging Award AG21612 (L.L.M.), NIH National Research Service Award NS045469 (C.L.S.), NIH Award P50AG16582 to the University of Alabama at Birmingham (UAB) Alzheimer's Disease Research Center, NIH Neuroscience Blueprint Core Grant NS57098 (UAB), and NIH–National Institute of Diabetes and Digestive and Kidney Diseases T32 Training Grant Award DK007545 (UAB). We thank Lindsey Vedder for her contribution to this work.
- Correspondence should be addressed to Dr. Lori L. McMahon, University of Alabama at Birmingham, 1918 University Boulevard, MCLM 964, Birmingham, AL 35294-0005. mcmahon{at}physiology.uab.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}